


CrnnCrispr: An Interpretable Deep Learning Method for CRISPR/Cas9 sgRNA On-Target Activity Prediction


Abstract
:1. Introduction
2. Results and Discussion
2.1. Ablation Study Shows the Importance of Convolution Module
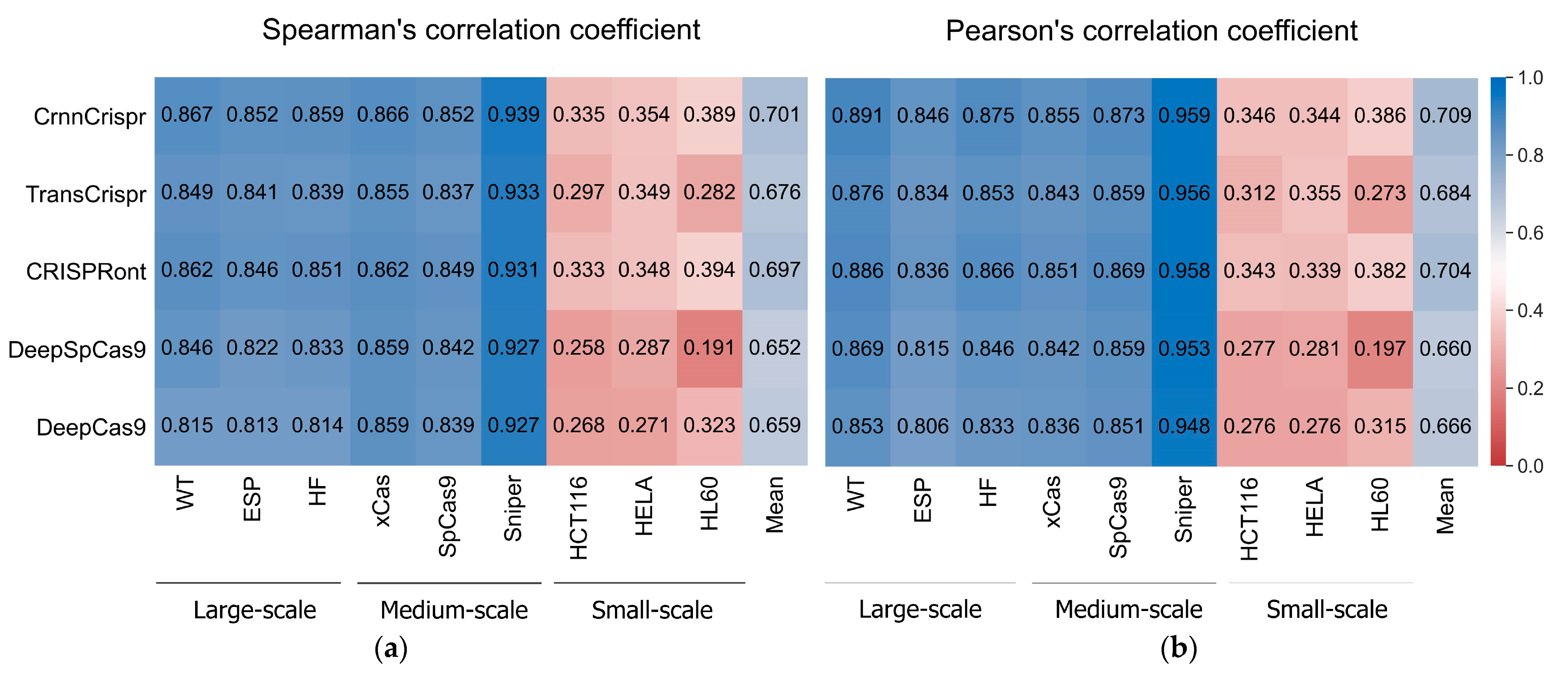
2.2. Performance Comparisons of State-of-the-Art Methods on Datasets with Various Scales under 5-Fold Cross-Validation
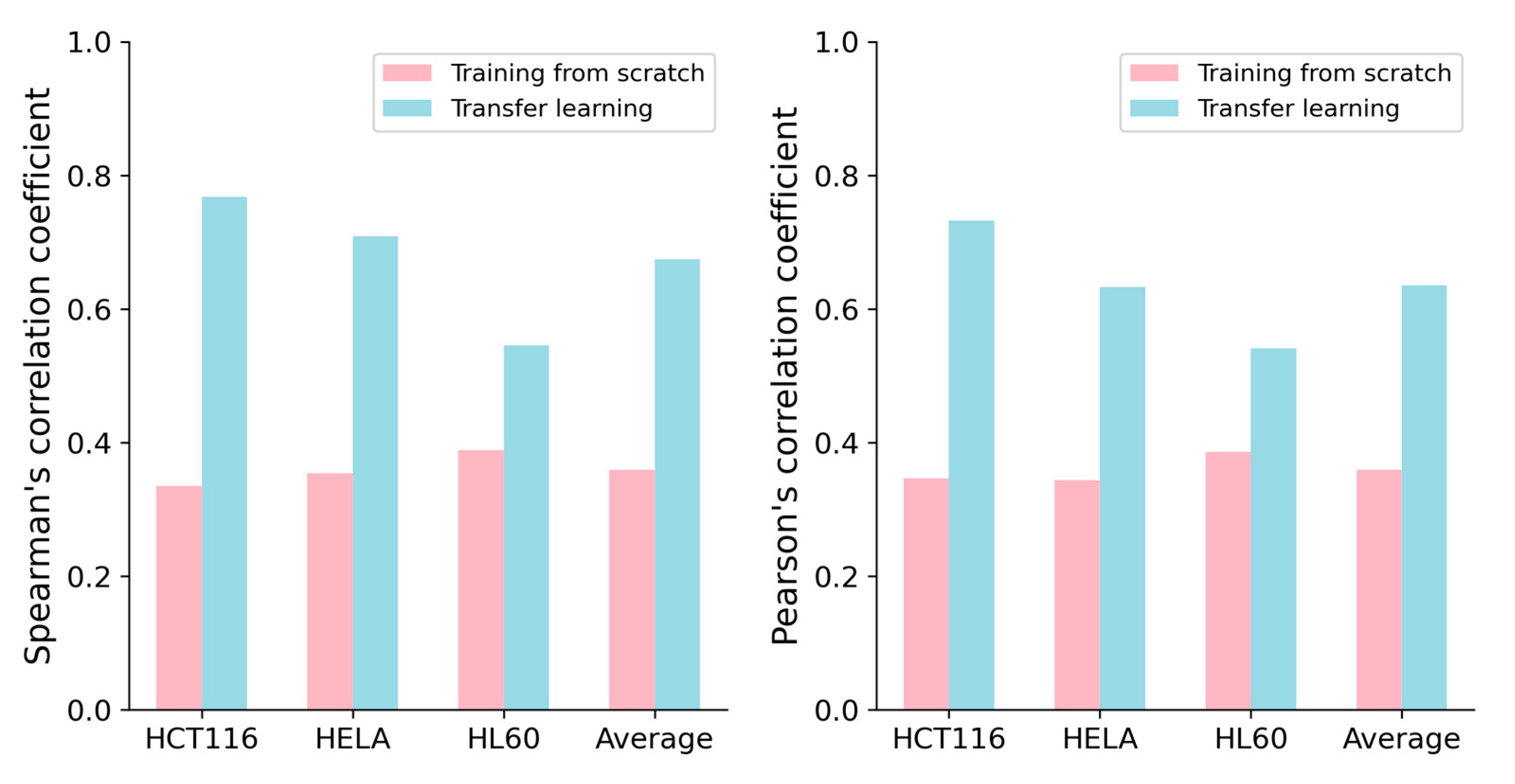
2.3. Transfer Learning Can Improve the Performance for Small-Scale Dataset Prediction
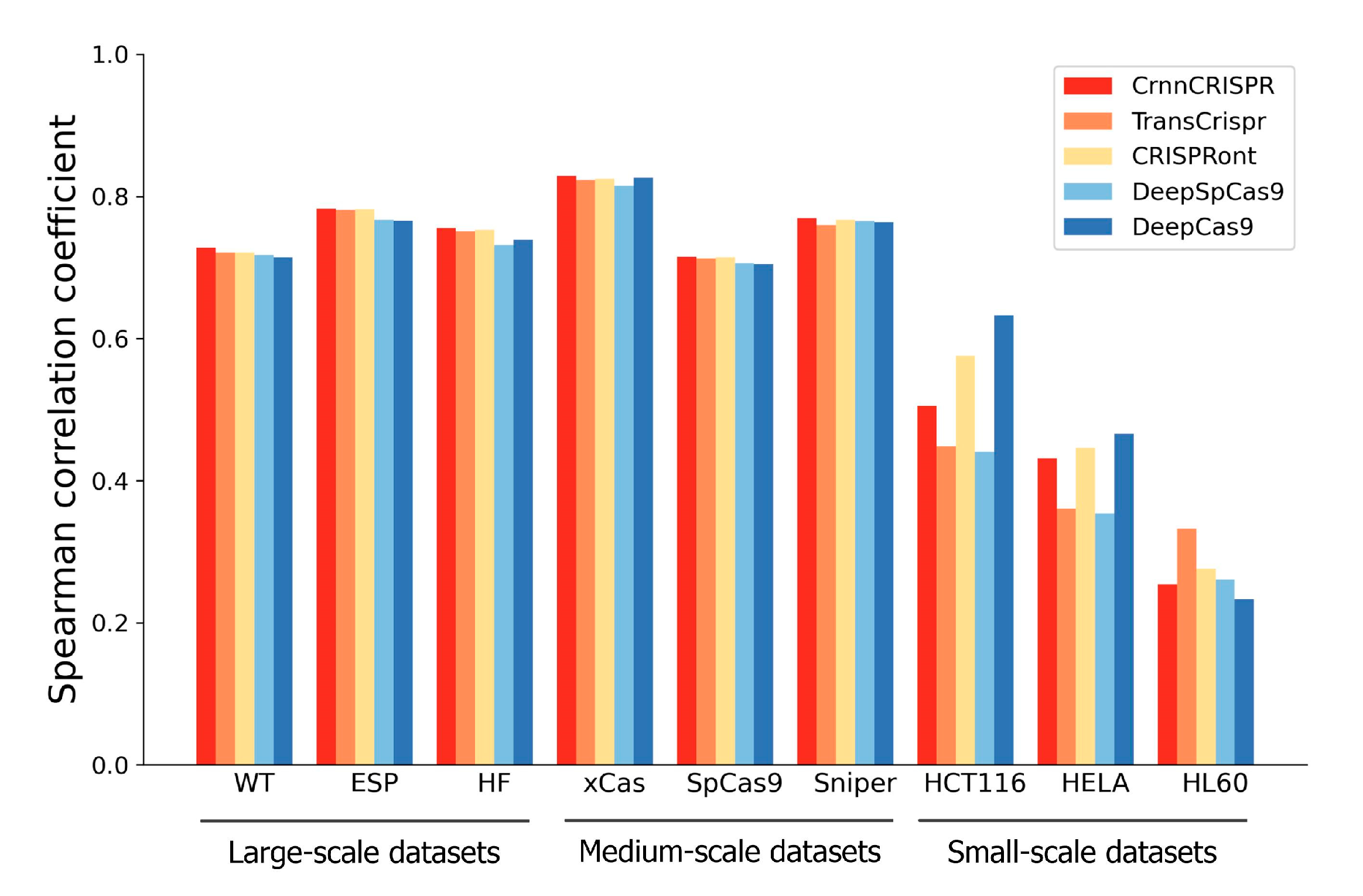
2.4. Generalizability Evaluation of Deep-Learning-Based Methods under a Leave-One-Cell-Out Procedure
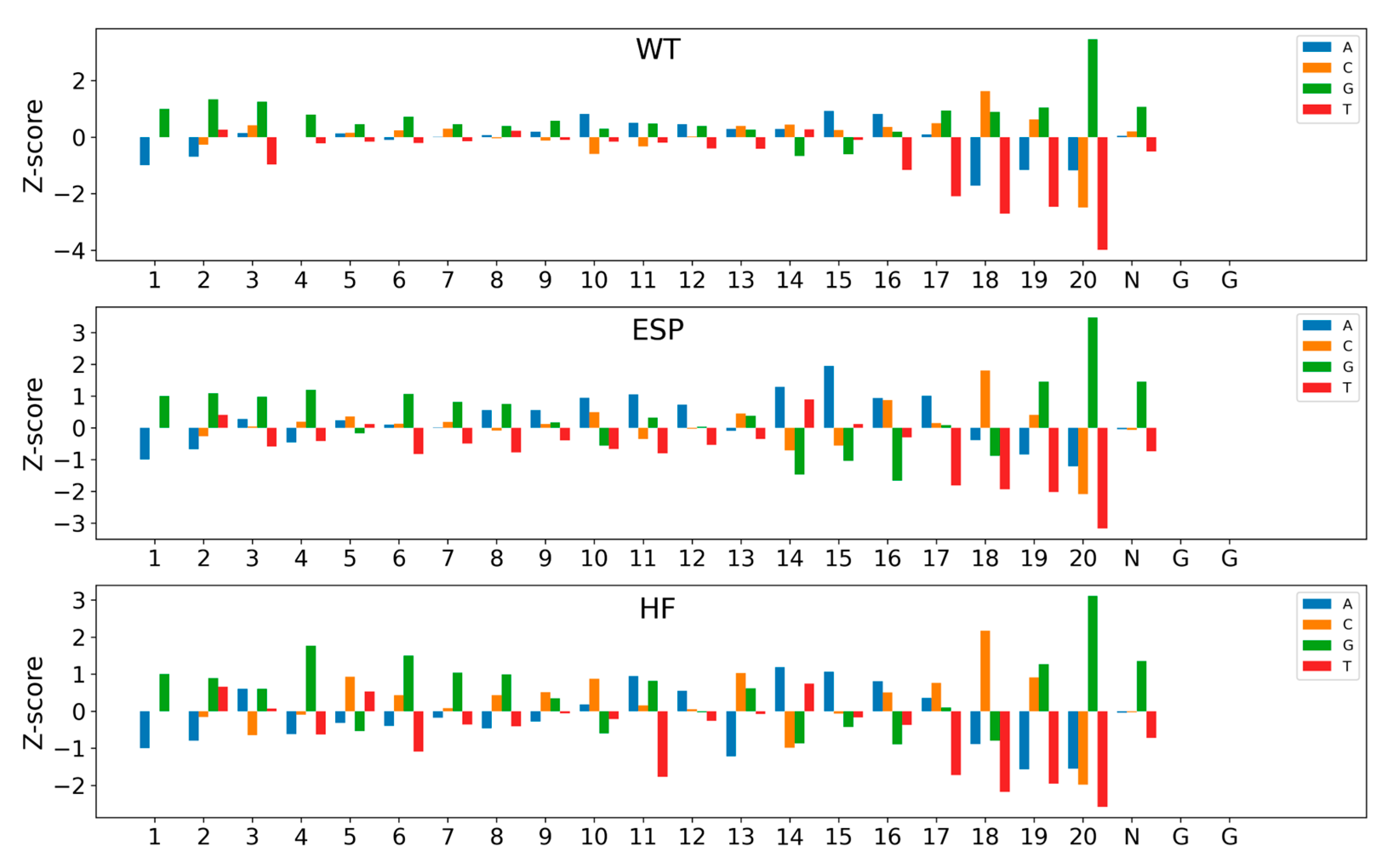
2.5. Model Interpretability
2.6. Discussion
3. Materials and Methods
3.1. Data Resources
3.2. Sequence Encoding
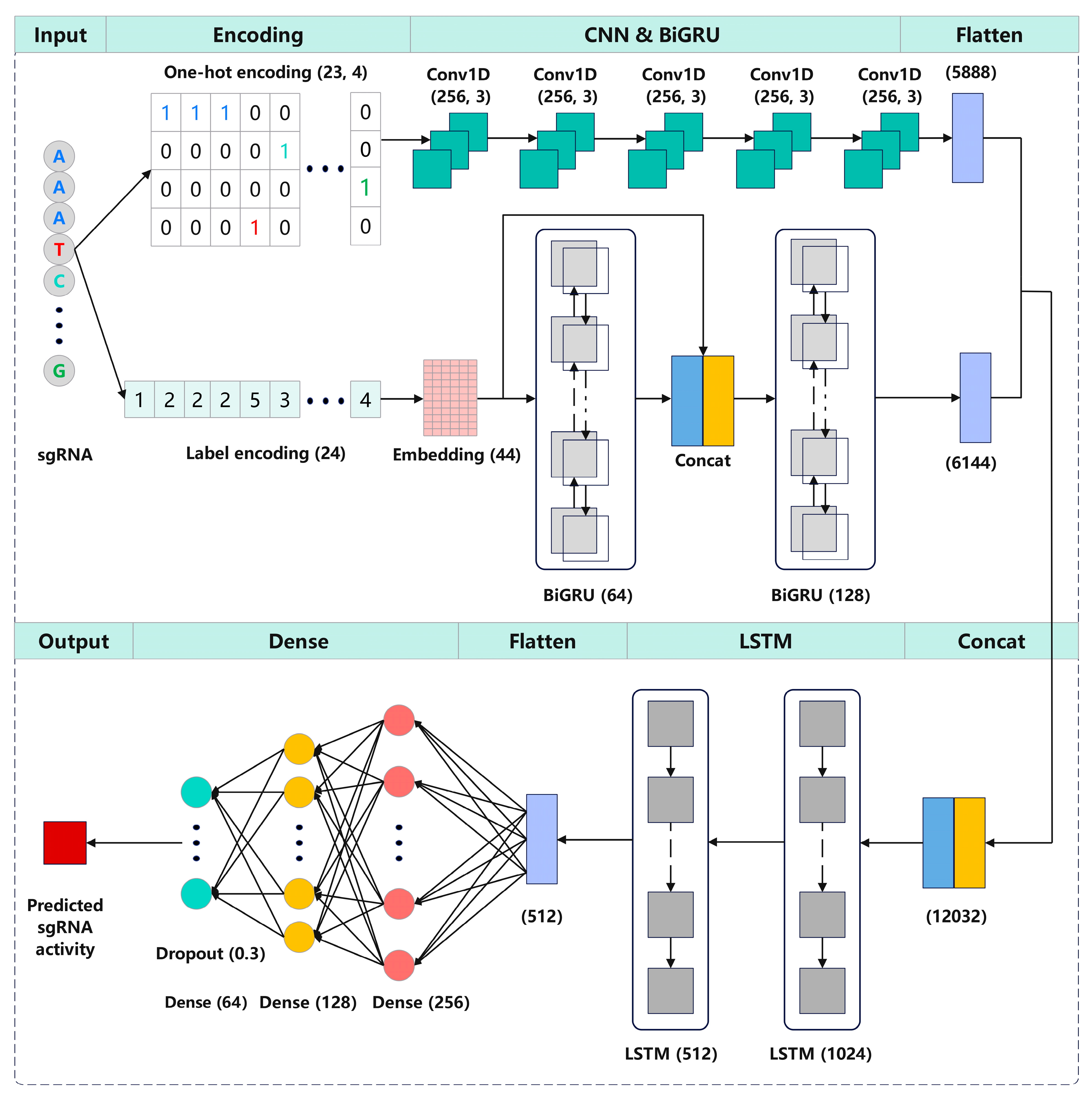
3.3. CrnnCrispr Model
3.4. Model Training
3.5. Performance Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-CAS9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Sagarbarria, M.G.S.; Caraan, J.A.M.; Layos, A.J.G. Usefulness of current sgRNA design guidelines and in vitro cleavage assays for plant CRISPR/Cas genome editing: A case targeting the polyphenol oxidase gene family in eggplant (Solanum melongena L.). Transgenic Res. 2023, 32, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Liu, W.; Wang, X. WU-CRISPR: Characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015, 16, 218. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Shi, Z.; Tao, D.; An, B. Recent advances in support vector machines. Neurocomputing 2016, 211, 1–3. [Google Scholar] [CrossRef]
- O’Shea, K.; Nash, R. An introduction to convolutional neural networks. arXiv 2015, arXiv:1511.08458. [Google Scholar]
- Alzubaidi, L.; Zhang, J.; Humaidi, A.J.; Al-Dujaili, A.; Duan, Y.; Al-Shamma, O.; Santamaría, J.; Fadhel, M.A.; Al-Amidie, M.; Farhan, L. Review of deep learning: Concepts, CNN architectures, challenges, applications, future directions. J. Big Data 2021, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Budach, S.; Marsico, A. pysster: Classification of biological sequences by learning sequence and structure motifs with convolutional neural networks. Bioinformatics 2018, 34, 3035–3037. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Tang, B.; Chen, W.; Luo, J. Prediction of CRISPR sgRNA activity using a deep convolutional neural network. J. Chem. Inf. Model. 2019, 59, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, G.; Mosquera, C.; Nápoles, G. A review on the long short-term memory model. Artif. Intell. Rev. 2020, 53, 5929–5955. [Google Scholar] [CrossRef]
- Salem, F.M. (Ed.) Gated RNN: The Gated Recurrent Unit (GRU) RNN. In Recurrent Neural Networks: From Simple to Gated Architectures; Springer International Publishing: Cham, Switzerland, 2022; pp. 85–100. [Google Scholar]
- Wang, D.; Zhang, C.; Wang, B.; Li, B.; Wang, Q.; Liu, D.; Wang, H.; Zhou, Y.; Shi, L.; Lan, F.; et al. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019, 10, 4284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Dai, Z.; Dai, X. C-RNNCrispr: Prediction of CRISPR/Cas9 sgRNA activity using convolutional and recurrent neural networks. Comput. Struct. Biotechnol. J. 2020, 18, 344–354. [Google Scholar] [CrossRef]
- Schuster, M.; Paliwal, K.K. Bidirectional recurrent neural networks. IEEE Trans. Signal Process. 1997, 45, 2673–2681. [Google Scholar] [CrossRef]
- Weiss, K.; Khoshgoftaar, T.M.; Wang, D. A survey of transfer learning. J. Big Data 2016, 3, 9. [Google Scholar] [CrossRef]
- Howard, J.; Ruder, S. Fine-tuned language models for text classification. arXiv 2018, arXiv:1801.06146. [Google Scholar]
- Zhang, G.; Dai, Z.; Dai, X. A novel hybrid CNN-SVR for CRISPR/Cas9 guide RNA activity prediction. Front. Genet. 2020, 10, 1303. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Jiang, Z. TransCrispr: Transformer based hybrid model for predicting CRISPR/Cas9 single guide RNA cleavage efficiency. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zeng, T.; Dai, Z.; Dai, X. Prediction of CRISPR/Cas9 single guide RNA cleavage efficiency and specificity by attention-based convolutional neural networks. Comput. Struct. Biotechnol. J. 2021, 19, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Kim, Y.; Lee, S.; Min, S.; Bae, J.Y.; Choi, J.W.; Park, J.; Jung, D.; Yoon, S.; Kim, H.H. SpCas9 activity prediction by DeepSpCas9, a deep learning–based model with high generalization performance. Sci. Adv. 2019, 5, eaax9249. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Xia, Y.; Yu, H.; Liu, Q.; Luo, Y.; Wang, G.; Chen, Z. Transferring ensemble representations using deep convolutional neural networks for small-scale image classification. IEEE Access 2019, 7, 168175–168186. [Google Scholar] [CrossRef]
- Li, B.; Ai, D.; Liu, X. CNN-XG: A hybrid framework for sgRNA on-target prediction. Biomolecules 2022, 12, 409. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Li, J.; Liu, Y.; Wu, Q. Precise and predictable CRISPR chromosomal rearrangements reveal principles of Cas9-mediated nucleotide insertion. Mol. Cell 2018, 71, 498–509.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Shen, X.; Pan, W. A simple convolutional neural network for prediction of enhancer–promoter interactions with DNA sequence data. Bioinformatics 2019, 35, 2899–2906. [Google Scholar] [CrossRef] [PubMed]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 80. [Google Scholar] [CrossRef]
