
Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
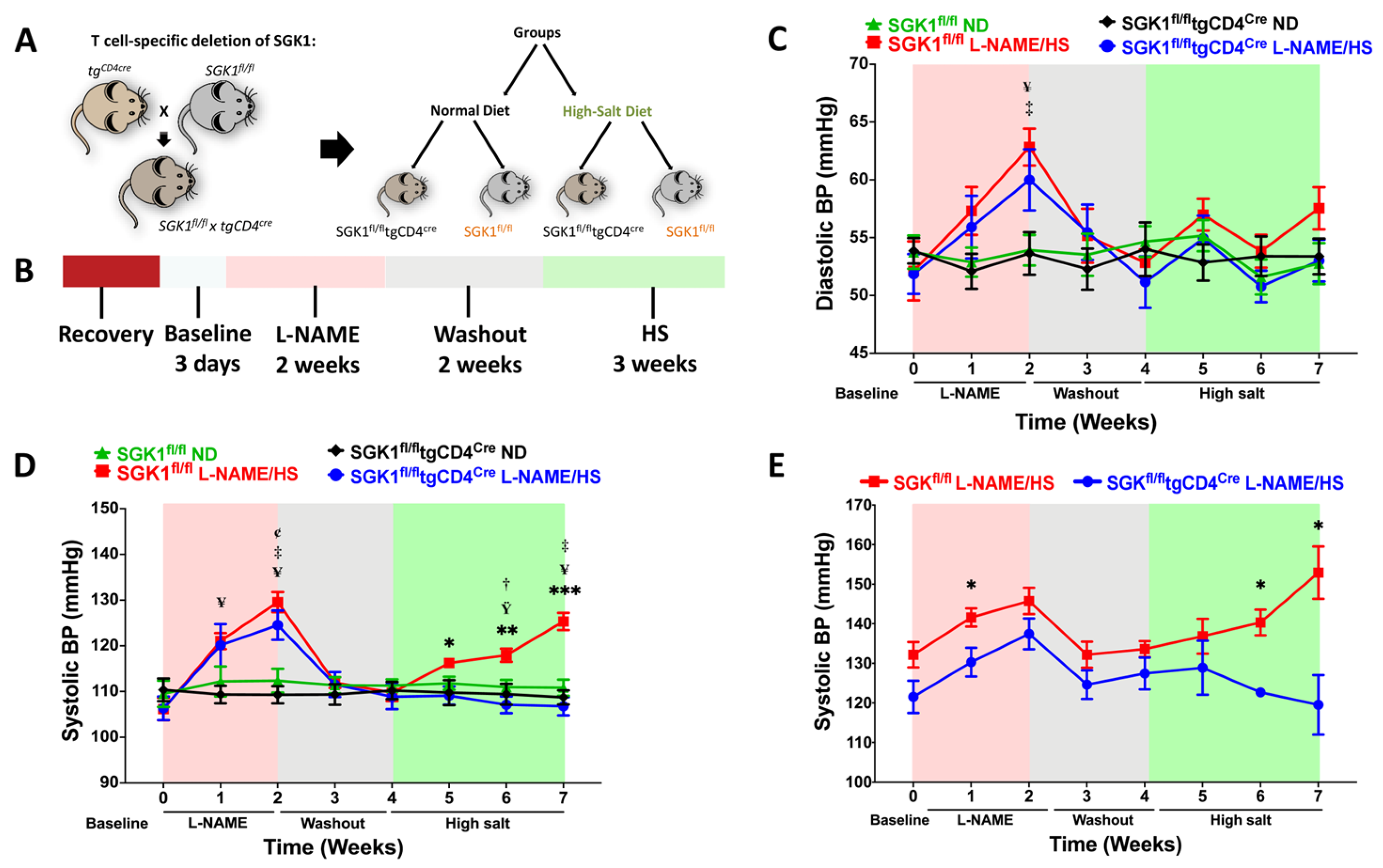
2.1. Specific Deletion of T Cell SGK1 Reduces L-NAME/HS-Induced Hypertension
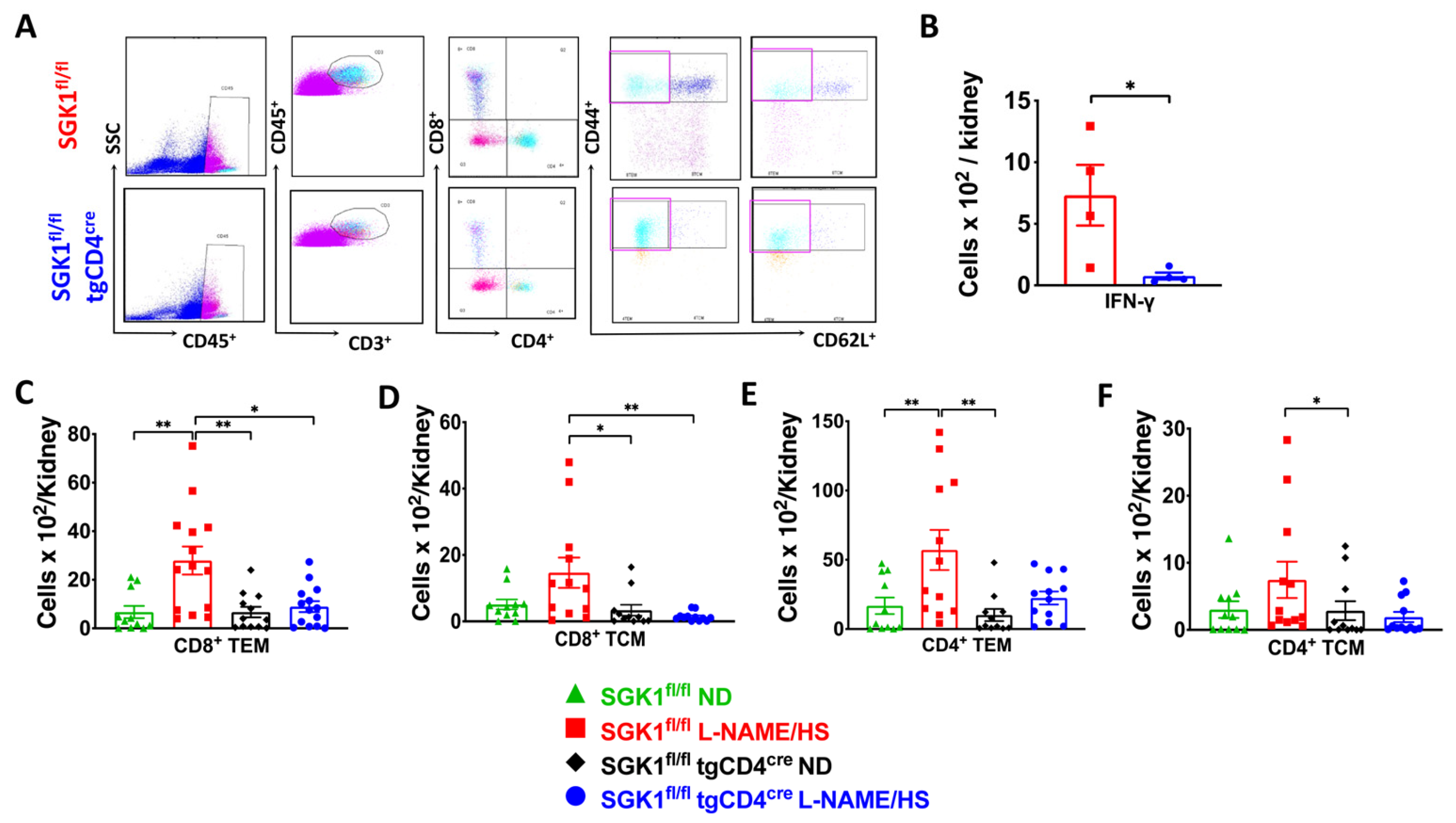
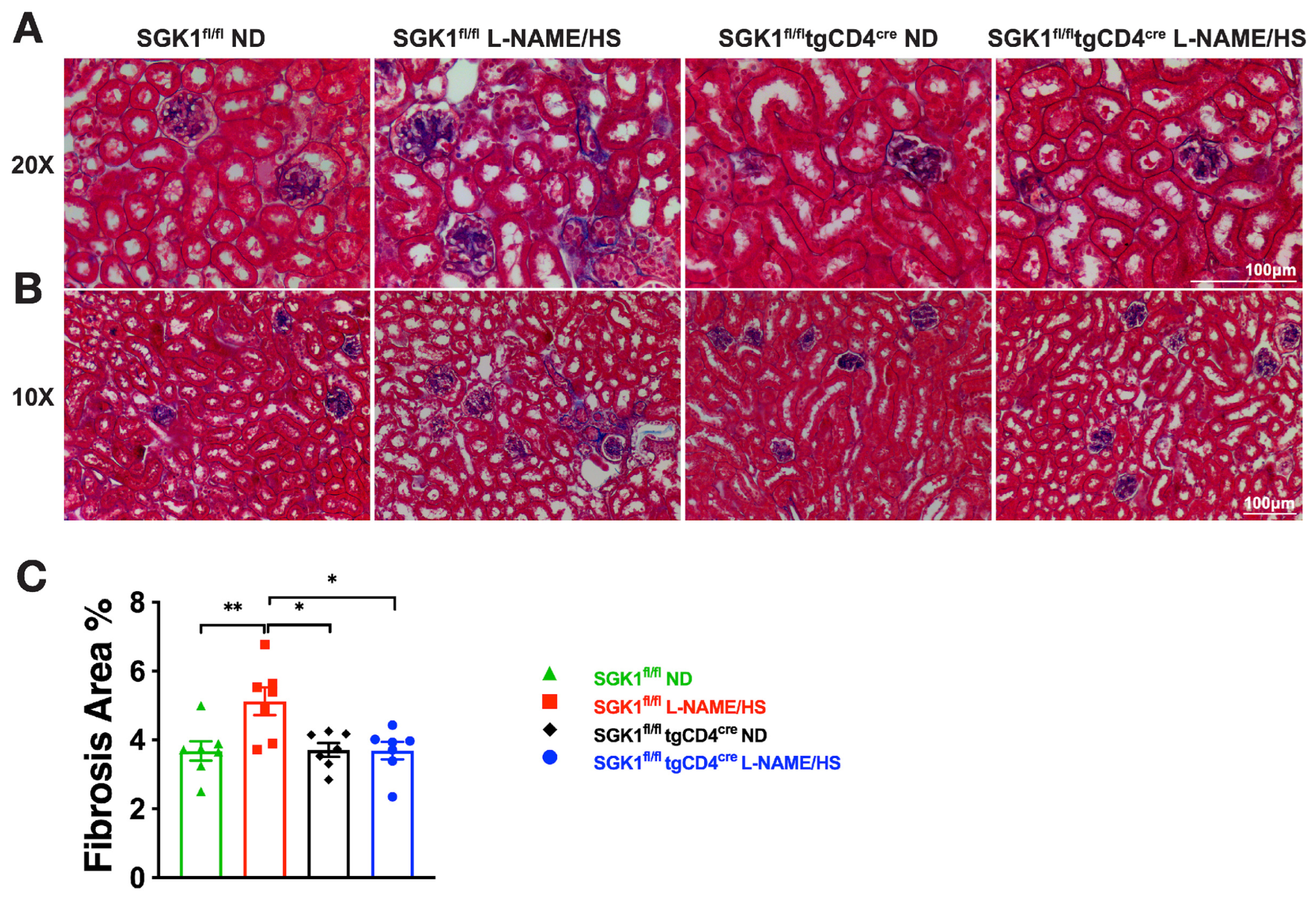
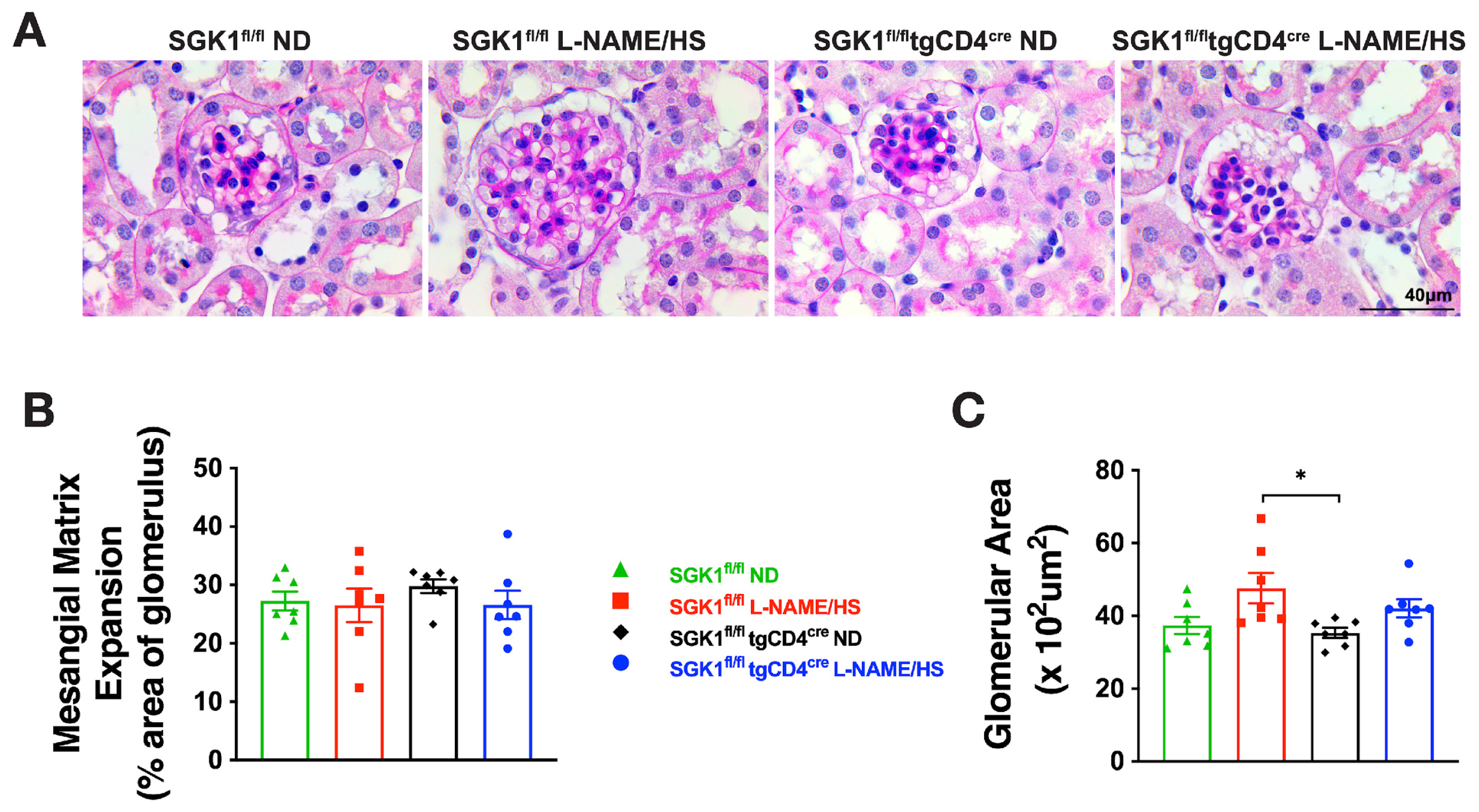
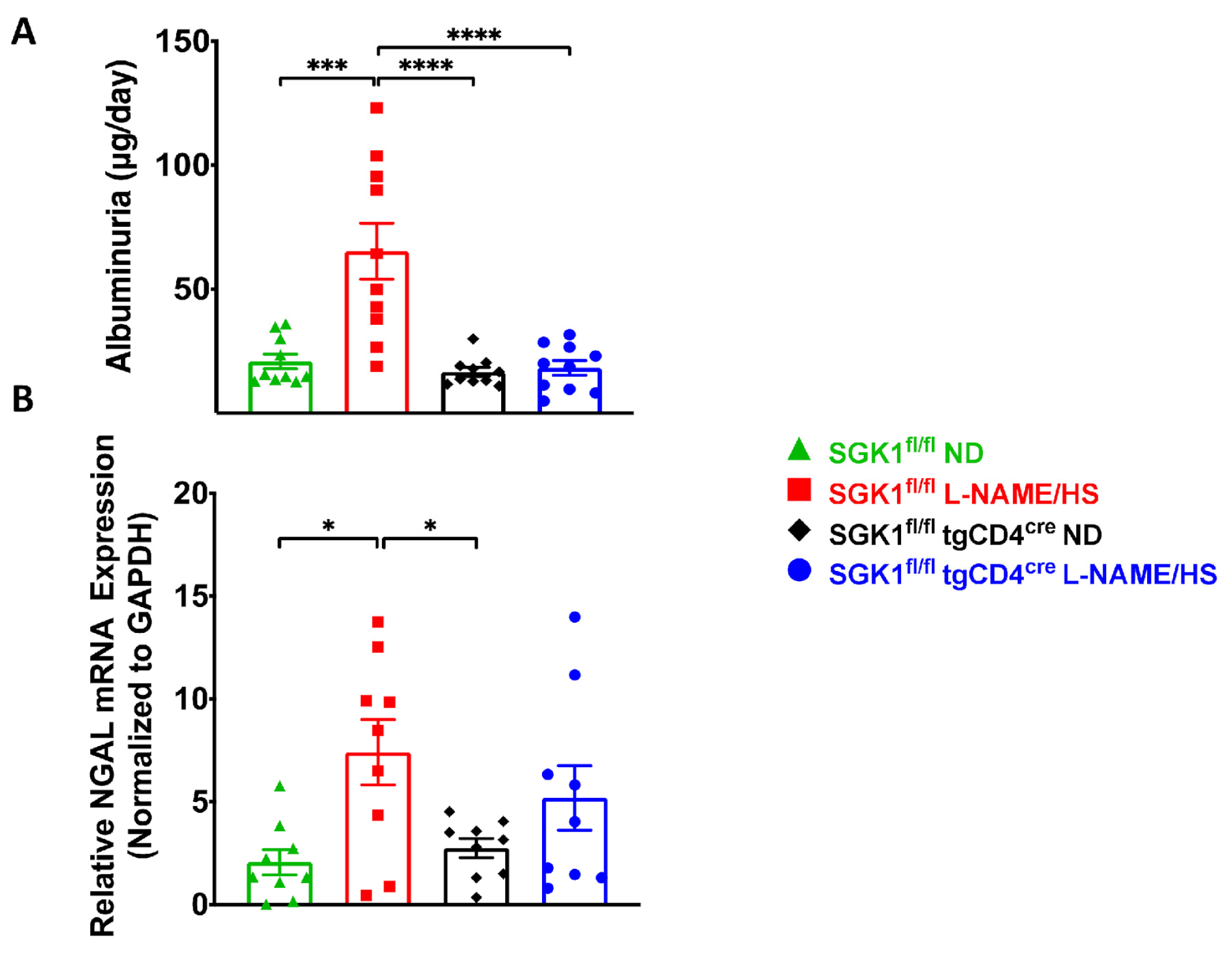
2.2. SGK1 Mediates Renal Inflammation and Promotes Renal Injury
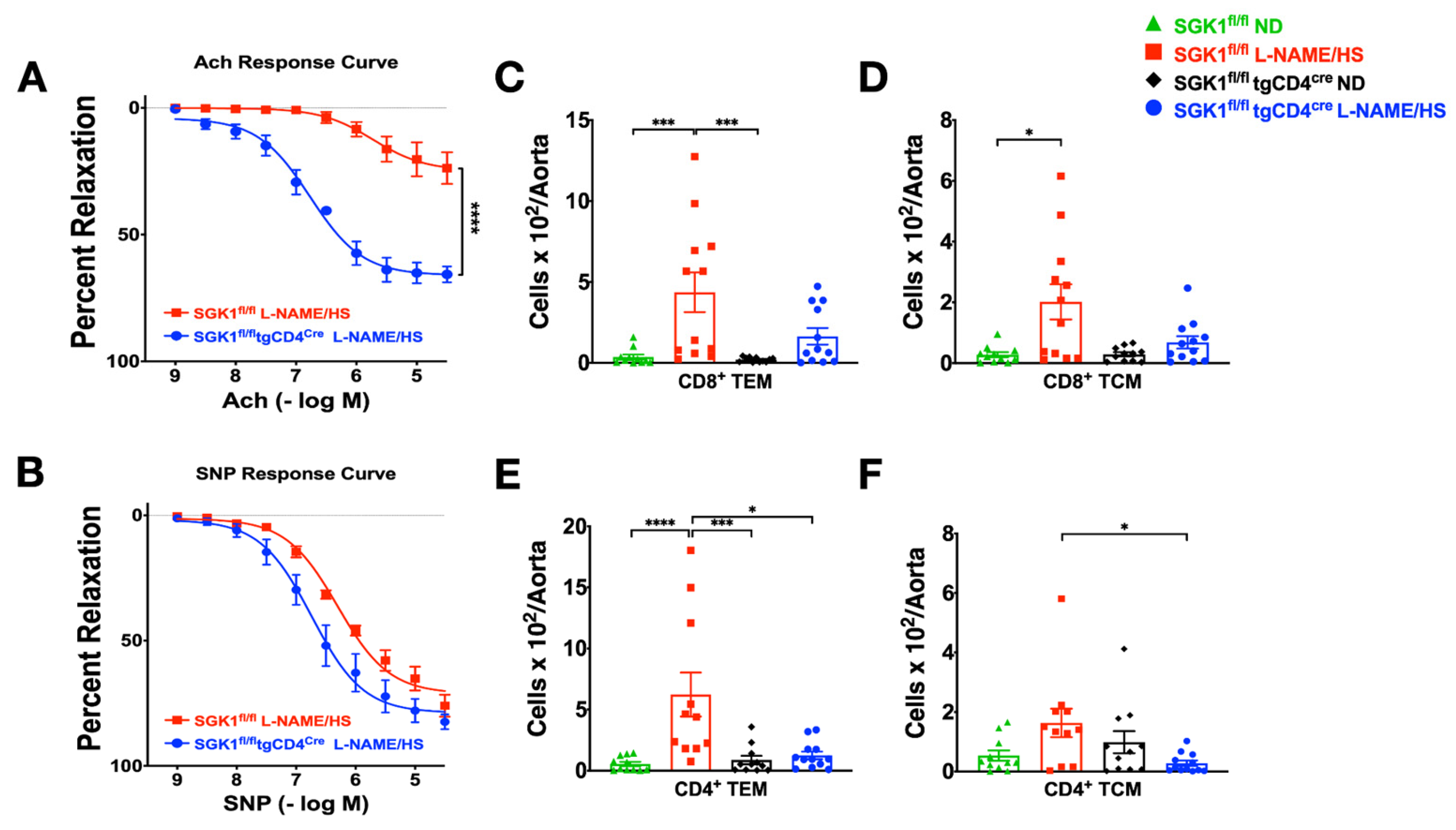
2.3. T Cell SGK1 Deficiency Protects against Vascular Inflammation and Injury
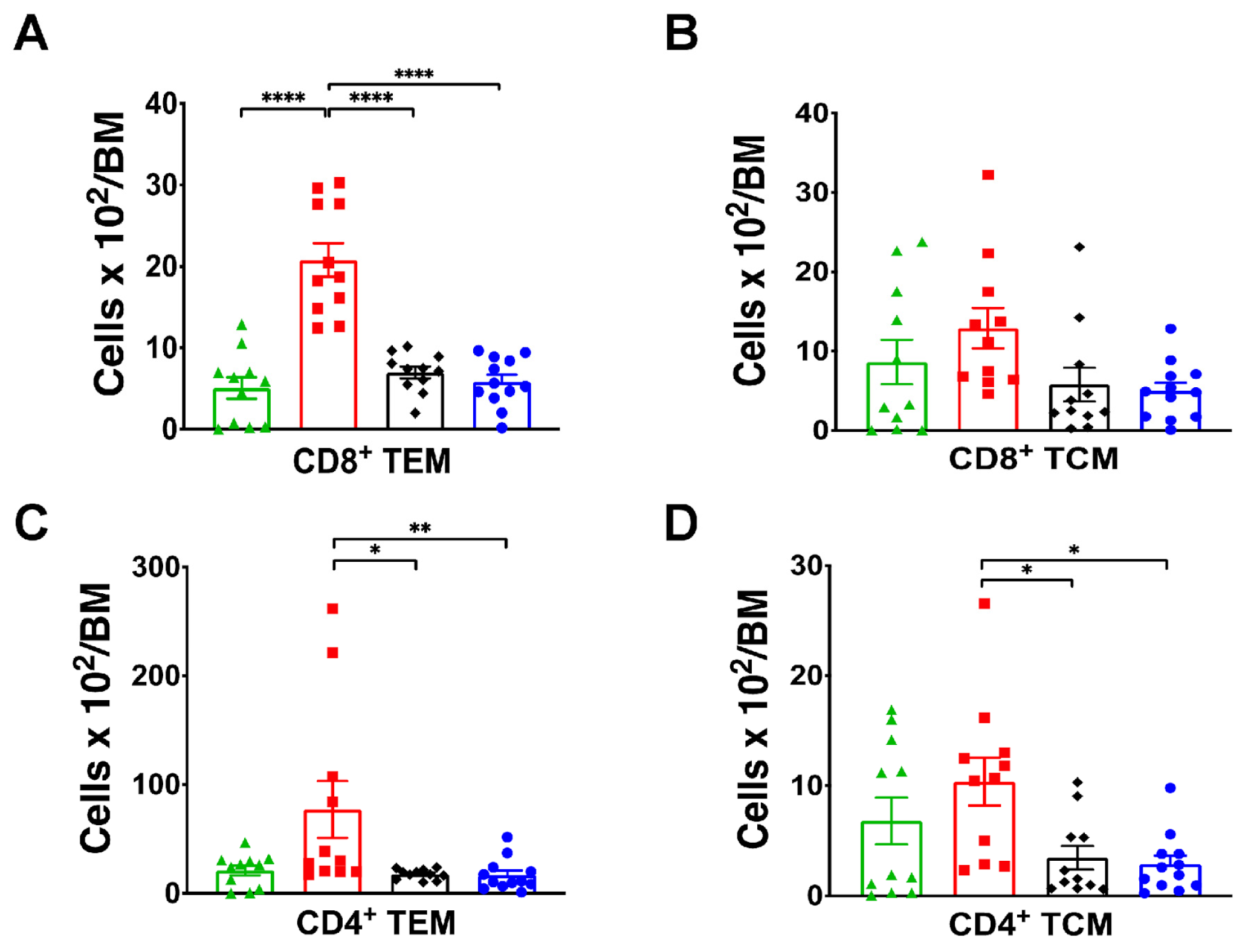
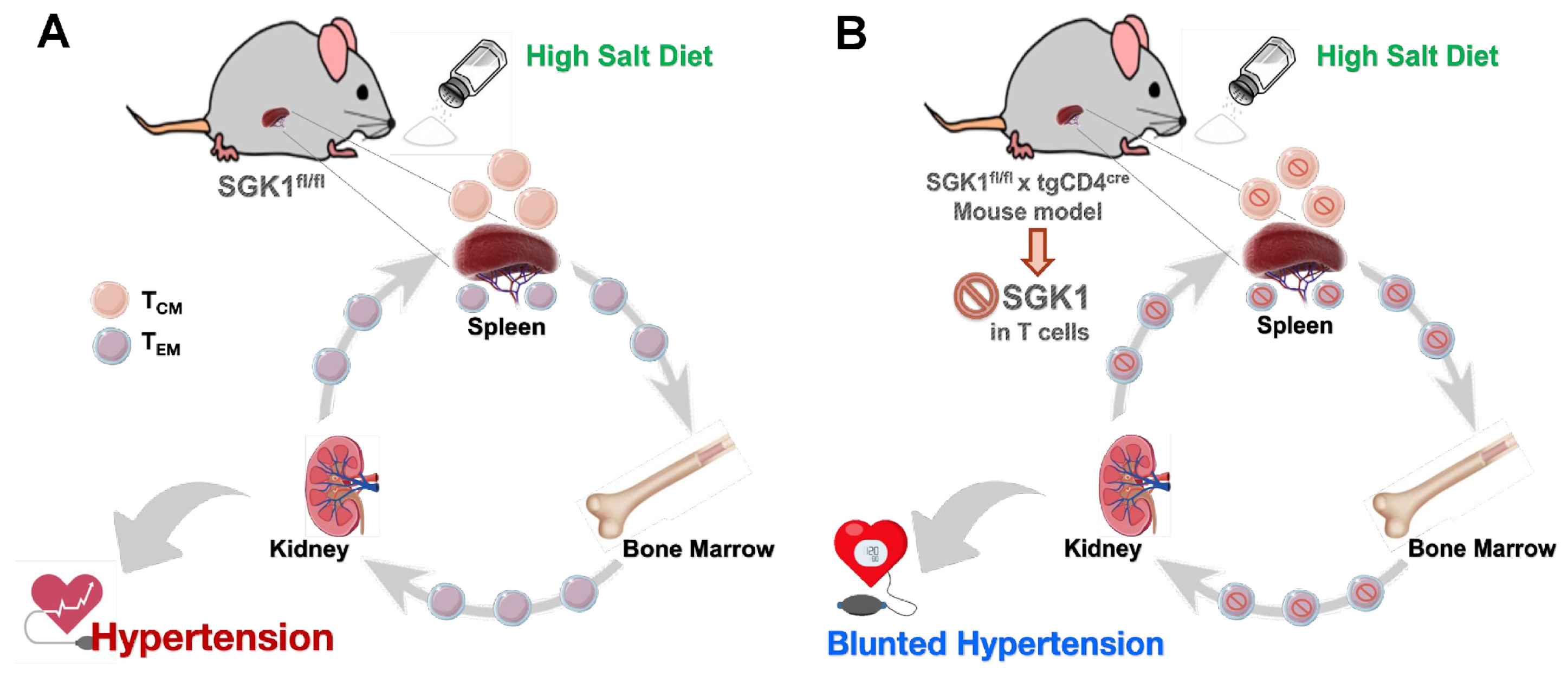
2.4. SGK1 Is Necessary for Bone Marrow Memory T Cells Accumulation
2.5. Adoptive Transfer of Bone Marrow TEM Cells from SGK1fl/fl Mice Promotes Salt Sensitivity
3. Discussion
4. Materials and Methods
4.1. Animals and Study Design
4.2. Blood Pressure Measurement
4.3. Flow Cytometry Analysis of Immune Cells
4.4. Mesenteric Vascular Reactivity
4.5. Measurements of Renal Injury Markers
Neutrophil Gelatinase-Associated Lipocalin (NGAL) mRNA Quantification
4.6. Kidney Histology
4.7. T Cells Adoptive Transfer Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Carretero, O.A.; Oparil, S. Essential hypertension: Part II: Treatment. Circulation 2000, 101, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, 1269–1324. [Google Scholar] [CrossRef] [PubMed]
- Ostchega, Y.; Fryar, C.D.; Nwankwo, T.; Nguyen, D.T. Hypertension Prevalence among Adults Aged 18 and over: United States, 2017–2018; NCHS Data Brief; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2020; pp. 1–8.
- Elijovich, F.; Weinberger, M.H.; Anderson, C.A.; Appel, L.J.; Bursztyn, M.; Cook, N.R.; Dart, R.A.; Newton-Cheh, C.H.; Sacks, F.M.; Laffer, C.L. Salt sensitivity of blood pressure: A scientific statement from the American Heart Association. Hypertension 2016, 68, e7–e46. [Google Scholar] [CrossRef] [PubMed]
- He, F.J.; Li, J.; Macgregor, G.A. Effect of longer-term modest salt reduction on blood pressure: Cochrane systematic review and meta-analysis of randomised trials. BMJ 2013, 346, f1325. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, M.H.; Fineberg, N.S.; Fineberg, S.E.; Weinberger, M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension 2001, 37, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.K.; Beck, F.X.; Müller, D.N.; Derer, W.; et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.; Love, R. Relation of sodium chloride intake to essential hypertension in humans. Fed. Proc. 1954, 13, 426. [Google Scholar]
- Stamler, J.; Elliott, P.; Kesteloot, H.; Nichols, R.; Claeys, G.; Dyer, A.R.; Stamler, R. Inverse relation of dietary protein markers with blood pressure. Findings for 10,020 men and women in the INTERSALT Study. Circulation 1996, 94, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Obarzanek, E.; Proschan, M.A.; Vollmer, W.M.; Moore, T.J.; Sacks, F.M.; Appel, L.J.; Svetkey, L.P.; Most-Windhauser, M.M.; Cutler, J.A. Individual blood pressure responses to changes in salt intake: Results from the DASH-Sodium trial. Hypertension 2003, 42, 459–467. [Google Scholar] [CrossRef] [PubMed]
