
Systematic Analysis of DNA Demethylase Gene Families in Foxtail Millet (Setaria italica L.) and Their Expression Variations after Abiotic Stresses

Abstract
:1. Introduction
2. Results
2.1. Genome-Wide Identification and Structural Analysis of DNA-deMTase Genes
2.2. Phylogenetic Tree Construction of DNA-deMTases Gene Family Proteins and Analysis of Conserved Structural Domains
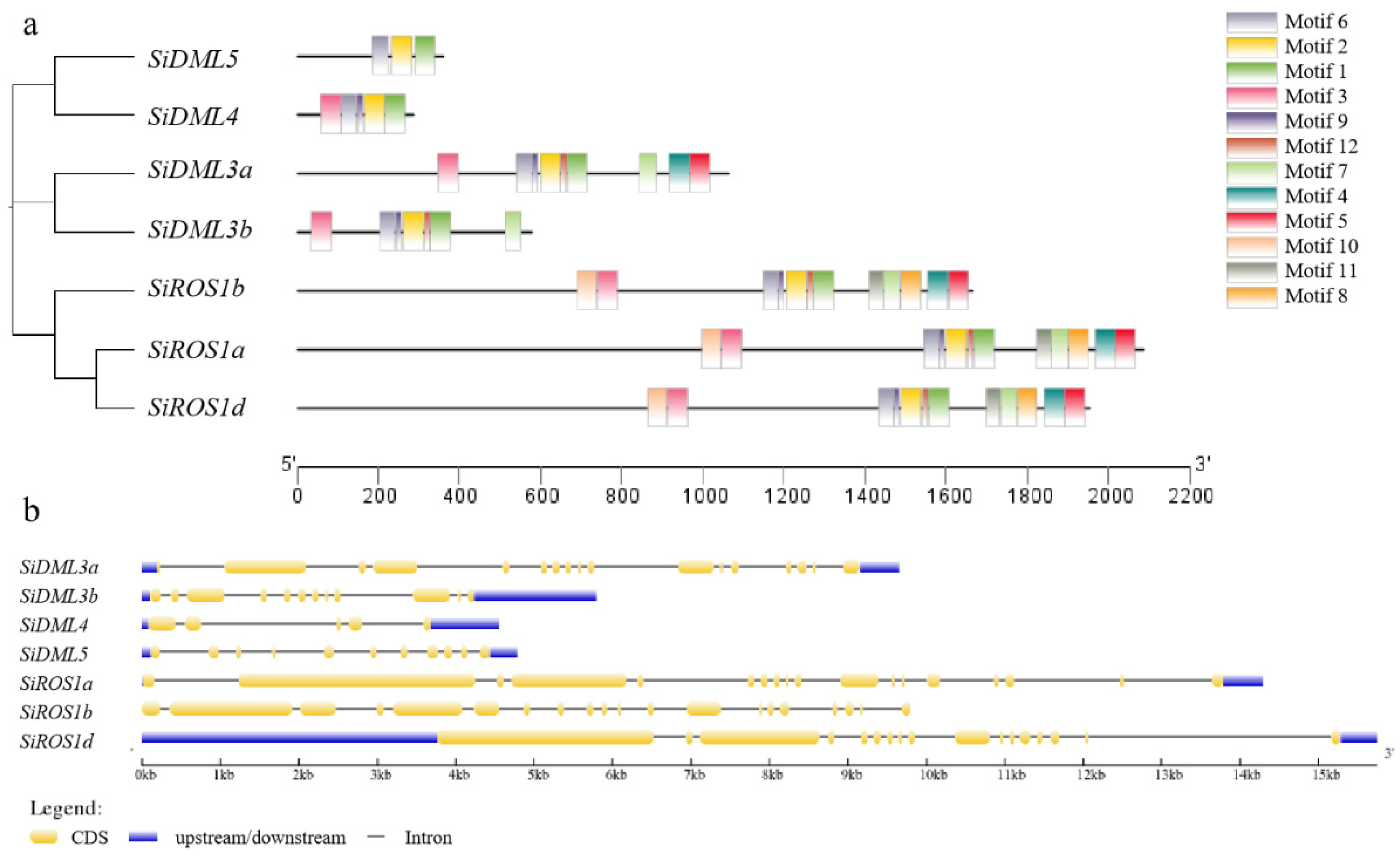
2.3. Observation of Conserved Motif and Gene Structure Analysis of DNA-deMTases in S. italica
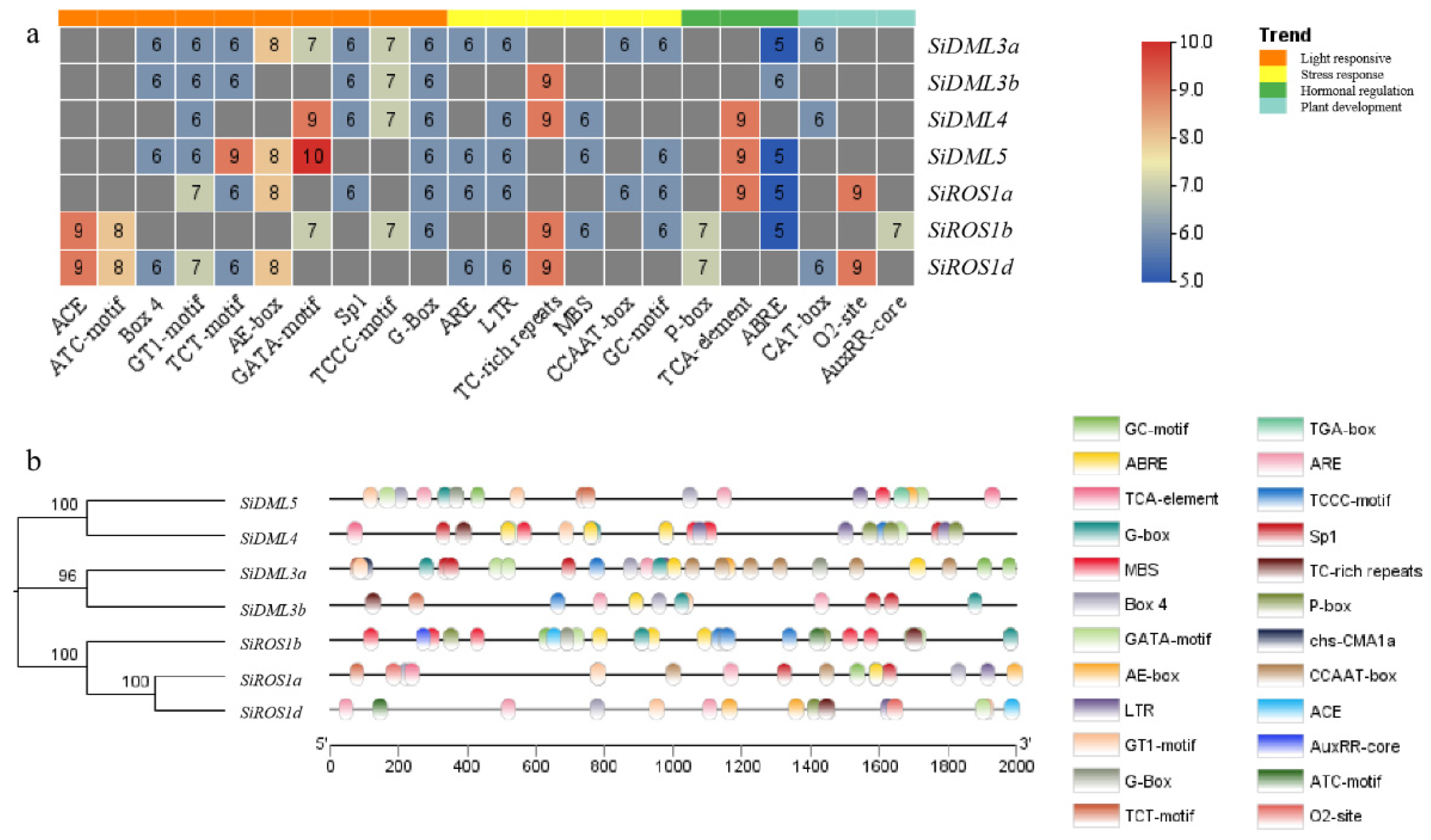
2.4. Analysis of Cis-Acting Elements in DNA-deMTase Genes
2.5. Chromosomal Location and Synteny Analysis
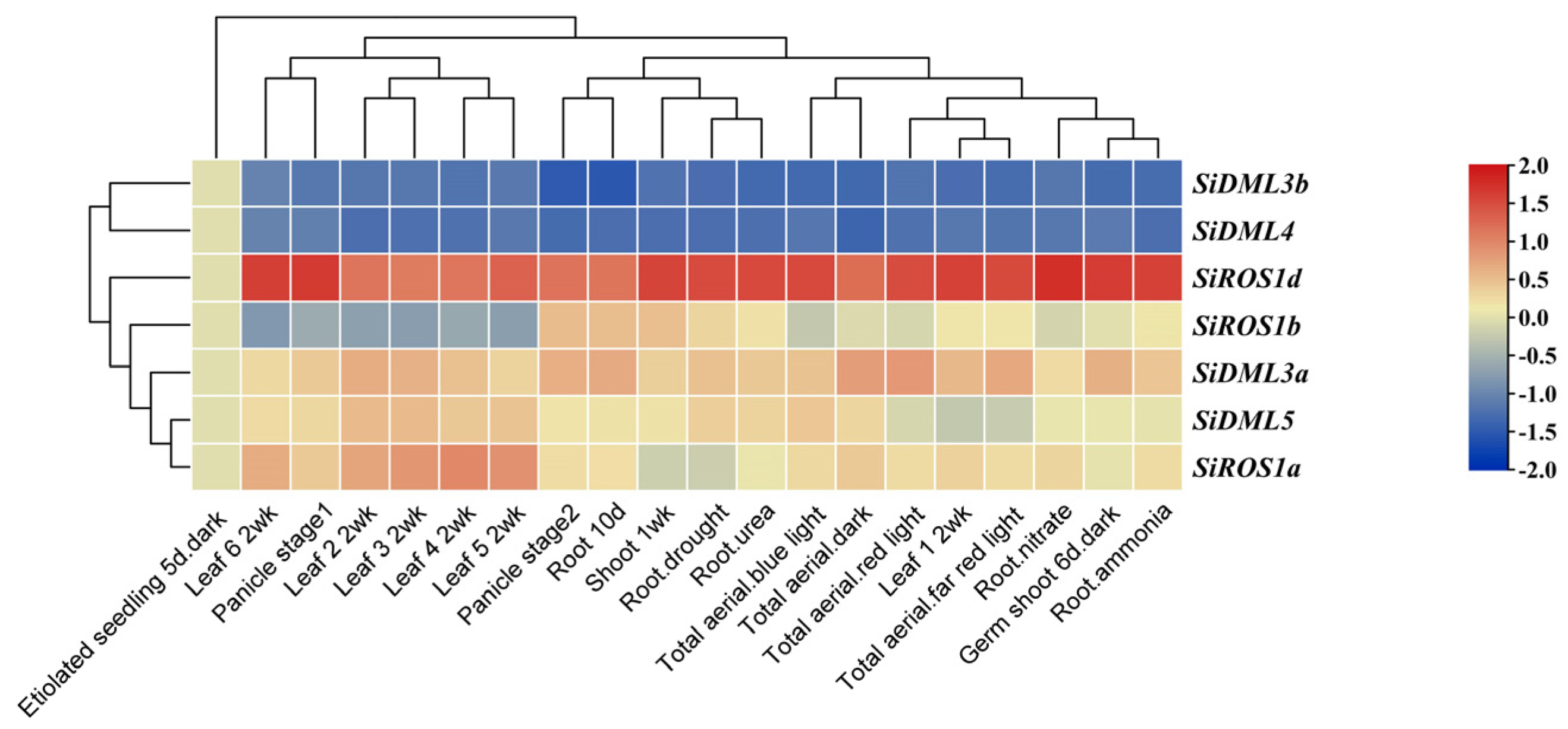
2.6. Expression Patterns of DNA-deMTase Genes in S. italica
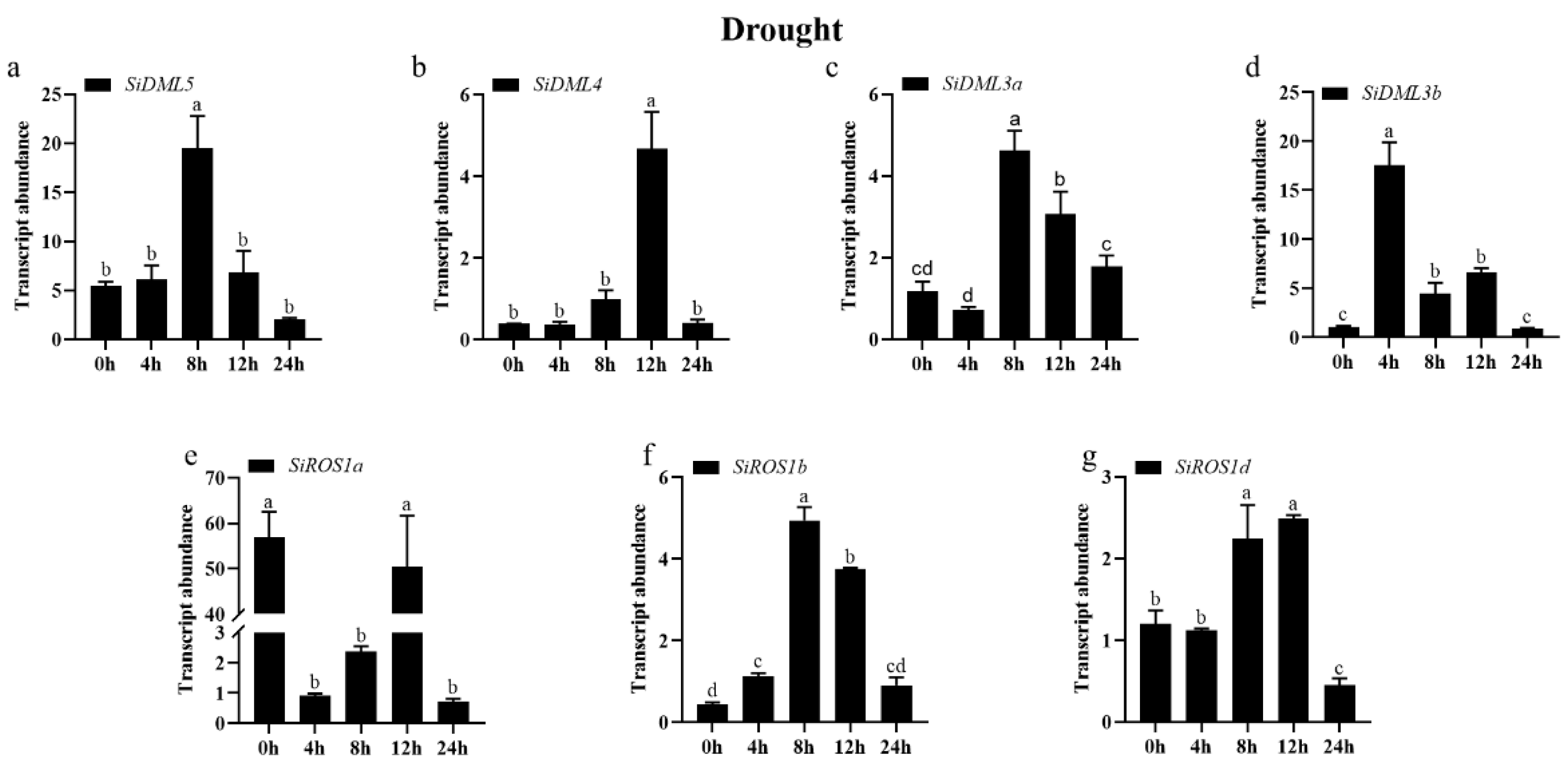
2.7. The Transcript Abundance of DNA-deMTase Genes in S. italica under Abiotic Stress
3. Discussion
3.1. Structural Features of DNA-deMTase in S. italica
3.2. Expression of the DNA-deMTase Gene in Abiotic Stress
4. Materials and Methods
4.1. Identification of DNA Demethylase Proteins in S. italica
4.2. Phylogenetic Tree and Evolutionarily Conserved Protein Domain Analysis
4.3. Conserved Motifs and Gene Structure
4.4. Analysis of Cis-Regulatory Elements and Chromosome Localization
4.5. Expression of DNA Demethylases
4.6. Plant Material, Growth Condition and Abiotic Stress Treatment
4.7. Total RNA Extraction and Transcript Abundance Analyzes of DNA-deMTase Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Ann. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.H.; Cokus, S.J.; Zhang, X.Y.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.X.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Du, J.M.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Omony, J.; Nussbaumer, T.; Gutzat, R. DNA methylation analysis in plants: Review of computational tools and future perspectives. Brief. Bioinf. 2020, 21, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Ashapkin, V.V.; Kutueva, L.I.; Aleksandrushkina, N.I.; Vanyushin, B.F. Epigenetic Mechanisms of Plant Adaptation to Biotic and Abiotic Stresses. Int. J. Mol. Sci. 2020, 21, 7457. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Kong, J.H.; Qin, C.; Yu, S.; Tan, J.J.; Chen, Y.R.; Wu, C.Q.; Wang, H.; Shi, Y.; Li, C.Y.; et al. Requirement of CHROMOMETHYLASE3 for somatic inheritance of the spontaneous tomato epimutation Colourless non-ripening. Sci. Rep. 2015, 5, 9192. [Google Scholar] [CrossRef] [PubMed]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.G.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]