- Kim, H.K.; Min, S.; Song, M.; Jung, S.; Choi, J.W.; Kim, Y.; Lee, S.; Yoon, S.; Kim, H.H. Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity. Nat. Biotechnol. 2018, 36, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Nishimasu, H.; Shi, X.; Ishiguro, S.; Gao, L.; Hirano, S.; Okazaki, S.; Noda, T.; Abudayyeh, O.O.; Gootenberg, J.S.; Mori, H.; et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 2018, 361, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.-h.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 2018, 9, 3048. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, H.K.; Lee, S.; Seo, J.H.; Choi, J.W.; Park, J.; Min, S.; Yoon, S.; Cho, S.-R.; Kim, H.H. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 2020, 38, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.M.; Iqbal, M.J. Classification of biological data using deep learning technique. NUML Int. J. Eng. Comput. 2022, 1, 13–26. [Google Scholar] [CrossRef]
- Bianchini, M.; Scarselli, F. On the Complexity of neural network classifiers: A comparison between shallow and deep architectures. IEEE Trans. Neural Netw. Learn. Syst. 2014, 25, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Iiduka, H. Appropriate Learning Rates of Adaptive learning rate optimization algorithms for training deep neural networks. IEEE Trans. Cybern. 2022, 52, 13250–13261. [Google Scholar] [CrossRef] [PubMed]