- WHO. Guideline: Sodium Intake for Adults and Children; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Wenzel, P.; Knorr, M.; Kossmann, S.; Stratmann, J.; Hausding, M.; Schuhmacher, S.; Karbach, S.H.; Schwenk, M.; Yogev, N.; Schulz, E.; et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011, 124, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Turner, J.E.; Krebs, C.; Kurts, C.; Harrison, D.G.; Ehmke, H. Immune Mechanisms in Arterial Hypertension. J. Am. Soc. Nephrol. JASN 2016, 27, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Binger, K.J.; Gebhardt, M.; Heinig, M.; Rintisch, C.; Schroeder, A.; Neuhofer, W.; Hilgers, K.; Manzel, A.; Schwartz, C.; Kleinewietfeld, M.; et al. High salt reduces the activation of IL-4- and IL-13-stimulated macrophages. J. Clin. Investig. 2015, 125, 4223–4238. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, immunity, and hypertension. Hypertension 2011, 57, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Jantsch, J.; Schatz, V.; Friedrich, D.; Schröder, A.; Kopp, C.; Siegert, I.; Maronna, A.; Wendelborn, D.; Linz, P.; Binger, K.J.; et al. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 2015, 21, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, N.R.; Foss, J.D.; Kryshtal, D.O.; Tsyba, N.; Kumaresan, S.; Xiao, L.; Mernaugh, R.L.; Itani, H.A.; Loperena, R.; Chen, W.; et al. Dendritic Cell Amiloride-Sensitive Channels Mediate Sodium-Induced Inflammation and Hypertension. Cell Rep. 2017, 21, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Kirabo, A.; Fontana, V.; de Faria, A.P.; Loperena, R.; Galindo, C.L.; Wu, J.; Bikineyeva, A.T.; Dikalov, S.; Xiao, L.; Chen, W.; et al. DC isoketal-modified proteins activate T cells and promote hypertension. J. Clin. Investig. 2014, 124, 4642–4656. [Google Scholar] [CrossRef] [PubMed]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Itani, H.A.; Xiao, L.; Saleh, M.A.; Wu, J.; Pilkinton, M.A.; Dale, B.L.; Barbaro, N.R.; Foss, J.D.; Kirabo, A.; Montaniel, K.R.; et al. CD70 Exacerbates Blood Pressure Elevation and Renal Damage in Response to Repeated Hypertensive Stimuli. Circ. Res. 2016, 118, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Samji, T.; Khanna, K.M. Understanding memory CD8+ T cells. Immunol. Lett. 2017, 185, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef] [PubMed]
- Van Beusecum, J.P.; Barbaro, N.R.; McDowell, Z.; Aden, L.A.; Xiao, L.; Pandey, A.K.; Itani, H.A.; Himmel, L.E.; Harrison, D.G.; Kirabo, A. High Salt Activates CD11c+ Antigen-Presenting Cells via SGK (Serum Glucocorticoid Kinase) 1 to Promote Renal Inflammation and Salt-Sensitive Hypertension. Hypertension 2019, 74, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Le Brocq, M.; Leslie, S.J.; Milliken, P.; Megson, I.L. Endothelial dysfunction: From molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1631–1674. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Beaulieu, S.; Lebecque, S.; Steinman, R.M.; Muller, W.A. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science 1998, 282, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kawakami-Mori, F.; Kang, L.; Ayuzawa, N.; Ogura, S.; Koid, S.S.; Reheman, L.; Yeerbolati, A.; Liu, B.; Yatomi, Y.; et al. Low-dose L-NAME induces salt sensitivity associated with sustained increased blood volume and sodium-chloride cotransporter activity in rodents. Kidney Int. 2020, 98, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Ghiadoni, L.; Virdis, A.; Taddei, S.; Gonzales, J.; Salazar, J.; Andersen, L.; Duranti, P.; Salvetti, A. Defective nitric oxide-pathway in salt-sensitive essential hypertensive patients. Am. J. Hypertens. 1997, 4, 20A. [Google Scholar] [CrossRef]
- Sverdlov, A.L.; Ngo, D.T.; Chan, W.P.; Chirkov, Y.Y.; Horowitz, J.D. Aging of the nitric oxide system: Are we as old as our NO? J. Am. Heart Assoc. 2014, 3, e000973. [Google Scholar] [CrossRef] [PubMed]
- Svetkey, L.P.; McKeown, S.P.; Wilson, A.F. Heritability of salt sensitivity in black Americans. Hypertension 1996, 28, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H. Ubiquitylation and the pathogenesis of hypertension. J. Clin. Investig. 2013, 123, 546–548. [Google Scholar] [CrossRef] [PubMed]
- Norlander, A.E.; Saleh, M.A.; Pandey, A.K.; Itani, H.A.; Wu, J.; Xiao, L.; Kang, J.; Dale, B.L.; Goleva, S.B.; Laroumanie, F.; et al. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight 2017, 2, e92801. [Google Scholar] [CrossRef] [PubMed]
- Aoi, W.; Niisato, N.; Sawabe, Y.; Miyazaki, H.; Tokuda, S.; Nishio, K.; Yoshikawa, T.; Marunaka, Y. Abnormal expression of ENaC and SGK1 mRNA induced by dietary sodium in Dahl salt-sensitively hypertensive rats. Cell Biol. Int. 2007, 31, 1288–1291. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yosef, N.; Thalhamer, T.; Zhu, C.; Xiao, S.; Kishi, Y.; Regev, A.; Kuchroo, V.K. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013, 496, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.L.; Kitz, A.; Wu, C.; Lowther, D.E.; Rodriguez, D.M.; Vudattu, N.; Deng, S.; Herold, K.C.; Kuchroo, V.K.; Kleinewietfeld, M.; et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J. Clin. Investig. 2015, 125, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Norlander, A.E.; Saleh, M.A.; Kamat, N.V.; Ko, B.; Gnecco, J.; Zhu, L.; Dale, B.L.; Iwakura, Y.; Hoover, R.S.; McDonough, A.A. Interleukin-17A regulates renal sodium transporters and renal injury in angiotensin II–induced hypertension. Hypertension 2016, 68, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.A.; Norlander, A.E.; Madhur, M.S. Inhibition of Interleukin 17-A but not Interleukin-17F Signaling Lowers Blood Pressure and Reduces End-organ Inflammation in Angiotensin II-induced Hypertension. JACC Basic. Transl. Sci. 2016, 1, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Madhur, M.S.; Lob, H.E.; McCann, L.A.; Iwakura, Y.; Blinder, Y.; Guzik, T.J.; Harrison, D.G. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 2010, 55, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Kamat, N.V.; Thabet, S.R.; Xiao, L.; Saleh, M.A.; Kirabo, A.; Madhur, M.S.; Delpire, E.; Harrison, D.G.; McDonough, A.A. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ−/− and interleukin-17A−/− mice. Hypertension 2015, 65, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Huang, D.Y.; Grahammer, F.; Wyatt, A.W.; Osswald, H.; Wulff, P.; Kuhl, D.; Lang, F. SGK1 as a determinant of kidney function and salt intake in response to mineralocorticoid excess. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2005, 289, R395–R401. [Google Scholar] [CrossRef] [PubMed]