- Zhong, S.L.; Fei, Z.J.; Chen, Y.R.; Zheng, Y.; Huang, M.Y.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef]
- Lee, S.; Choi, J.; Park, J.C.; Choi, J.Y.; Park, J.W.; Hong, C.P.; Choi, D.; Han, S.; Choi, K.U.; Roh, T.Y.; et al. DDM1-mediated gene body DNA methylation is associated with inducible activation of defense-related genes in Arabidopsis. Genome Biol. 2023, 24, 106. [Google Scholar] [CrossRef]
- Macdonald, W.A. Epigenetic mechanisms of genomic imprinting: Common themes in the regulation of imprinted regions in mammals, plants, and insects. Genet. Res. Int. 2012, 2012, 585024. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.F.; An, Y.Q.C.; et al. Dynamic DNA Methylation in Plant Growth and Development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Zhu, C.; Zhao, J.H.; Liu, T.; Gao, F.; Zhang, Y.C.; Duan, C.G. DNA methylation-dependent epigenetic regulation of Verticillium dahliae virulence in plants. aBIOTECH. 2023, 4, 185–201. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Zhang, X.; Zhang, M.; Han, D.; Qiu, C.; Han, Z. Dynamics of phytohormone and DNA methylation patterns changes during dormancy induction in strawberry (Fragaria × ananassa Duch.). Plant Cell Rep. 2012, 31, 155–165. [Google Scholar] [CrossRef]
- Deleris, A.; Halter, T.; Navarro, L. DNA Methylation and Demethylation in Plant Immunity. Annu. Rev. Phytopathol. 2016, 54, 579–603. [Google Scholar] [CrossRef] [PubMed]
- Abdulraheem, M.I.; Xiong, Y.; Moshood, A.Y.; Cadenas-Pliego, G.; Zhang, H.; Hu, J. Mechanisms of Plant Epigenetic Regulation in Response to Plant Stress: Recent Discoveries and Implications. Plants 2024, 13, 163. [Google Scholar] [CrossRef]
- Wang, R.H.; Xu, J.H. Genomic DNA methylation and histone methylation. Yi Chuan Hered. 2014, 36, 191–199. [Google Scholar] [CrossRef]
- Zhang, H.M.; Lang, Z.B.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.H.; Zhang, X.Y.; Chen, Z.G.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Kerr, L.; Kafetzopoulos, I.; Grima, R.; Sproul, D. Genome-wide single-molecule analysis of long-read DNA methylation reveals heterogeneous patterns at heterochromatin that reflect nucleosome organisation. PLoS Genet. 2023, 19, e1010958. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.H.; Liu, H.F.; Liu, J.; Hua, W.; Xu, S.M.; Li, J. Systematic Analysis of the DNA Methylase and Demethylase Gene Families in Rapeseed (Brassica napus L.) and Their Expression Variations After Salt and Heat stresses. Int. J. Mol. Sci. 2020, 21, 953. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem 2002, 3, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Active DNA demethylation mediated by DNA glycosylases. Ann. Rev. Genet. 2009, 43, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.K.; Li, Y.; Duan, W.K.; Huang, F.Y.; Hou, X.L. Cold acclimation alters DNA methylation patterns and confers tolerance to heat and increases growth rate in Brassica rapa. J. Exp. Bot. 2017, 68, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA Methylome Is Stable under Transgenerational Drought Stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Z.; Min, L.; Wang, M.J.; Wang, C.Z.; Zhao, Y.L.; Li, Y.Y.; Fang, Q.D.; Wu, Y.L.; Xie, S.; Ding, Y.H.; et al. Disrupted Genome Methylation in Response to High Temperature Has Distinct Affects on Microspore Abortion and Anther Indehiscence. Plant Cell. 2018, 30, 1387–1403. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schï Nberger, B.; Menz, J.; Ludewig, U. Plasticity of DNA methylation and gene expression under zinc deficiency in Arabidopsis roots. Plant Cell Physiol. 2018, 59, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, M.; Zhang, H.Y.; Duan, L.J.; Sun, X.J.; Jiang, Q.Y.; Zhang, H.; Hu, Z. Continuous salt stress-induced long non-coding RNAs and DNA methylation patterns in soybean roots. BMC Genom. 2019, 20, 730. [Google Scholar] [CrossRef]
- Marfil, C.; Ibañez, V.; Alonso, R.; Varela, A.; Bottini, R.; Masuelli, R.; Fontana, A.; Berli, F. Changes in grapevine DNA methylation and polyphenols content induced by solar ultraviolet-B radiation, water deficit and abscisic acid spray treatments. Plant Physiol. Biochem. PPB 2019, 135, 287–294. [Google Scholar] [CrossRef]
- Song, Q.X.; Zhang, T.Z.; Stelly, D.M.; Chen, Z.J. Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 2017, 18, 99. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev. 2000, 10, 217–223. [Google Scholar] [CrossRef]
- Yang, L.; Lang, C.; Wu, Y.; Meng, D.; Yang, T.; Li, D.; Jin, T.; Zhou, X. ROS1-mediated decrease in DNA methylation and increase in expression of defense genes and stress response genes in Arabidopsis thaliana due to abiotic stresses. BMC Plant Biol. 2022, 22, 104. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Narayana Chevala, V.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 5, 14922. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, W.F.; Ren, H.Z.; Xiong, F.; Zhang, H.J.; Yao, L.N.; Liu, Y.; Wang, L.; Wang, X.C.; Yang, Y.J.; et al. Genome-wide Investigation and Expression Analysis of DNA Demethylase Genes in Tea Plant (Camellia Sinensis). J. Tea Sci. 2021, 41, 28–39. [Google Scholar] [CrossRef]