- Robeson, S.M.; Willmott, C.J. Decomposition of the mean absolute error (MAE) into systematic and unsystematic components. PLoS ONE 2023, 18, e0279774. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bovik, A.C. Mean squared error: Love it or leave it? A new look at signal fidelity measures. IEEE Signal Process. Mag. 2009, 26, 98–117. [Google Scholar] [CrossRef]
- Sedgwick, P. Spearman’s rank correlation coefficient. BMJ 2018, 362, k4131. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Marsman, M.; Wagenmakers, E.-J. Analytic posteriors for Pearson’s correlation coefficient. Stat. Neerl. 2018, 72, 4–13. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Architecture |
|---|---|
| CrnnCrispr-onehot | Only uses one-hot encoding to encode the sgRNA sequence and inputs the encoded matrix into both the CNN and BiGRU branches |
| CrnnCrispr-label | Only uses label encoding to encode the sgRNA sequence and inputs the encoded matrix into both the CNN and BiGRU branches |
| CrnnCrispr-w/oConv | Eliminates the CNN branch in the model |
| CrnnCrispr-w/oBiGRU | Eliminates the BiGRU branch in the model |
| Model | SCC | PCC |
|---|---|---|
| CrnnCrispr | 0.867 ± 0.007 | 0.892 ± 0.007 |
| CrnnCrispr-onehot | 0.863 ± 0.005 | 0.885 ± 0.006 |
