
Microcephaly Gene Mcph1 Deficiency Induces p19ARF-Dependent Cell Cycle Arrest and Senescence

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Mcph1 Knockout Results in Delayed Growth Early in the Embryonic Stage
2.2. Mcph1 Deletion Affected Metabolism and Development Processes
2.3. The DEGs Caused by Mcph1 Depletion Are Not Enriched for Transcription Factor E2F1
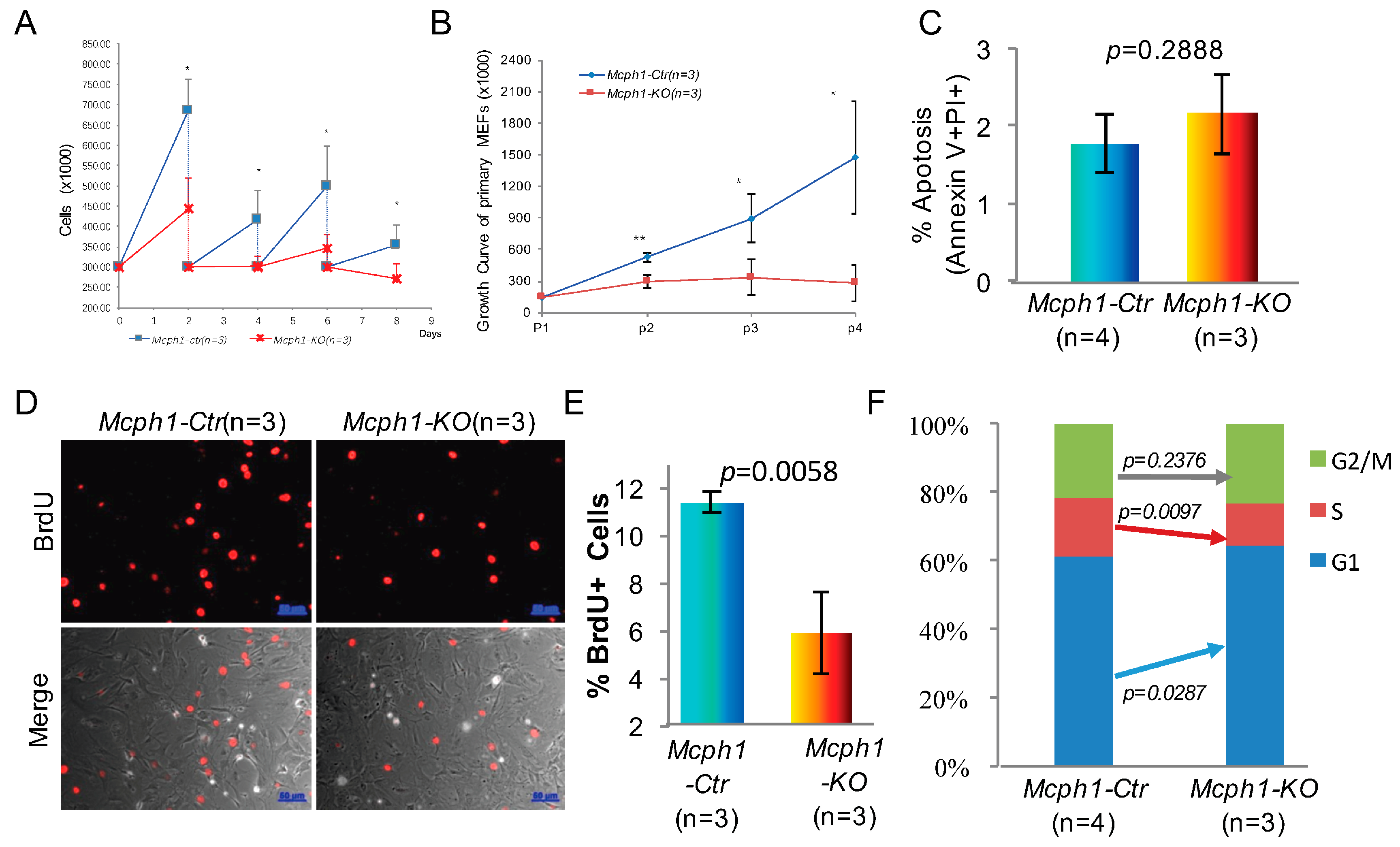
2.4. Mcph1 Knockout Blocks Proliferation and Increases Senescence in MEFs
2.5. Growth Inhibition in the Mcph1-KO Cells Can Be Rescued by the Depletion of p19Arf
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. RNA-Seq and Data Analysis
4.3. RNA Isolation and PCR Analysis
4.4. Primary MEFs Isolation and Culture
4.5. Proliferation Assay of Primary MEFs
4.6. BrdU Labeling and Staining
4.7. Cell Cycle Analysis
4.8. Annexin Apoptosis Assays
4.9. Senescence-Associated β-Galactosidase Staining
4.10. Western Blotting
4.11. Luciferase Reporter Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, A.; Markandaya, M.; Girimaji, S.C. Primary microcephaly: Microcephalin and ASPM determine the size of the human brain. J. Biosci. 2002, 27, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G.; Bond, J.; Enard, W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005, 76, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, Z.W.; Wang, Z.Q. The DNA damage response molecule MCPH1 in brain development and beyond. Acta. Biochim. Biophys. Sin. 2016, 48, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Pulvers, J.N.; Journiac, N.; Arai, Y.; Nardelli, J. MCPH1: A window into brain development and evolution. Front. Cell. Neurosci. 2015, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Gruber, R.; Zhou, Z.; Sukchev, M.; Joerss, T.; Frappart, P.O.; Wang, Z.Q. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat. Cell Biol. 2011, 13, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.P.; McHale, D.P.; Campbell, D.A.; Jafri, H.; Rashid, Y.; Mannan, J.; Karbani, G.; Corry, P.; Levene, M.I.; Mueller, R.F.; et al. Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am. J. Hum. Genet. 1998, 63, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.P.; Eastwood, H.; Bell, S.M.; Adu, J.; Toomes, C.; Carr, I.M.; Roberts, E.; Hampshire, D.J.; Crow, Y.J.; Mighell, A.J.; et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 2002, 71, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, H.; Neumann, L.M.; Schindler, D.; Wirges, A.; Tönnies, H.; Trimborn, M.; Krebsova, A.; Richter, R.; Sperling, K. Premature chromosome condensation in humans associated with microcephaly and mental retardation: A novel autosomal recessive condition. Am. J. Hum. Genet. 2002, 70, 1015–1022. [Google Scholar] [CrossRef]