- Liu, Q.X.; Xue, Q.Z.; Xu, J.H. Phylogenetic analysis of DNA demethylase genes in angiosperm. Hereditas 2014, 36, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Mauro-Herrera, M.; Doust, A.N. Domestication and Improvement in the Model C4 Grass, Setaria. Front. Plant Sci. 2018, 9, 719. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, J.; Liu, K.B.; Wu, N.; Li, Y.; Zhou, K.; Ye, M.; Zhang, T.; Zhang, H.; Yang, X.; et al. Earliest domestication of common millet (Panicum miliaceum) in East Asia extended to 10,000 years ago. Proc. Natl. Acad. Sci. USA 2009, 106, 7367–7372. [Google Scholar] [CrossRef] [PubMed]
- Li, P.H.; Brutnell, T.P. Setaria viridis and Setaria italica, model genetic systems for the Panicoid grasses. J. Exp. Bot. 2011, 62, 3031–3037. [Google Scholar] [CrossRef]
- Krishnamurthy, L.; Upadhyaya, H.D.; Gowda, L.L.; Kashiwagi, J.; Purushothaman, R.; Sube, S.; Vadez, V. Large variation for salinity tolerance in the core collection of foxtail millet (Setaria italica (L.) P. Beauv.) germplasm. Crop Pasture Sci. 2013, 65, 353–361. [Google Scholar] [CrossRef]
- Sudhakar, C.; Veeranagamallaiah, G.; Nareshkumar, A.; Sudhakarbabu, O.; Sivakumar, M.; Pandurangaiah, M.; Kiranmai, K.; Lokesh, U. Polyamine metabolism influences antioxidant defense mechanism in foxtail millet (Setaria italica L.) cultivars with different salinity tolerance. Plant Cell Rep. 2015, 34, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Genger, R.K.; Kovac, K.A.; Dennis, E.S.; Peacock, W.J.; Finnegan, E.J. Multiple DNA methyltransferase genes in Arabidopsis thaliana. Plant Mol. Biol. 1999, 41, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Huang, X.; Lan, H.X.; Huma, T.; Bao, Y.M.; Huang, J.; Zhang, H.S. Comprehensive gene expression analysis of the DNA (cytosine-5) methyltransferase family in rice (Oryza sativa L.). Genet. Mol. Res. GMR 2014, 13, 5159–5172. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, G.; Mattsson, O.; Okkels, F.T. Specific Levels of DNA Methylation in Various Tissues, Cell Lines, and Cell Types of Daucus carota. Plant Physiol. 