
Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
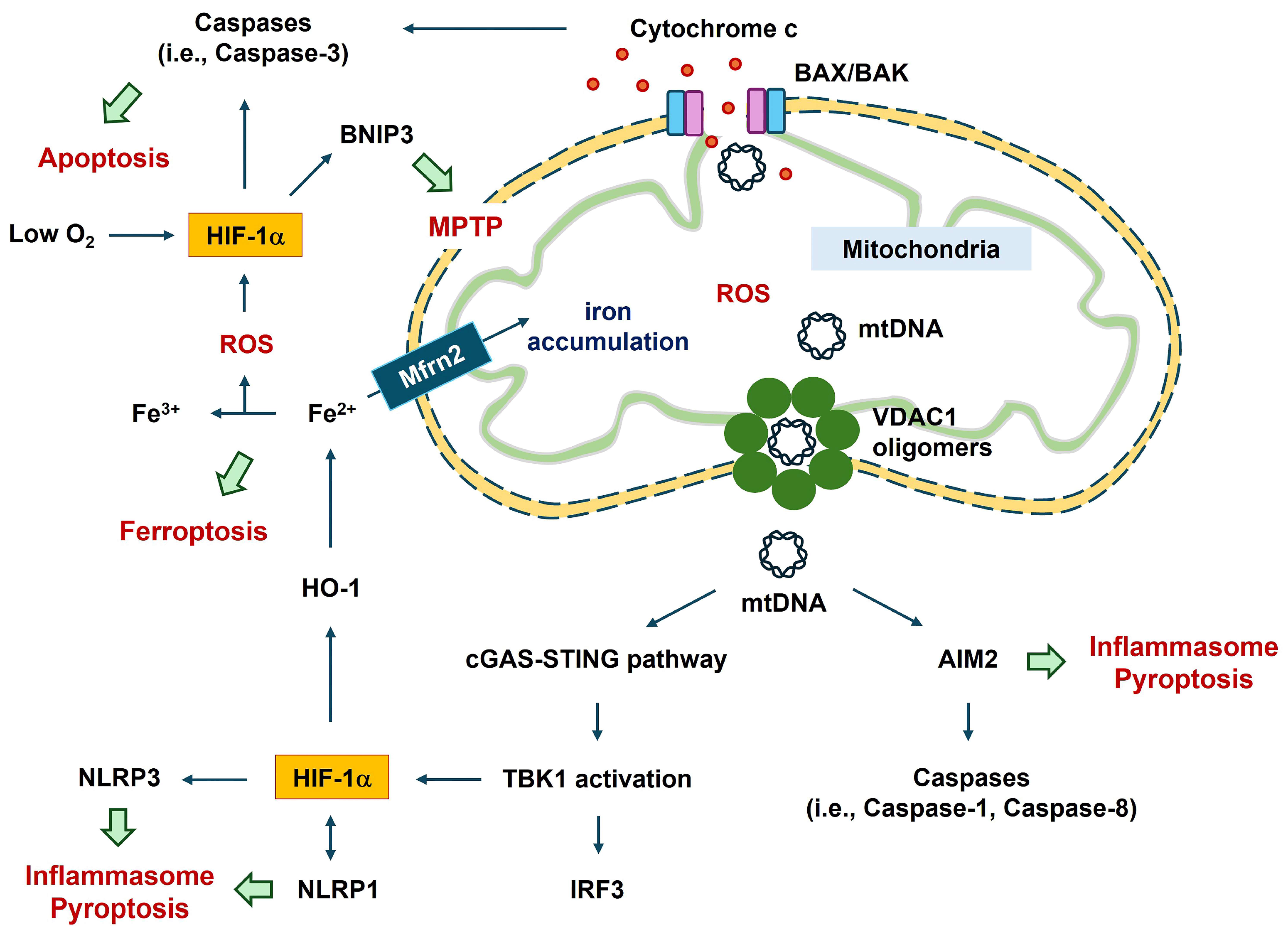
:1. Introduction
2. Role of HIF in Cell Damage
2.1. Apoptosis
2.2. Ferroptosis
3. Role of HIF in Inflammasomes
3.1. Role of HIF in Stroke, with a Focus on Inflammasomes
3.2. Role of HIF in TBI, with a Focus on Inflammasomes
3.3. Role of HIF in AD, Focusing on Inflammasomes
4. Role of HIF-1α in Mitochondrial Functions
4.1. Mitochondrial DNA
4.2. HIF-1α–BNIP3 Axis in Mitochondrial Functions
4.3. Role of HIF-1α in VDAC1-Mediated Mitochondrial Functions
5. Role of HIF-1α in Cellular Activation
5.1. Astrocyte Activation
5.2. Oligodendrocyte Activation
5.3. Microglia/Macrophage Activation
5.4. Vascular Cells
6. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Zhang, F.; Niu, L.; Li, S.; Le, W. Pathological Impacts of Chronic Hypoxia on Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Shapira, R.; Gdalyahu, A.; Gottfried, I.; Sasson, E.; Hadanny, A.; Efrati, S.; Blinder, P.; Ashery, U. Hyperbaric oxygen therapy alleviates vascular dysfunction and amyloid burden in an Alzheimer’s disease mouse model and in elderly patients. Aging 2021, 13, 20935–20961. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Liu, G.; Zeng, X.; Xiang, Y.; Chen, X.; Le, W. Therapeutic effects of long-term HBOT on Alzheimer’s disease neuropathologies and cognitive impairment in APP(swe)/PS1(dE9) mice. Redox Biol. 2024, 70, 103006. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Life with oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Barteczek, P.; Li, L.; Ernst, A.S.; Bohler, L.I.; Marti, H.H.; Kunze, R. Neuronal HIF-1alpha and HIF-2alpha deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Maki, T.; Mandeville, E.T.; Koh, S.H.; Hayakawa, K.; Arai, K.; Kim, Y.M.; Whalen, M.J.; Xing, C.; Wang, X.; et al. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat. Med. 2016, 22, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.L.; Park, J.S.; Manzanero, S.; Choi, Y.; Baik, S.H.; Okun, E.; Gelderblom, M.; Fann, D.Y.; Magnus, T.; Launikonis, B.S.; et al. Evidence that collaboration between HIF-1alpha and Notch-1 promotes neuronal cell death in ischemic stroke. Neurobiol. Dis. 2014, 62, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tao, T.; Xu, J.; Liu, Z.; Zou, Z.; Jin, M. HIF-1alpha attenuates neuronal apoptosis by upregulating EPO expression following cerebral ischemia-reperfusion injury in a rat MCAO model. Int. J. Mol. Med. 2020, 45, 1027–1036. [Google Scholar] [PubMed]
- Hassan, H.; Chen, R. Hypoxia in Alzheimer’s disease: Effects of hypoxia inducible factors. Neural Regen. Res. 2021, 16, 310–311. [Google Scholar] [PubMed]
- Amalia, L.; Sadeli, H.A.; Parwati, I.; Rizal, A.; Panigoro, R. Hypoxia-inducible factor-1alpha in acute ischemic stroke: Neuroprotection for better clinical outcome. Heliyon 2020, 6, e04286. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Kim, Y.E.; Jeon, H.S.; Yoo, M.; Kim, M.; Kim, Y.M.; Koh, S.H.; Choi, Y.K. Chronic hypoxia of endothelial cells boosts HIF-1alpha-NLRP1 circuit in Alzheimer’s disease. Free Radic. Biol. Med. 2023, 204, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Mitroshina, E.V.; Savyuk, M.O.; Ponimaskin, E.; Vedunova, M.V. Hypoxia-Inducible Factor (HIF) in Ischemic Stroke and Neurodegenerative Disease. Front. Cell Dev. Biol. 2021, 9, 703084. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, M.; Choi, Y.K. The Role of a Neurovascular Signaling Pathway Involving Hypoxia-Inducible Factor and Notch in the Function of the Central Nervous System. Biomol. Ther. 2020, 28, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Chang, M.S.; Koh, S.H.; Choi, Y.K. Repair Mechanisms of the Neurovascular Unit after Ischemic Stroke with a Focus on VEGF. Int. J. Mol. Sci. 2021, 22, 8543. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Tyson, D.W.; Rosevear, H.M.; Brosius, F.C., 3rd. Hypoxia-inducible factor-1alpha is a critical mediator of hypoxia induced apoptosis in cardiac H9c2 and kidney epithelial HK-2 cells. BMC Cardiovasc. Disord. 