- Artunc, F.; Amann, K.; Nasir, O.; Friedrich, B.; Sandulache, D.; Jahovic, N.; Risler, T.; Vallon, V.; Wulff, P.; Kuhl, D.; et al. Blunted DOCA/high salt induced albuminuria and renal tubulointerstitial damage in gene-targeted mice lacking SGK1. J. Mol. Med. 2006, 84, 737–746. [Google Scholar] [CrossRef]
- Sierra-Ramos, C.; Velazquez-Garcia, S.; Keskus, A.G.; Vastola-Mascolo, A.; Rodríguez-Rodríguez, A.E.; Luis-Lima, S.; Hernández, G.; Navarro-González, J.F.; Porrini, E.; Konu, O.; et al. Increased SGK1 activity potentiates mineralocorticoid/NaCl-induced kidney injury. Am. J. Physiol. Renal Physiol. 2021, 320, F628–F643. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Kirabo, A.; Wu, J.; Saleh, M.A.; Zhu, L.; Wang, F.; Takahashi, T.; Loperena, R.; Foss, J.D.; Mernaugh, R.L. Renal denervation prevents immune cell activation and renal inflammation in angiotensin II–induced hypertension. Circ. Res. 2015, 117, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Maaliki, D.; Itani, M.M.; Itani, H.A. Pathophysiology and genetics of salt-sensitive hypertension. Front. Physiol. 2022, 13, 1001434. [Google Scholar] [CrossRef] [PubMed]
- Itani, M.M.; Jarrah, H.; Maaliki, D.; Radwan, Z.; Farhat, R.; Itani, H.A. Sphingosine 1 phosphate promotes hypertension specific memory T cell trafficking in response to repeated hypertensive challenges. Front. Physiol. 2022, 13, 930487. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.C.; Vassallo, P.F.; Crajoinas Rde, O.; Oliveira, M.L.; Martins, F.L.; Nogueira, B.V.; Motta-Santos, D.; Araújo, I.B.; Forechi, L.; Girardi, A.C.; et al. Renal Effects and Underlying Molecular Mechanisms of Long-Term Salt Content Diets in Spontaneously Hypertensive Rats. PLoS ONE 2015, 10, e0141288. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kumagai, H.; Suzuki, A.; Kobayashi, N.; Ohkawa, S.; Odamaki, M.; Kohsaka, T.; Yamamoto, T.; Ikegaya, N. Relaxin ameliorates salt-sensitive hypertension and renal fibrosis. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2012, 27, 2190–2197. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.C.; Burrell, L.M.; Black, M.J.; Wu, L.L.; Dilley, R.J.; Cooper, M.E.; Johnston, C.I. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation 1998, 98, 2621–2628. [Google Scholar] [CrossRef] [PubMed]