| CrnnCrispr-label | 0.863 ± 0.004 | 0.887 ± 0.004 |
| CrnnCrispr-w/oConv | 0.836 ± 0.005 | 0.865 ± 0.007 |
| CrnnCrispr-w/oBiGRU | 0.861 ± 0.006 | 0.884 ± 0.007 |
| Method | HCT116 | HELA | HL60 | Average |
|---|---|---|---|---|
| ||||
| CrnnCrispr | 0.768 ± 0.055 | 0.709 ± 0.055 | 0.545 ± 0.049 | 0.674 ± 0.053 |
| TransCrispr | 0.629 ± 0.019 | 0.625 ± 0.020 | 0.535 ± 0.050 | 0.596 ± 0.030 |
| CRISPRont | 0.765 ± 0.067 | 0.706 ± 0.052 | 0.527 ± 0.030 | 0.666 ± 0.050 |
| DeepSpCas9 | 0.646 ± 0.031 | 0.615 ± 0.041 | 0.471 ± 0.035 | 0.577 ± 0.036 |
| DeepCas9 | 0.698 ± 0.027 | 0.667 ± 0.026 | 0.518 ± 0.043 | 0.628 ± 0.032 |
| ||||
| CrnnCrispr | 0.732 ± 0.053 | 0.633 ± 0.049 | 0.541 ± 0.047 | 0.635 ± 0.050 |
| TransCrispr | 0.633 ± 0.024 | 0.595 ± 0.019 | 0.561 ± 0.031 | 0.596 ± 0.025 |
| CRISPRont | 0.735 ± 0.053 | 0.649 ± 0.042 | 0.509 ± 0.023 | 0.631 ± 0.039 |
| DeepSpCas9 | 0.548 ± 0.026 | 0.506 ± 0.043 | 0.341 ± 0.053 | 0.465 ± 0.041 |
| DeepCas9 | 0.649 ± 0.035 | 0.598 ± 0.033 | 0.499 ± 0.057 | 0.582 ± 0.042 |
| Dataset | Training | Validation | Testing | Total | Scale Level | Ref. |
|---|---|---|---|---|---|---|
| Benchmark | 115,528 | 28,882 | 36,102 | 180,512 | Large | [25] |
| WT | 42,536 | 4726 | 8341 | 55,603 | Large | [27] |
| ESP | 44,841 | 4982 | 8793 | 58,616 | Large | [28] |
| HF | 43,519 | 4835 | 8533 | 56,887 | Large | [29] |