2008, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, Y.; Qin, L.; Xu, X.; Sun, Y.; Zhong, M.; Lu, Y.; Hu, K.; Wei, L.; Chen, J. Oxidative stress drives vascular smooth muscle cell damage in acute Stanford type A aortic dissection through HIF-1alpha/HO-1 mediated ferroptosis. Heliyon 2023, 9, e22857. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zheng, Z.; Guo, Q.; Tian, M.; Yang, J.; Liu, X.; Zhu, X.; Liu, S. The role of HIF-1alpha/HO-1 pathway in hippocampal neuronal ferroptosis in epilepsy. iScience 2023, 26, 108098. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, J.; Zhao, T.; Chen, J.; Kang, L.; Wei, Y.; Han, L.; Shen, L.; Long, C.; Wu, S.; et al. Di-(2-ethylhexyl) phthalate exposure leads to ferroptosis via the HIF-1alpha/HO-1 signaling pathway in mouse testes. J. Hazard. Mater. 2022, 426, 127807. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Guan, S.; Wang, Z.; Ni, H.; Ding, D.; Xu, W.; Li, G. HIF-1alpha aggravated traumatic brain injury by NLRP3 inflammasome-mediated pyroptosis and activation of microglia. J. Chem. Neuroanat. 2021, 116, 101994. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Hong, J.; Lee, J.W.; Kim, S.S.; Sim, H.; Lee, J.C.; Kim, D.W.; Lim, S.S.; Kang, I.J.; Won, M.H. Ischemia-Induced Cognitive Impairment Is Improved via Remyelination and Restoration of Synaptic Density in the Hippocampus after Treatment with COG-Up((R)) in a Gerbil Model of Ischemic Stroke. Vet. Sci. 2021, 8, 321. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Chan, P.H. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid. Redox Signal. 2003, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Ikonomovic, M.D.; Mi, Z.; Abrahamson, E.E. Disordered APP metabolism and neurovasculature in trauma and aging: Combined risks for chronic neurodegenerative disorders. Ageing Res. Rev. 2017, 34, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Desmond, D.W.; Moroney, J.T.; Sano, M.; Stern, Y. Incidence of dementia after ischemic stroke: Results of a longitudinal study. Stroke J. Cereb. Circ. 2002, 33, 2254–2260. [Google Scholar] [CrossRef] [PubMed]
- Snowdon, D.A.; Greiner, L.H.; Mortimer, J.A.; Riley, K.P.; Greiner, P.A.; Markesbery, W.R. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997, 277, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Diener, H.C.; Cunha, L.; Forbes, C.; Sivenius, J.; Smets, P.; Lowenthal, A. European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J. Neurol. Sci. 1996, 143, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhang, J.; Huang, G.; Yan, J.; Xu, C.; Dou, Z.; Sun, C.; Zhang, H. The crosstalk between HIFs and mitochondrial dysfunctions in cancer development. Cell Death Dis. 2021, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Booth, C.; Potten, C.S. What is apoptosis, and why is it important? BMJ 2001, 322, 1536–1538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Liu, H.; Prabhakaran, K.; Zhang, X.; Borowitz, J.L.; Isom, G.E. HIF-1alpha activation by a redox-sensitive pathway mediates cyanide-induced BNIP3 upregulation and mitochondrial-dependent cell death. Free Radic. Biol. Med. 2007, 43, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, Z.; Sandhu, G.; Ma, X.; Yang, X.; Geiger, J.D.; Kong, J. Evidence of oxidative stress-induced BNIP3 expression in amyloid beta neurotoxicity. Brain Res. 2007, 1138, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Lu, J.; Wang, X.; Wu, H.; Mei, S.; Zheng, J.; Xu, S.; Lenahan, C.; Chen, S.; Zhang, J.; et al. HIF-1alpha Mediates TRAIL-Induced Neuronal Apoptosis via Regulating DcR1 Expression Following Traumatic Brain Injury. Front. Cell Neurosci. 