1991, 95, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, M.R.; Venora, G.; Ravalli, C.; Stoilov, L.; Gecheff, K.; Cremonini, R. DNA methylation and chromosomal rearrangements in reconstructed karyotypes of Hordeum vulgare L. Protoplasma 2008, 232, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.X.; Xi, Y.L.; Cheng, B.J.; Zhu, S.W. Genome-wide identification and expression profiling of DNA methyltransferase gene family in maize. Plant Cell Rep. 2014, 33, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Teyssier, E.; Bernacchia, G.; Maury, S.; How Kit, A.; Stammitti-Bert, L.; Rolin, D.; Gallusci, P. Tissue dependent variations of DNA methylation and endoreduplication levels during tomato fruit development and ripening. Planta 2008, 228, 391–399. [Google Scholar] [CrossRef]
- Nakano, Y.; Steward, N.; Sekine, M.; Kusano, T.; Sano, H. A tobacco NtMET1 cDNA encoding a DNA methyltransferase: Molecular characterization and abnormal phenotypes of transgenic tobacco plants. Plant Cell Physiol. 2000, 41, 448–457. [Google Scholar] [CrossRef]
- Garg, R.; Kumari, R.; Tiwari, S.; Goyal, S. Genomic survey, gene expression analysis and structural modeling suggest diverse roles of DNA methyltransferases in legumes. PLoS ONE 2014, 9, e88947. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Cao, D.Y.; Ju, Z.J.; Gao, C.; Mei, X.H.; Fu, D.Q.; Zhu, H.L.; Luo, Y.B.; Zhu, B.Z. Genome-wide identification of cytosine-5 DNA methyltransferases and demethylases in Solanum lycopersicum. Gene 2014, 550, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Victoria, D.; Aliki, K.; Venetia, K.; Georgios, M.; Zoe, H. Spatial and temporal expression of cytosine-5 DNA methyltransferase and DNA demethylase gene families of the Ricinus communis during seed development and drought stress. Plant Growth Regul. 2018, 84, 81–94. [Google Scholar] [CrossRef]
- Gu, T.T.; Ren, S.; Wang, Y.H.; Han, Y.H.; Li, Y. Characterization of DNA methyltransferase and demethylase genes in Fragaria vesca. Mol. Genet. Genom. MGG 2016, 291, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Gao, C.; Bian, X.T.; Zhao, S.Z.; Zhao, C.Z.; Xia, H.; Song, H.; Hou, L.; Wan, S.B.; Wang, X.J. Genome-Wide Identification and Comparative Analysis of Cytosine-5 DNA Methyltransferase and Demethylase Families in Wild and Cultivated Peanut. Front. Plant Sci. 2016, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.M.; Zhang, G.H.; Teixeira da Silva, J.A.; Li, M.Z.; Zhao, C.H.; He, C.M.; Si, C.; Zhang, M.Z.; Duan, J. Genome-wide identification and analysis of DNA methyltransferase and demethylase gene families in Dendrobium officinale reveal