- Hartner, A.; Cordasic, N.; Klanke, B.; Veelken, R.; Hilgers, K.F. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2003, 18, 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Kren, S.; Hostetter, T.H. The course of the remnant kidney model in mice. Kidney Int. 1999, 56, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, F.; Krebs, C.; Abdulhag, U.N.; Meyer-Schwesinger, C.; Maas, R.; Helmchen, U.; Hilgers, K.F.; Wolf, G.; Stahl, R.A.; Wenzel, U. Rapid development of severe end-organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int. 2008, 73, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, S.; Ishola, D.A., Jr.; Joles, J.A.; Bluyssen, H.A.; Koomans, H.A.; Braam, B. Resistance to oxidative stress by chronic infusion of angiotensin II in mouse kidney is not mediated by the AT2 receptor. Am. J. Physiol. Renal Physiol. 2005, 288, F1191–F1200. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczyk, T.P.; Nosalski, R.; Szczepaniak, P.; Budzyn, K.; Osmenda, G.; Skiba, D.; Sagan, A.; Wu, J.; Vinh, A.; Marvar, P.J.; et al. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. Faseb. J. 2016, 30, 1987–1999. [Google Scholar] [CrossRef] [PubMed]
- Laffer, C.L.; Scott, R.C., 3rd; Titze, J.M.; Luft, F.C.; Elijovich, F. Hemodynamics and Salt-and-Water Balance Link Sodium Storage and Vascular Dysfunction in Salt-Sensitive Subjects. Hypertension 2016, 68, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.A.; McMaster, W.G.; Wu, J.; Norlander, A.E.; Funt, S.A.; Thabet, S.R.; Kirabo, A.; Xiao, L.; Chen, W.; Itani, H.A. Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation. J. Clin. Investig. 2015, 125, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M.M.; Ahmari, N.; Carvajal, J.M.; Zingler, M.B.; Qi, Y.; Kim, S.; Joseph, J.; Garcia-Pereira, F.; Johnson, R.D.; Shenoy, V. Involvement of bone marrow cells and neuroinflammation in hypertension. Circ. Res. 2015, 117, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Sercan Alp, Ö.; Durlanik, S.; Schulz, D.; McGrath, M.; Grün, J.R.; Bardua, M.; Ikuta, K.; Sgouroudis, E.; Riedel, R.; Zehentmeier, S. Memory CD8+ T cells colocalize with IL-7+ stromal cells in bone marrow and rest in terms of proliferation and transcription. Eur. J. Immunol. 2015, 45, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Kopp, C.; Linz, P.; Dahlmann, A.; Hammon, M.; Jantsch, J.; Müller, D.N.; Schmieder, R.E.; Cavallaro, A.; Eckardt, K.U.; Uder, M.; et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 2013, 61, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Titze, J. Sodium balance is not just a renal affair. Curr. Opin. Nephrol. Hypertens. 2014, 23, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.; Dinarello, C.A. Osmotic regulation of cytokine synthesis in vitro. Proc. Natl. Acad. Sci. USA 1995, 92, 12230–12234. [Google Scholar] [CrossRef] [PubMed]
- Pang, D.J.; Neves, J.F.; Sumaria, N.; Pennington, D.J. Understanding the complexity of γδ T-cell subsets in mouse and human. Immunology 2012, 136, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.G.; Wilson, R.M.; Heo, J.; Murthy, N.R.; Baid, S.; Ouchi, N.; Sam, F. Interferon-γ ablation exacerbates myocardial hypertrophy in diastolic heart failure. Am. J. Physiol. Heart C 2012, 303, H587–H596. [Google Scholar] [CrossRef] [PubMed]