2020, 14, 192. [Google Scholar] [CrossRef] [PubMed]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, K.; Hertlein, V.; Jenner, A.; Dellmann, T.; Gojkovic, M.; Pena-Blanco, A.; Dadsena, S.; Wajngarten, N.; Danial, J.S.H.; Thevathasan, J.V.; et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell 2022, 82, 933–949.e9. [Google Scholar] [CrossRef]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- Wincewicz, A.; Sulkowska, M.; Koda, M.; Sulkowski, S. Cumulative expression of HIF-1-alpha, Bax, Bcl-xL and P53 in human colorectal cancer. Pathology 2007, 39, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Wang, M.; Wu, Y.; Du, J.; Li, Y.; Su, A.; Zhong, L.; Xie, Z.; Gong, M.; Liang, J.; et al. The mechanisms of Huangqi Guizhi Wuwu decoction in treating ischaemic stroke based on network pharmacology and experiment verification. Pharm. Biol. 2023, 61, 1014–1029. [Google Scholar] [CrossRef]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Heme oxygenase-1 in Alzheimer disease: A tribute to Moussa Youdim. J. Neural Transm. 2011, 118, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Glial HO-1 expression, iron deposition and oxidative stress in neurodegenerative diseases. Neurotox. Res. 1999, 1, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Kim, Y.M. Beneficial and Detrimental Roles of Heme Oxygenase-1 in the Neurovascular System. Int. J. Mol. Sci. 2022, 23, 7041. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Cressatti, M.; Zukor, H.; Liberman, A.; Galindez, C.; Schipper, H.M. Parkinsonian features in aging GFAP.HMOX1 transgenic mice overexpressing human HO-1 in the astroglial compartment. Neurobiol. Aging 2017, 58, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ye, P.; Kong, C.; Chao, Y.; Yu, W.; Jiang, X.; Luo, J.; Gu, Y.; Chen, S.L. Mitoferrin 2 deficiency prevents mitochondrial iron overload-induced endothelial injury and alleviates atherosclerosis. Exp. Cell Res. 2021, 402, 112552. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, S.; Sun, Z.; Dong, H.; Yu, H.; Huang, M.; Gao, X. Ferroptosis Enhanced Diabetic Renal Tubular Injury via HIF-1alpha/HO-1 Pathway in db/db Mice. Front. Endocrinol. 2021, 12, 626390. [Google Scholar] [CrossRef] [PubMed]
- Henning, Y.; Blind, U.S.; Larafa, S.; Matschke, J.; Fandrey, J. Hypoxia aggravates ferroptosis in RPE cells by promoting the Fenton reaction. Cell Death Dis. 2022, 13, 662. [Google Scholar] [CrossRef] [PubMed]
- Vara-Perez, M.; Rossi, M.; Van den Haute, C.; Maes, H.; Sassano, M.L.; Venkataramani, V.; Michalke, B.; Romano, E.; Rillaerts, K.; Garg, A.D.; et al. BNIP3 promotes HIF-1alpha-driven melanoma growth by curbing intracellular iron homeostasis. EMBO J. 2021, 40, e106214. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef] [PubMed]
- Quiles Del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lozovatsky, L.; Sukumaran, A.; Gonzalez, L.; Jain, A.; Liu, D.; Ayala-Lopez, N.; Finberg, K.E. NCOA4 is regulated by HIF and mediates mobilization of murine hepatic iron stores after blood loss. Blood 2020, 136, 2691–2702. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Coutts, G.; Lenart, N.; Cruickshank, S.M.; Pelegrin, P.; Skinner, J.; Rothwell, N.; Allan, S.M.; Brough, D. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. USA 2015, 112, 4050–4055. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Gerlic, M.; Metcalf, D.; Preston, S.; Pellegrini, M.; O’Donnell, J.A.; McArthur, K.; Baldwin, T.M.; Chevrier, S.; Nowell, C.J.; et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 2012, 37, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Karki, R.; Kanneganti, T.D. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Han, L.; Guo, J.; Wang, X.; Liu, D.; Tian, J.; Zhang, M.; An, F. AIM2 accelerates the atherosclerotic plaque progressions in ApoE-/- mice. Biochem. Biophys. Res. Commun. 2018, 498, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2023, 23, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, G.; Samal, D.; Khandayataray, P.; Murthy, M.K. A Review on Caspases: Key Regulators of Biological Activities and Apoptosis. Mol. Neurobiol. 2023, 60, 5805–5837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, L.; Li, R.; Wei, X.; Luan, W.; Liu, P.; Zhao, J. Hypoxia-Inducible Factor 1-alpha (HIF-1alpha) Induces Apoptosis of Human Uterosacral Ligament Fibroblasts through the Death Receptor and Mitochondrial Pathways. Med. Sci. Monit. 2018, 24, 8722–8733. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Stone, C.R.; Geng, X.; Ding, Y. Hypoxia-inducible factor-1 alpha and RIP3 triggers NLRP3 inflammasome in ischemic stroke. Brain Circ. 2018, 4, 191–192. [Google Scholar] [PubMed]