their potential functions in polysaccharide accumulation. BMC Plant Biol. 2021, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zhang, S.T.; Zhou, C.Z.; Chen, L.; Fu, H.F.; Li, X.Z.; Lin, Y.L.; Lai, Z.X.; Guo, Y.Q. Genome-wide investigation and transcriptional analysis of cytosine-5 DNA methyltransferase and DNA demethylase gene families in tea plant (Camellia sinensis) under abiotic stress and withering processing. PeerJ. 2020, 8, e8432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; He, X.Q.; Zhao, H.C.; Xu, W.C.; Deng, H.; Wang, H.; Wang, S.Y.; Su, D.; Zheng, Z.L.; Yang, B.; et al. Genome-Wide Identification of DNA Methylases and Demethylases in Kiwifruit (Actinidia chinensis). Front. Plant Sci. 2020, 11, 514993. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.H.; Paszkowski, J. Heat-induced release of epigenetic silencing reveals the concealed role of an imprinted plant gene. PLoS Genet. 2014, 10, e1004806. [Google Scholar] [CrossRef]
- Xu, R.; Wang, Y.H.; Zheng, H.; Lu, W.; Wu, C.G.; Huang, J.G.; Yan, K.; Yang, G.D.; Zheng, C.C. Salt-induced transcription factor MYB74 is regulated by the RNA-directed DNA methylation pathway in Arabidopsis. J. Exp. Bot. 2015, 66, 5997–6008. [Google Scholar] [CrossRef]
- Chang, Y.N.; Zhu, C.; Jiang, J.; Zhang, H.; Zhu, J.K.; Duan, C.G. Epigenetic regulation in plant abiotic stress responses. J. Integr. Plant Biol. 2020, 62, 563–580. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Tan, M.; Wang, L.; Zhao, W.; You, J.; Wang, L.; Yan, X.; Wang, W. The pattern of alternative splicing and DNA methylation alteration and their interaction in linseed (Linum usitatissimum L.) response to repeated drought stresses. Biol. Res. 2023, 56, 12. [Google Scholar] [CrossRef]
- Cui, X.G.; Shi, L.R. Effect of Salt Stress on Different Seed Germination and Seedling Growth of Setaria italica (L.). Heilongjiang Agric. Sci. 2011, 14–16. [Google Scholar]
- Pei, S.S.; Wang, Y.G.; Yin, M.Q.; Yuan, X.Y.; Huang, M.J.; Wen, Y.Y.; Zhao, J. Effect of Compound Seed Soaking on Seed Germination and Seedling Growth in Millet(Setaria italica)under Drought Stress. J. Shanxi Agricult. Sci. 2013, 41, 676–679. [Google Scholar]
- Li, C.; Yue, J.; Wu, X.; Xu, C.; Yu, J. An ABA-responsive DRE-binding protein gene from Setaria italica, SiARDP, the target gene of SiAREB, plays a critical role under drought stress. J. Exp. Bot. 2014, 65, 5415–5427. [Google Scholar] [CrossRef] [PubMed]