- Trimborn, M.; Bell, S.M.; Felix, C.; Rashid, Y.; Jafri, H.; Griffiths, P.D.; Neumann, L.M.; Krebs, A.; Reis, A.; Sperling, K.; et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am. J. Hum. Genet. 2004, 75, 261–266. [Google Scholar] [CrossRef]
- Ke, Q.; Li, W.; Lai, X.; Chen, H.; Huang, L.; Kang, Z.; Li, K.; Ren, J.; Lin, X.; Zheng, H.; et al. TALEN-based generation of a cynomolgus monkey disease model for human microcephaly. Cell Res. 2016, 26, 1048–1061. [Google Scholar] [CrossRef]
- Liang, Y.; Gao, H.; Lin, S.Y.; Peng, G.; Huang, X.; Zhang, P.; Goss, J.A.; Brunicardi, F.C.; Multani, A.S.; Chang, S.; et al. BRIT1/MCPH1 is essential for mitotic and meiotic recombination DNA repair and maintaining genomic stability in mice. PLoS Genet. 2010, 6, e1000826. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.W.; Tapias, A.; Bruhn, C.; Gruber, R.; Sukchev, M.; Wang, Z.Q. DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair 2013, 12, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Elledge, S.J. Multiple tumor suppressor pathways negatively regulate telomerase. Cell 2003, 113, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Yim, E.K.; Dai, H.; Jackson, A.P.; Burgt, I.; Pan, M.R.; Hu, R.; Li, K.; Lin, S.Y. BRIT1/MCPH1 links chromatin remodelling to DNA damage response. Nat. Cell Biol. 2009, 11, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Li, M.; Lin, Q.; Qi, X.; Su, B. Functional divergence of the brain-size regulating gene MCPH1 during primate evolution and the origin of humans. BMC Biol. 2013, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Z.; Lin, F.T.; Lin, W.C. MCPH1/BRIT1 cooperates with E2F1 in the activation of checkpoint, DNA repair and apoptosis. EMBO Rep. 2008, 9, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar]
- Wagner, K.D.; Wagner, N. The Senescence Markers p16INK4A, p14ARF/p19ARF, and p21 in Organ Development and Homeostasis. Cells 2022, 11, 1966. [Google Scholar] [CrossRef]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef]
- Sherr, C.J.; DePinho, R.A. Cellular senescence: Mitotic clock or culture shock? Cell 2000, 102, 407–410. [Google Scholar] [CrossRef]
- Liu, X.; Schneble-Löhnert, N.; Kristofova, M.; Qing, X.; Labisch, J.; Hofmann, S.; Ehrenberg, S.; Sannai, M.; Jörß, T.; Ori, A.; et al. The N-terminal BRCT domain determines MCPH1 function in brain development and fertility. Cell Death Dis. 2021, 12, 143. [Google Scholar] [CrossRef]
- Biswas, A.K.; Johnson, D.G. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res. 2012, 72, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Stanelle, J.; Pützer, B.M. E2F1-induced apoptosis: Turning killers into therapeutics. Trends Mol. Med. 2006, 12, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Denechaud, P.D.; Fajas, L.; Giralt, A. E2F1, a Novel Regulator of Metabolism. Front. Endocrinol. 2017, 8, 311. [Google Scholar] [CrossRef] [PubMed]