- Jiang, Q.; Geng, X.; Warren, J.; Eugene Paul Cosky, E.; Kaura, S.; Stone, C.; Li, F.; Ding, Y. Hypoxia Inducible Factor-1alpha (HIF-1alpha) Mediates NLRP3 Inflammasome-Dependent-Pyroptotic and Apoptotic Cell Death Following Ischemic Stroke. Neuroscience 2020, 448, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Min, X.; Chai, Y.; Zhang, J.; Chen, X. NLRP1 Inflammasomes: A Potential Target for the Treatment of Several Types of Brain Injury. Front. Immunol. 2022, 13, 863774. [Google Scholar] [CrossRef]
- Fann, D.Y.; Lim, Y.A.; Cheng, Y.L.; Lok, K.Z.; Chunduri, P.; Baik, S.H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.G.; et al. Evidence that NF-kappaB and MAPK Signaling Promotes NLRP Inflammasome Activation in Neurons Following Ischemic Stroke. Mol. Neurobiol. 2018, 55, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, H.; Lu, X.; Wang, J.; Zhang, X.; Sun, S.; Bao, Z.; Tian, W.; Ning, S.; Wang, L.; et al. Overexpression of MicroRNA-9a-5p Ameliorates NLRP1 Inflammasome-mediated Ischemic Injury in Rats Following Ischemic Stroke. Neuroscience 2020, 444, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, H.; Shindo, A.; Shimada, T.; Yata, K.; Wakita, H.; Takahashi, R.; Tomimoto, H. Chronic cerebral hypoperfusion activates AIM2 and NLRP3 inflammasome. Brain Res. 2020, 1736, 146779. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Tabassum, H.; Parvez, S. NLRP3 inflammasome in traumatic brain injury: Its implication in the disease pathophysiology and potential as a therapeutic target. Life Sci. 2023, 314, 121352. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Li, W.; Huang, S.; Yin, Z.; Xu, X.; Chen, F.; Kong, X.; Wang, H.; Zhang, J.; Lei, P. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res. 2018, 1697, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Brickler, T.; Gresham, K.; Meza, A.; Coutermarsh-Ott, S.; Williams, T.M.; Rothschild, D.E.; Allen, I.C.; Theus, M.H. Nonessential Role for the NLRP1 Inflammasome Complex in a Murine Model of Traumatic Brain Injury. Mediators Inflamm. 2016, 2016, 6373506. [Google Scholar] [CrossRef]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef] [PubMed]
- Ashok, B.S.; Ajith, T.A.; Sivanesan, S. Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin. Exp. Pharmacol. Physiol. 2017, 44, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.F.; Xu, H.; Zhang, Y.W. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Y.W.; Zhang, X.; Liu, R.; Zhang, X.; Hong, S.; Xia, K.; Xia, J.; Zhang, Z.; Xu, H. Transcriptional regulation of APH-1A and increased gamma-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J. 2006, 20, 1275–1277. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Xu, T.; Fang, Q.; Zhang, H.; Yue, L.; Hu, G.; Sun, L. Quercetin hinders microglial activation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy. Redox Biol. 2021, 44, 102010. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Miao, Y.; Xiong, X.; Tan, J.; Han, Z.; Chen, F.; Lei, P.; Zhang, Q. Microglial exosomes alleviate intermittent hypoxia-induced cognitive deficits by suppressing NLRP3 inflammasome. Biol. Direct 2023, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Rapino, C.; Bianchi, G.; Di Giulio, C.; Centurione, L.; Cacchio, M.; Antonucci, A.; Cataldi, A. HIF-1alpha cytoplasmic accumulation is associated with cell death in old rat cerebral cortex exposed to intermittent hypoxia. Aging Cell 2005, 4, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Kung-Chun Chiu, D.; Pui-Wah Tse, A.; Law, C.T.; Ming-Jing Xu, I.; Lee, D.; Chen, M.; Kit-Ho Lai, R.; Wai-Hin Yuen, V.; Wing-Sum Cheu, J.; Wai-Hung