- Bilichak, A.; Ilnystkyy, Y.; Hollunder, J.; Kovalchuk, I. The progeny of Arabidopsis thaliana plants exposed to salt exhibit changes in DNA methylation, histone modifications and gene expression. PLoS ONE 2012, 7, e30515. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Zhang, Z.J.; Wu, H.L.; Huang, C.Y.; Shuai, P.; Ye, C.Y.; Tang, S.; Wang, Y.J.; Yang, L.; Wang, J.; et al. Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014, 15 (Suppl 1), S9. [Google Scholar] [CrossRef] [PubMed]
- Rakei, A.; Maali-Amiri, R.; Zeinali, H.; Ranjbar, M. DNA methylation and physio-biochemical analysis of chickpea in response to cold stress. Protoplasma 2016, 253, 61–76. [Google Scholar] [CrossRef]
- Song, Y.; Jia, Z.; Hou, Y.; Ma, X.; Li, L.; Jin, X.; An, L. Roles of DNA Methylation in Cold Priming in Tartary Buckwheat. Front. Plant Sci. 2020, 11, 608540. [Google Scholar] [CrossRef]
- Sun, M.; Yang, Z.; Liu, L.; Duan, L. DNA Methylation in Plant Responses and Adaption to Abiotic Stresses. Int. J. Mol. Sci. 2022, 23, 6910. [Google Scholar] [CrossRef]
- Yu, A.; Lepère, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.L.; Penterman, J.; Fischer, R.L.; Voinnet, O.; et al. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.N.; Liu, D.T.; Zhang, X.; Jiang, W.; Wang, L.; Li, D.S.; Cheng, X.M.; Gao, D.R. Genome-Wide Investigation and Expression Analysis of DNA Demethylase Genes in Wheat (Triticum aestivum) during the Grain Development. J. Triticeae Crops. 2023, 43, 1361–1371. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Kayum, M.A.; Park, J.I.; Nath, U.K.; Saha, G.; Biswas, M.K.; Kim, H.T.; Nou, I.S. Genome-wide characterization and expression profiling of PDI family gene reveals function as abiotic and biotic stress tolerance in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC Genom. 2017, 18, 885. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Fan, Y.; Lai, D.; Yang, H.; Xue, G.; He, A.; Chen, L.; Feng, L.; Ruan, J.; Xiang, D.; Yan, J.; et al. Genome-wide identification and expression analysis of the bHLH transcription factor family and its response to abiotic stress in foxtail millet (Setaria italica L.). BMC Genom. 2021, 22, 778. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Li, J.; Jiao, L.; Li, C.; Zhu, D.; Yu, J. A Non-specific Setaria italica Lipid Transfer Protein Gene Plays a Critical Role under Abiotic Stress. Front. Plant Sci. 2016, 7, 1752. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, A.; Du, Y.; Wang, G.; Li, Y.; Zhao, G.; Wang, X.; Zhang, W.; Cheng, K.; Liu, X.; et al. Foxtail millet (Setaria italica (L.) P. Beauv) CIPKs are responsive to ABA and abiotic stresses. PLoS ONE 2019, 14, e0225091. [Google Scholar] [CrossRef]