| xCas | 28,869 | 3208 | 5661 | 37,738 | Medium | [30] |
| SpCas9 | 23,397 | 2600 | 4588 | 30,585 | Medium | [31] |
| Sniper | 28,912 | 3213 | 5669 | 37,794 | Medium | [32] |
| HCT116 | 3243 | 360 | 636 | 4239 | Small | [33] |
| HELA | 6197 | 689 | 1215 | 8101 | Small | [33] |
| HL60 | 1588 | 177 | 311 | 2076 | Small | [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Xie, H.; Chen, Y.; Zhang, G. CrnnCrispr: An Interpretable Deep Learning Method for CRISPR/Cas9 sgRNA On-Target Activity Prediction. Int. J. Mol. Sci. 2024, 25, 4429. https://doi.org/10.3390/ijms25084429
Zhu W, Xie H, Chen Y, Zhang G. CrnnCrispr: An Interpretable Deep Learning Method for CRISPR/Cas9 sgRNA On-Target Activity Prediction. International Journal of Molecular Sciences. 2024; 25(8):4429. https://doi.org/10.3390/ijms25084429
Chicago/Turabian StyleZhu, Wentao, Huanzeng Xie, Yaowen Chen, and Guishan Zhang. 2024. "CrnnCrispr: An Interpretable Deep Learning Method for CRISPR/Cas9 sgRNA On-Target Activity Prediction" International Journal of Molecular Sciences 25, no. 8: 4429. https://doi.org/10.3390/ijms25084429
APA StyleZhu, W., Xie, H., Chen, Y., & Zhang, G. (2024). CrnnCrispr: An Interpretable Deep Learning Method for CRISPR/Cas9 sgRNA On-Target Activity Prediction. International Journal of Molecular Sciences, 25(8), 4429. https://doi.org/10.3390/ijms25084429