- Leavy, O. T cells: Salt promotes pathogenic TH17 cells. Nat. Rev. Immunol. 2013, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Chiasson, V.L.; Chatterjee, P.; Kopriva, S.E.; Young, K.J.; Mitchell, B.M. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc. Res. 2013, 97, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Small, H.Y.; Migliarino, S.; Czesnikiewicz-Guzik, M.; Guzik, T.J. Hypertension: Focus on autoimmunity and oxidative stress. Free. Radic. Biol. Med. 2018, 125, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Thabet, S.R.; Kirabo, A.; Trott, D.W.; Saleh, M.A.; Xiao, L.; Madhur, M.S.; Chen, W.; Harrison, D.G. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ. Res. 2014, 114, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; You, B.; Chen, S.-P.; Liao, J.K.; Sun, J. Tumor necrosis factor-α downregulates endothelial nitric oxide synthase mRNA stability via translation elongation factor 1-α 1. Circ. Res. 2008, 103, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.C.; Chen, Y.; Lang, F. Glucocorticoid activation of Na+/H+ exchanger isoform 3 revisited. The roles of SGK1 and NHERF2. J. Biol. Chem. 2002, 277, 7676–7683. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawachi, H.; Fujita, T. Podocyte as the target for aldosterone: Roles of oxidative stress and Sgk1. Hypertension 2007, 49, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Kuwana, H.; Kobayashi, T.; Okado, T.; Suzuki, N.; Yoshimoto, T.; Hirata, Y.; Sasaki, S. Aldosterone-stimulated SGK1 activity mediates profibrotic signaling in the mesangium. J. Am. Soc. Nephrol. JASN 2008, 19, 298–309. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F.; Esler, M. Translational medicine: The antihypertensive effect of renal denervation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R245–R253. [Google Scholar] [CrossRef]
- Cheng, M.H.; Anderson, M.S. Monogenic autoimmunity. Annu. Rev. Immunol. 2012, 30, 393–427. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of γδ T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, J.L.; Kennard, S.; Huby, A.C.; Antonova, G.; Lu, Q.; Jaffe, I.Z.; Patel, V.S.; Fulton, D.J.R.; Belin de Chantemèle, E.J. Progesterone Predisposes Females to Obesity-Associated Leptin-Mediated Endothelial Dysfunction via Upregulating Endothelial MR (Mineralocorticoid Receptor) Expression. Hypertension 2019, 74, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Grabek, A.; Dolfi, B.; Klein, B.; Jian-Motamedi, F.; Chaboissier, M.C.; Schedl, A. The Adult Adrenal Cortex Undergoes Rapid Tissue Renewal in a Sex-Specific Manner. Cell Stem Cell 2019, 25, 290–296.e2. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Dyer, A.; Stamler, R. The INTERSALT study: Results for 24-h sodium and potassium, by age and sex. INTERSALT Co-operative Research Group. J. Hum. Hypertens. 1989, 3, 323–330. [Google Scholar] [PubMed]