Ho, D.; et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, T.U.; Yazihan, N.; Dalgic, A.; Kaya, E.E.; Salman, B.; Kocak, M.; Akcil, E. Role of ATP-dependent K channels in the effects of erythropoietin in renal ischaemia injury. Indian. J. Med. Res. 2015, 141, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, W.; Chen, W.; Wang, H.; Zhang, Y.; Yu, T. Mechanism of the hypoxia inducible factor 1/hypoxic response element pathway in rat myocardial ischemia/diazoxide post-conditioning. Mol. Med. Rep. 2020, 21, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Dimova, E.Y.; Petry, A.; Martinez-Ruiz, A.; Hernansanz-Agustin, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T. The role of the nitric oxide pathway in brain injury and its treatment--from bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Wang, C.; Jin, S.W.; Labrecque, M.P.; Beischlag, T.V.; Brockman, M.A.; Choy, J.C. Expression of human inducible nitric oxide synthase in response to cytokines is regulated by hypoxia-inducible factor-1. Free Radic. Biol. Med. 2019, 130, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sri Swetha Victoria, V.; Praneeth Kumar, P.; Karmakar, S.; Swetha, M.; Reddy, A. Cross-talk between insulin resistance and nitrogen species in hypoxia leads to deterioration of tissue and homeostasis. Int. Immunopharmacol. 2023, 122, 110472. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Y.; Qi, Z.; Cao, L.; Ding, S. Mitochondrial transfer/transplantation: An emerging therapeutic approach for multiple diseases. Cell Biosci. 2022, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Norat, P.; Sokolowski, J.D.; Gorick, C.M.; Soldozy, S.; Kumar, J.S.; Chae, Y.; Yagmurlu, K.; Nilak, J.; Sharifi, K.A.; Walker, M.; et al. Intraarterial Transplantation of Mitochondria after Ischemic Stroke Reduces Cerebral Infarction. Stroke Vasc. Interv. Neurol. 2023, 3, e000644. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimers Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Qu, D.; Xi, Z.; Huan, Y.; Zhang, K.; Yu, C.; Yang, D.; Kang, J.; Lin, W.; Wu, S.; et al. Mitochondria transplantation protects traumatic brain injury via promoting neuronal survival and astrocytic BDNF. Transl. Res. 2021, 235, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Perez-Trevino, P.; Velasquez, M.; Garcia, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Yao, W.; Alsiddig, M.C.; Huo, L.; Liu, H.; Miao, Y.L. Hypoxia-inducible factor-1alpha-dependent autophagy plays a role in glycolysis switch in mouse granulosa cells. Biol. Reprod. 2018, 99, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Li, X.; Tan, R.; An, J.; Cai, Z.; Hu, X.; Wang, F.; Wang, H.; Lu, C.; Lu, H. HIF-1alpha/Beclin1-Mediated Autophagy Is Involved in Neuroprotection Induced by Hypoxic Preconditioning. J. Mol. Neurosci. 2018, 66, 238–250. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1alpha to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar] [PubMed]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1alpha protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.J.; Wang, Z.Y.; Xu, L.; Chen, X.H.; Li, X.X.; Liao, W.T.; Ma, H.K.; Jiang, M.D.; Xu, T.T.; Xu, J.; et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020, 36, 101671. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, A.; Betto, R.M.; Diamante, L.; Tesoriere, A.; Ghirardo, R.; Cioccarelli, C.; Meneghetti, G.; Peron, M.; Laquatra, C.; Tiso, N.; et al. STAT3 and HIF1alpha cooperatively mediate the transcriptional and physiological responses to hypoxia. Cell Death Discov. 2023, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Jiang, J.; Xie, F.; Chen, L. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 2020, 506, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Sun, X.; Zheng, C.; Xue, C.; Jin, Y.; Zhou, N.; Sun, S. The evolutionarily conserved hif-1/bnip3 pathway promotes mitophagy and mitochondrial fission in crustacean testes during hypoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2023, 324, R128–R142. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Mallet, R.T.; Burtscher, M.; Millet, G.P. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing Res. Rev. 2021, 68, 101343. [Google Scholar] [CrossRef] [PubMed]