- Inoshita, S.; Terada, Y.; Nakashima, O.; Kuwahara, M.; Sasaki, S.; Marumo, F. Regulation of the G1/S transition phase in mesangial cells by E2F1. Kidney Int. 1999, 56, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Hasbaoui, B.E.; Elyajouri, A.; Abilkassem, R.; Agadr, A. Nijmegen breakage syndrome: Case report and review of literature. Pan Afr. Med. J. 2020, 35, 85. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Jeggo, P.A. The role of the DNA damage response pathways in brain development and microcephaly: Insight from human disorders. DNA Repair 2008, 7, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Gavvovidis, I.; Pöhlmann, C.; Marchal, J.A.; Stumm, M.; Yamashita, D.; Hirano, T.; Schindler, D.; Neitzel, H.; Trimborn, M. MCPH1 patient cells exhibit delayed release from DNA damage-induced G2/M checkpoint arrest. Cell Cycle 2010, 9, 4893–4899. [Google Scholar] [CrossRef] [PubMed]
- Journiac, N.; Gilabert-Juan, J.; Cipriani, S.; Benit, P.; Liu, X.; Jacquier, S.; Faivre, V.; Delahaye-Duriez, A.; Csaba, Z.; Hourcade, T.; et al. Cell Metabolic Alterations due to Mcph1 Mutation in Microcephaly. Cell Rep. 2020, 31, 107506. [Google Scholar] [CrossRef]
- Cheung, A.H.; Hui, C.H.; Wong, K.Y.; Liu, X.; Chen, B.; Kang, W.; To, K.F. Out of the cycle: Impact of cell cycle aberrations on cancer metabolism and metastasis. Int. J. Cancer 2023, 152, 1510–1525. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Shi, L.; Wan, L.; Inuzuka, H.; Long, J.; Guo, J.; Zhang, J.; Yuan, M.; Zhang, S.; et al. Skp2 dictates cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle. Cell Res. 2021, 31, 80–93. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Roussel, M.F. The INK4 family of cell cycle inhibitors in cancer. Oncogene 1999, 18, 5311–5317. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules 2019, 9, 87. [Google Scholar] [CrossRef]
- Westbury, J.; Watkins, M.; Ferguson-Smith, A.C.; Smith, J. Dynamic temporal and spatial regulation of the cdk inhibitor p57(kip2) during embryo morphogenesis. Mech. Dev. 2001, 109, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, L.; Wright, G.M.; Pillai, S.R.; Chellappan, S.P.; Cress, W.D. CDKN1C negatively regulates RNA polymerase II C-terminal domain phosphorylation in an E2F1-dependent manner. J. Biol. Chem. 2010, 285, 9813–9822. [Google Scholar] [CrossRef]
- Berland, S.; Haukanes, B.I.; Juliusson, P.B.; Houge, G. Deep exploration of a CDKN1C mutation causing a mixture of Beckwith-Wiedemann and IMAGe syndromes revealed a novel transcript associated with developmental delay. J. Med. Genet. 2022, 59, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef]