- Shukri, M.Z.; Tan, J.W.; Manosroi, W.; Pojoga, L.H.; Rivera, A.; Williams, J.S.; Seely, E.W.; Adler, G.K.; Jaffe, I.Z.; Karas, R.H. Biological sex modulates the adrenal and blood pressure responses to angiotensin II. Hypertension 2018, 71, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. Sodium sensitivity of blood pressure in Chinese populations. Curr. Hypertens. Rep. 2010, 12, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, J.L.; Harwood, D.; Bender, L.; Shrestha, L.; Brands, M.W.; Morwitzer, M.J.; Kennard, S.; Antonova, G.; Belin de Chantemèle, E.J. Lack of Suppression of Aldosterone Production Leads to Salt-Sensitive Hypertension in Female but Not Male Balb/C Mice. Hypertension 2018, 72, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, O.; Pizzolo, F.; Ciacciarelli, A.; Corrocher, R.; Signorelli, D.; Falcone, S.; Blengio, G.S. Menopause not aldosterone-to-renin ratio predicts blood pressure response to a mineralocorticoid receptor antagonist in primary care hypertensive patients. Am. J. Hypertens. 2008, 21, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Gwoo, S.; Kim, Y.N.; Shin, H.S.; Jung, Y.S.; Rim, H. Predictors of hyperkalemia risk after hypertension control with aldosterone blockade according to the presence or absence of chronic kidney disease. Nephron Clin. Pract. 2014, 128, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Whelton, P.K.; He, J.; Appel, L.J.; Cutler, J.A.; Havas, S.; Kotchen, T.A.; Roccella, E.J.; Stout, R.; Vallbona, C.; Winston, M.C.; et al. Primary prevention of hypertension: Clinical and public health advisory from The National High Blood Pressure Education Program. JAMA 2002, 288, 1882–1888. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.L.; Cheung, B.M.; Man, Y.B.; Lau, C.P.; Lam, K.S. Prevalence, awareness, treatment, and control of hypertension among United States adults 1999–2004. Hypertension 2007, 49, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Oshima, T.; Matsuura, H.; Watanabe, M.; Ishida, M.; Ishida, T.; Ozono, R.; Kajiyama, G.; Kanbe, M. Effects of age and sex on sodium chloride sensitivity: Association with plasma renin activity. Clin. Nephrol. 1994, 42, 376–380. [Google Scholar] [PubMed]
- Weinberger, M.H.; Miller, J.Z.; Luft, F.C.; Grim, C.E.; Fineberg, N.S. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 1986, 8, Ii127–Ii134. [Google Scholar] [CrossRef] [PubMed]
- De Moudt, S.; Hendrickx, J.O.; Neutel, C.; De Munck, D.; Leloup, A.; De Meyer, G.R.Y.; Martinet, W.; Fransen, P. Progressive aortic stiffness in aging C57Bl/6 mice displays altered contractile behaviour and extracellular matrix changes. Commun. Biol. 2022, 5, 605. [Google Scholar] [CrossRef] [PubMed]
- Wirth, A.; Wang, S.; Takefuji, M.; Tang, C.; Althoff, T.F.; Schweda, F.; Wettschureck, N.; Offermanns, S. Age-dependent blood pressure elevation is due to increased vascular smooth muscle tone mediated by G-protein signalling. Cardiovasc. Res. 2016, 109, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, K.; Lei, H.; Sun, Z. Klotho gene deficiency causes salt-sensitive hypertension via monocyte chemotactic protein-1/CC chemokine receptor 2-mediated inflammation. J. Am. Soc. Nephrol. JASN 2015, 26, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, T.; Inoue, T.; Miyazaki, T.; Kobori, H.; Nishiyama, A.; Ishii, N.; Hayashi, M.; Suzuki, H. Klotho Ameliorates Medullary Fibrosis and Pressure Natriuresis in Hypertensive Rat Kidneys. Hypertension 2018, 72, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Imura, A.; Urakawa, I.; Shimada, T.; Murakami, J.; Aono, Y.; Hasegawa, H.; Yamashita, T.; Nakatani, K.; Saito, Y.; et al. Establishment of sandwich ELISA for soluble alpha-Klotho measurement: Age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem. Biophys. Res. Commun. 