- Madhu, V.; Boneski, P.K.; Silagi, E.; Qiu, Y.; Kurland, I.; Guntur, A.R.; Shapiro, I.M.; Risbud, M.V. Hypoxic Regulation of Mitochondrial Metabolism and Mitophagy in Nucleus Pulposus Cells Is Dependent on HIF-1alpha-BNIP3 Axis. J. Bone Miner. Res. 2020, 35, 1504–1524. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, L.; Peng, R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules 2022, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, A.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, Y.; Xia, X.; Sun, H.; Gao, J.; Ren, Q.; Zhou, T.; Ma, C.; Xia, J.; Yin, C. Activation of macrophage TBK1-HIF-1alpha-mediated IL-17/IL-10 signaling by hyperglycemia aggravates the complexity of coronary atherosclerosis: An in vivo and in vitro study. FASEB J. 2021, 35, e21609. [Google Scholar] [PubMed]
- Hu, H.; Guo, L.; Overholser, J.; Wang, X. Mitochondrial VDAC1: A Potential Therapeutic Target of Inflammation-Related Diseases and Clinical Opportunities. Cells 2022, 11, 3174. [Google Scholar] [CrossRef] [PubMed]
- Zinghirino, F.; Pappalardo, X.G.; Messina, A.; Nicosia, G.; De Pinto, V.; Guarino, F. VDAC Genes Expression and Regulation in Mammals. Front. Physiol. 2021, 12, 708695. [Google Scholar] [CrossRef] [PubMed]
- Guarino, F.; Zinghirino, F.; Mela, L.; Pappalardo, X.G.; Ichas, F.; De Pinto, V.; Messina, A. NRF-1 and HIF-1alpha contribute to modulation of human VDAC1 gene promoter during starvation and hypoxia in HeLa cells. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148289. [Google Scholar] [CrossRef] [PubMed]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Mizrachi, D.; Keinan, N. Oligomerization of the mitochondrial protein VDAC1: From structure to function and cancer therapy. Prog. Mol. Biol. Transl. Sci. 2013, 117, 303–334. [Google Scholar]
- Riddle, S.R.; Ahmad, A.; Ahmad, S.; Deeb, S.S.; Malkki, M.; Schneider, B.K.; Allen, C.B.; White, C.W. Hypoxia induces hexokinase II gene expression in human lung cell line A549. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L407–L416. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 2004, 19, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Arii, S.; Mori, A.; Isobe, N.; Yang, W.; Oe, H.; Fujimoto, A.; Yonenaga, Y.; Sakashita, H.; Imamura, M. Hexokinase II and VEGF expression in liver tumors: Correlation with hypoxia-inducible factor 1 alpha and its significance. J. Hepatol. 2004, 40, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 2008, 40, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Ramanujan, V.K.; Becker, C.; Fett, S.; Underhill, D.M.; Wolf, A.J. Hexokinase dissociation from mitochondria promotes oligomerization of VDAC that facilitates NLRP3 inflammasome assembly and activation. Sci. Immunol. 2023, 8, eade7652. [Google Scholar] [CrossRef] [PubMed]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The Voltage-dependent Anion Channel 1 Mediates Amyloid beta Toxicity and Represents a Potential Target for Alzheimer Disease Therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. RNA silencing of genes involved in Alzheimer’s disease enhances mitochondrial function and synaptic activity. Biochim. Biophys. Acta 2013, 1832, 2368–2378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; Levin, E.R. Deciphering vascular endothelial cell growth factor/vascular permeability factor signaling to vascular permeability. Inhibition by atrial natriuretic peptide. J. Biol. Chem. 2002, 277, 44385–44398. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Harper, S.J. Regulation of vascular permeability by vascular endothelial growth factors. Vascul Pharmacol. 2002, 39, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: Implications for drug therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Famakin, B.M.; Tsymbalyuk, O.; Tsymbalyuk, N.; Ivanova, S.; Woo, S.K.; Kwon, M.S.; Gerzanich, V.; Simard, J.M. HMGB1 is a Potential Mediator of Astrocytic TLR4 Signaling Activation following Acute and Chronic Focal Cerebral Ischemia. Neurol. Res. Int. 2020, 2020, 3929438. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Li, H.; Li, Y.; Zhang, Y.; Liu, G.; Mi, H.; Li, H.; Xiao, Q.; Niu, L.; Yu, X. Hypoxia-induced HMGB1 promotes glioma stem cells self-renewal and tumorigenicity via RAGE. iScience 2022, 25, 104872. [Google Scholar] [CrossRef]