- Ge, C.; Che, L.; Ren, J.; Pandita, R.K.; Lu, J.; Li, K.; Pandita, T.K.; Du, C. BRUCE regulates DNA double-strand break response by promoting USP8 deubiquitination of BRIT1. Proc. Natl. Acad. Sci. USA 2015, 112, E1210–E1219. [Google Scholar] [CrossRef]
- Khan, S.H.; Moritsugu, J.; Wahl, G.M. Differential requirement for p19ARF in the p53-dependent arrest induced by DNA damage, microtubule disruption, and ribonucleotide depletion. Proc. Natl. Acad. Sci. USA 2000, 97, 3266–3271. [Google Scholar] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Yevshin, I.; Sharipov, R.; Kolmykov, S.; Kondrakhin, Y.; Kolpakov, F. GTRD: A database on gene transcription regulation-2019 update. Nucleic Acids Res. 2019, 47, D100–D105. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Yao, S.; Xu, M.D.; Wang, Y.; Zhao, S.T.; Wang, J.; Chen, G.F.; Chen, W.B.; Liu, J.; Huang, G.B.; Sun, W.J.; et al. Astrocytic lactate dehydrogenase A regulates neuronal excitability and depressive-like behaviors through lactate homeostasis in mice. Nat. Commun. 2023, 14, 729. [Google Scholar] [CrossRef]
- Casas, B.S.; Vitória, G.; do Costa, M.N.; Madeiro da Costa, R.; Trindade, P.; Maciel, R.; Navarrete, N.; Rehen, S.K.; Palma, V. hiPSC-derived neural stem cells from patients with schizophrenia induce an impaired angiogenesis. Transl. Psychiatry 2018, 8, 48. [Google Scholar] [CrossRef]
- Hoffman, J.L.; Faccidomo, S.; Kim, M.; Taylor, S.M.; Agoglia, A.E.; May, A.M.; Smith, E.N.; Wong, L.C.; Hodge, C.W. Alcohol drinking exacerbates neural and behavioral pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Int. Rev. Neurobiol. 2019, 148, 169–230. [Google Scholar]
- Moreau, J.L.; Artap, S.T.; Shi, H.; Chapman, G.; Leone, G.; Sparrow, D.B.; Dunwoodie, S.L. Cited2 is required in trophoblasts for correct placental capillary patterning. Dev. Biol. 2014, 392, 62–79. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Bosanko, K.A.; Caffrey, A.R.; Bernstein, J.A.; Martin, D.M.; Williams, M.S.; Berry-Kravis, E.M.; Mark, P.R.; Manning, M.A.; Bhambhani, V.; et al. Mutation update for the SATB2 gene. Hum. Mutat. 2019, 40, 1013–1029. [Google Scholar] [CrossRef] [PubMed]
- El Waly, B.; Mignon-Ravix, C.; Cacciagli, P.; Buhler, E.; Ben Zeev, B.; Villard, L. Molecular characterization of a 1p36 chromosomal duplication and in utero interference define ENO1 as a candidate gene for polymicrogyria. Eur. J. Hum. Genet. 2020, 28, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Mu, H.; Zheng, L.; Li, B.; Wu, C.; Niu, B.; Shen, Q.; He, X.; Hua, J. EIF2S3Y suppresses the pluripotency state and promotes the proliferation of mouse embryonic stem cells. Oncotarget 2016, 7, 11321–11331. [Google Scholar] [CrossRef] [PubMed]