2010, 398, 513–518. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, X. Klotho: A novel biomarker for cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Sun, Z. Activation of DNA demethylases attenuates aging-associated arterial stiffening and hypertension. Aging Cell 2018, 17, e12762. [Google Scholar] [CrossRef] [PubMed]
- Kawarazaki, W.; Mizuno, R.; Nishimoto, M.; Ayuzawa, N.; Hirohama, D.; Ueda, K.; Kawakami-Mori, F.; Oba, S.; Marumo, T.; Fujita, T. Salt causes aging-associated hypertension via vascular Wnt5a under Klotho deficiency. J. Clin. Investig. 2020, 130, 4152–4166. [Google Scholar] [CrossRef] [PubMed]
- Trott, D.W.; Thabet, S.R.; Kirabo, A.; Saleh, M.A.; Itani, H.; Norlander, A.E.; Wu, J.; Goldstein, A.; Arendshorst, W.J.; Madhur, M.S.; et al. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension 2014, 64, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Fejes-Tóth, G.; Frindt, G.; Náray-Fejes-Tóth, A.; Palmer, L.G. Epithelial Na+ channel activation and processing in mice lacking SGK1. Am. J. Physiol. Renal Physiol. 2008, 294, F1298–F1305. [Google Scholar] [CrossRef] [PubMed]
- Muntner, P.; Carey, R.M.; Gidding, S.; Jones, D.W.; Taler, S.J.; Wright, J.T., Jr.; Whelton, P.K. Potential US Population Impact of the 2017 ACC/AHA High Blood Pressure Guideline. Circulation 2018, 137, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Bridges, L.E.; Williams, C.L.; Pointer, M.A.; Awumey, E.M. Mesenteric artery contraction and relaxation studies using automated wire myography. J. Vis. Exp. JoVE 2011, 55, e3119. [Google Scholar] [CrossRef]
- Rangan, G.K.; Tesch, G.H. Quantification of renal pathology by image analysis. Nephrology 2007, 12, 553–558. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maaliki, D.; Itani, M.; Jarrah, H.; El-Mallah, C.; Ismail, D.; El Atie, Y.E.; Obeid, O.; Jaffa, M.A.; Itani, H.A. Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension. Int. J. Mol. Sci. 2024, 25, 4402. https://doi.org/10.3390/ijms25084402
Maaliki D, Itani M, Jarrah H, El-Mallah C, Ismail D, El Atie YE, Obeid O, Jaffa MA, Itani HA. Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension. International Journal of Molecular Sciences. 2024; 25(8):4402. https://doi.org/10.3390/ijms25084402
Chicago/Turabian StyleMaaliki, Dina, Maha Itani, Hala Jarrah, Carla El-Mallah, Diana Ismail, Yara E. El Atie, Omar Obeid, Miran A. Jaffa, and Hana A. Itani. 2024. "Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension" International Journal of Molecular Sciences 25, no. 8: 4402. https://doi.org/10.3390/ijms25084402
APA StyleMaaliki, D., Itani, M., Jarrah, H., El-Mallah, C., Ismail, D., El Atie, Y. E., Obeid, O., Jaffa, M. A., & Itani, H. A. (2024). Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension. International Journal of Molecular Sciences, 25(8), 4402. https://doi.org/10.3390/ijms25084402