- Yang, R.; Gao, Y.; Li, H.; Huang, W.; Tu, D.; Yang, M.; Liu, X.; Hong, J.S.; Gao, H.M. Posttranslational S-nitrosylation modification regulates HMGB1 secretion and promotes its proinflammatory and neurodegenerative effects. Cell Rep. 2022, 40, 111330. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Wu, M.; Lu, Y.; Wu, X.; Yu, B.; Chen, R.; Lu, J.; Tong, H. HMGB1-activatied NLRP3 inflammasome induces thrombocytopenia in heatstroke rat. PeerJ 2022, 10, e13799. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Relja, B.; Land, W.G. Damage-associated molecular patterns in trauma. Eur. J. Trauma. Emerg. Surg. 2020, 46, 751–775. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Rinholm, J.E. Mitochondria in Myelinating Oligodendrocytes: Slow and Out of Breath? Metabolites 2021, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Sams, E.C. Oligodendrocytes in the aging brain. Neuronal Signal 2021, 5, NS20210008. [Google Scholar] [CrossRef] [PubMed]
- Manukjan, N.; Majcher, D.; Leenders, P.; Caiment, F.; van Herwijnen, M.; Smeets, H.J.; Suidgeest, E.; van der Weerd, L.; Vanmierlo, T.; Jansen, J.F.A.; et al. Hypoxic oligodendrocyte precursor cell-derived VEGFA is associated with blood-brain barrier impairment. Acta Neuropathol. Commun. 2023, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Allan, K.C.; Hu, L.R.; Scavuzzo, M.A.; Morton, A.R.; Gevorgyan, A.S.; Cohn, E.F.; Clayton, B.L.L.; Bederman, I.R.; Hung, S.; Bartels, C.F.; et al. Non-canonical Targets of HIF1a Impair Oligodendrocyte Progenitor Cell Function. Cell Stem Cell 2021, 28, 257–272 e11. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, M.E.; Hu, J.; Sutter, P.A.; Kim, H.W.; Huang, J.K.; Crocker, S.J. The Cellular Senescence Factor Extracellular HMGB1 Directly Inhibits Oligodendrocyte Progenitor Cell Differentiation and Impairs CNS Remyelination. Front. Cell Neurosci. 2022, 16, 833186. [Google Scholar] [CrossRef]
- Bai, W.; Zhou, J.; Zhou, N.; Liu, Q.; Cui, J.; Zou, W.; Zhang, W. Hypoxia-increased RAGE expression regulates chemotaxis and pro-inflammatory cytokines release through nuclear translocation of NF-kappa B and HIF1alpha in THP-1 cells. Biochem. Biophys. Res. Commun. 2018, 495, 2282–2288. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [PubMed]
- Amersfoort, J.; Eelen, G.; Carmeliet, P. Immunomodulation by endothelial cells—Partnering up with the immune system? Nat. Rev. Immunol. 2022, 22, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Hegdekar, N.; Sarkar, C.; Bustos, S.; Ritzel, R.M.; Hanscom, M.; Ravishankar, P.; Philkana, D.; Wu, J.; Loane, D.J.; Lipinski, M.M. Inhibition of autophagy in microglia and macrophages exacerbates innate immune responses and worsens brain injury outcomes. Autophagy 2023, 19, 2026–2044. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Jing, Y.; Zhao, X.; Wang, M.; Zhang, M.; Ma, R.; Ma, W.; Lv, Y.; Zhu, L. Modulation of the HMGB1/TLR4/NF-kappaB signaling pathway in the CNS by matrine in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2021, 352, 577480. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Lee, D.H.; Jeong, H.S.; Kim, S.H.; Ye, S.K.; Cho, C.H. STAT3 activation in microglia increases pericyte apoptosis in diabetic retinas through TNF-a/AKT/p70S6 kinase signaling. Biochem. Biophys. Res. Commun. 2022, 613, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Kurobe, H.; Sugasawa, N.; Kinoshita, H.; Higashida, M.; Matsuoka, Y.; Yoshida, Y.; Hirata, Y.; Sakata, M.; Maxfield, M.W.; et al. Role of macrophage-derived hypoxia-inducible factor (HIF)-1alpha as a mediator of vascular remodelling. Cardiovasc. Res. 2013, 99, 705–715. [Google Scholar] [CrossRef]