- Ogneva, Z.V.; Dubrovina, A.S.; Kiselev, K.V. Age-associated alterations in DNA methylation and expression of methyltransferase and demethylase genes in Arabidopsis thaliana. Biol. Plant. 2016, 60, 628–634. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | No. of Introns | Length (Aa) | MW (kDa) | pI | Instability Index | GRAVY Value | Subcellular Localization |
|---|---|---|---|---|---|---|---|---|
| SiDML5 | Seita.8G104300 | 10 | 358 | 39.74 | 9.67 | 41.68 | −0.455 | Chloroplas. Nucleus. |
| SiDML3a | Seita.1G164500 | 16 | 1060 | 118.04 | 7.84 | 49.08 | −0.549 | Nucleus. |
| SiROS1b | Seita.3G223900 | 19 | 1663 | 185.59 | 6.20 | 56.29 | −0.640 | Nucleus. |
| SiROS1a | Seita.2G152900 | 17 | 2082 | 230.61 | 6.79 | 46.03 | −0.605 | Nucleus. |
| SiROS1d | Seita.1G164200 | 16 | 1954 | 216.94 | 6.45 | 47.77 | −0.672 | Nucleus. |
| SiDML3b | Seita.9G185500 | 11 | 578 | 65.77 | 5.97 | 42.44 | −0.665 | Nucleus. |
| SiDML4 | Seita.4G105300 | 4 | 286 | 31.52 | 9.10 | 56.21 | −0.300 | Nucleus. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Wang, X.; Di, Y.; Li, J.; Li, K.; Wei, H.; Zhang, F.; Su, Z. Systematic Analysis of DNA Demethylase Gene Families in Foxtail Millet (Setaria italica L.) and Their Expression Variations after Abiotic Stresses. Int. J. Mol. Sci. 2024, 25, 4464. https://doi.org/10.3390/ijms25084464
Sun Y, Wang X, Di Y, Li J, Li K, Wei H, Zhang F, Su Z. Systematic Analysis of DNA Demethylase Gene Families in Foxtail Millet (Setaria italica L.) and Their Expression Variations after Abiotic Stresses. International Journal of Molecular Sciences. 2024; 25(8):4464. https://doi.org/10.3390/ijms25084464
Chicago/Turabian StyleSun, Yingying, Xin Wang, Yunfei Di, Jinxiu Li, Keyu Li, Huanhuan Wei, Fan Zhang, and Zhenxia Su. 2024. "Systematic Analysis of DNA Demethylase Gene Families in Foxtail Millet (Setaria italica L.) and Their Expression Variations after Abiotic Stresses" International Journal of Molecular Sciences 25, no. 8: 4464. https://doi.org/10.3390/ijms25084464
APA StyleSun, Y., Wang, X., Di, Y., Li, J., Li, K., Wei, H., Zhang, F., & Su, Z. (2024). Systematic Analysis of DNA Demethylase Gene Families in Foxtail Millet (Setaria italica L.) and Their Expression Variations after Abiotic Stresses. International Journal of Molecular Sciences, 25(8), 4464. https://doi.org/10.3390/ijms25084464