- Halder, S.K.; Milner, R. Mild hypoxia triggers transient blood-brain barrier disruption: A fundamental protective role for microglia. Acta Neuropathol. Commun. 2020, 8, 175. [Google Scholar] [CrossRef]
- Iadecola, C.; Gottesman, R.F. Cerebrovascular Alterations in Alzheimer Disease. Circ. Res. 2018, 123, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J. Brain microvasculature and hypoxia-related proteins in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2011, 4, 616–627. [Google Scholar] [PubMed]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimers Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Brohi, K.; Calfee, C.S.; Rahn, P.; Chesebro, B.B.; Christiaans, S.C.; Carles, M.; Howard, M.; Pittet, J.F. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: Role of injury severity and tissue hypoperfusion. Crit. Care 2009, 13, R174. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zheng, Y.; Sun, L.; Badea, S.R.; Jin, Y.; Liu, Y.; Rolfe, A.J.; Sun, H.; Wang, X.; Cheng, Z.; et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat. Neurosci. 2019, 22, 421–435. [Google Scholar] [CrossRef]
- Hong, K.H.; Ryu, J.; Han, K.H. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 2005, 105, 1405–1407. [Google Scholar] [CrossRef] [PubMed]
- Sandsmark, D.K.; Bashir, A.; Wellington, C.L.; Diaz-Arrastia, R. Cerebral Microvascular Injury: A Potentially Treatable Endophenotype of Traumatic Brain Injury-Induced Neurodegeneration. Neuron 2019, 103, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Ovase, R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Thirumangalakudi, L.; Samany, P.G.; Owoso, A.; Wiskar, B.; Grammas, P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Alluri, H.; Wilson, R.L.; Anasooya Shaji, C.; Wiggins-Dohlvik, K.; Patel, S.; Liu, Y.; Peng, X.; Beeram, M.R.; Davis, M.L.; Huang, J.H.; et al. Melatonin Preserves Blood-Brain Barrier Integrity and Permeability via Matrix Metalloproteinase-9 Inhibition. PLoS ONE 2016, 11, e0154427. [Google Scholar] [CrossRef] [PubMed]
- Muradashvili, N.; Benton, R.L.; Saatman, K.E.; Tyagi, S.C.; Lominadze, D. Ablation of matrix metalloproteinase-9 gene decreases cerebrovascular permeability and fibrinogen deposition post traumatic brain injury in mice. Metab. Brain Dis. 2015, 30, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.Y.; Guo, Z.N.; Zhang, D.H.; Qu, Y.; Jin, H. The Role of Pericytes in Ischemic Stroke: Fom Cellular Functions to Therapeutic Targets. Front. Mol. Neurosci. 2022, 15, 866700. [Google Scholar] [CrossRef] [PubMed]
- Zozulya, A.; Weidenfeller, C.; Galla, H.J. Pericyte-endothelial cell interaction increases MMP-9 secretion at the blood-brain barrier in vitro. Brain Res. 2008, 1189, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guan, C.; Liu, C.; Li, H.; Wu, J.; Sun, C. Targeting hypoxia-inducible factor-1alpha: A new strategy for triple-negative breast cancer therapy. Biomed. Pharmacother. 2022, 156, 113861. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, F.; Yang, H.; Wang, Z. Action Sites and Clinical Application of HIF-1alpha Inhibitors. Molecules 2022, 27, 3426. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, Y.K. Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases. Int. J. Mol. Sci. 2024, 25, 4465. https://doi.org/10.3390/ijms25084465
Choi YK. Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases. International Journal of Molecular Sciences. 2024; 25(8):4465. https://doi.org/10.3390/ijms25084465
Chicago/Turabian StyleChoi, Yoon Kyung. 2024. "Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases" International Journal of Molecular Sciences 25, no. 8: 4465. https://doi.org/10.3390/ijms25084465
APA StyleChoi, Y. K. (2024). Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases. International Journal of Molecular Sciences, 25(8), 4465. https://doi.org/10.3390/ijms25084465