- Paduano, F.; Colao, E.; Loddo, S.; Orlando, V.; Trapasso, F.; Novelli, A.; Perrotti, N.; Iuliano, R. 7q35 Microdeletion and 15q13.3 and Xp22.33 Microduplications in a Patient with Severe Myoclonic Epilepsy, Microcephaly, Dysmorphisms, Severe Psychomotor Delay and Intellectual Disability. Genes 2020, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- George, E.L.; Georges-Labouesse, E.N.; Patel-King, R.S.; Rayburn, H.; Hynes, R.O. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 1993, 119, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.; Hu, J.; Cai, D.; Chen, S.; Dunn, T.W.; Pearce, K.; Glanzman, D.L.; Schacher, S.; Sossin, W.S. Isoform Specificity of PKMs during Long-Term Facilitation in Aplysia Is Mediated through Stabilization by KIBRA. J. Neurosci. 2019, 39, 8632–8644. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.; Neri, J.; Souza, C.R.M.; Valverde, J.G.; De Araújo, J.M.G.; Nascimento, M.; Branco, R.C.C.; Arrais, N.M.R.; Lassmann, T.; Blackwell, J.M.; et al. Zika Virus Changes Methylation of Genes Involved in Immune Response and Neural Development in Brazilian Babies Born with Congenital Microcephaly. J. Infect. Dis. 2021, 223, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, H.; Cho, M.T.; Retterer, K.; Person, R.; Schaefer, G.B.; Danylchuk, N.; Malik, S.; Wechsler, S.B.; Wheeler, P.G.; van Gassen, K.L.; et al. Mutations in HIVEP2 are associated with developmental delay, intellectual disability, and dysmorphic features. Neurogenetics 2016, 17, 159–164. [Google Scholar] [CrossRef]
- Kerns, S.L.; Guevara-Aguirre, J.; Andrew, S.; Geng, J.; Guevara, C.; Guevara-Aguirre, M.; Guo, M.; Oddoux, C.; Shen, Y.; Zurita, A.; et al. A novel variant in CDKN1C is associated with intrauterine growth restriction, short stature, and early-adulthood-onset diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E2117–E2122. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.-N.; Gao, Y.; Lai, X.; Li, X.; Liu, G.; Ding, M.; Wang, Z.; Guo, Z.; Qin, Y.; Li, X.; et al. Microcephaly Gene Mcph1 Deficiency Induces p19ARF-Dependent Cell Cycle Arrest and Senescence. Int. J. Mol. Sci. 2024, 25, 4597. https://doi.org/10.3390/ijms25094597
Jiang Y-N, Gao Y, Lai X, Li X, Liu G, Ding M, Wang Z, Guo Z, Qin Y, Li X, et al. Microcephaly Gene Mcph1 Deficiency Induces p19ARF-Dependent Cell Cycle Arrest and Senescence. International Journal of Molecular Sciences. 2024; 25(9):4597. https://doi.org/10.3390/ijms25094597
Chicago/Turabian StyleJiang, Yi-Nan, Yizhen Gao, Xianxin Lai, Xinjie Li, Gen Liu, Mingmei Ding, Zhiyi Wang, Zixiang Guo, Yinying Qin, Xin Li, and et al. 2024. "Microcephaly Gene Mcph1 Deficiency Induces p19ARF-Dependent Cell Cycle Arrest and Senescence" International Journal of Molecular Sciences 25, no. 9: 4597. https://doi.org/10.3390/ijms25094597
APA StyleJiang, Y. -N., Gao, Y., Lai, X., Li, X., Liu, G., Ding, M., Wang, Z., Guo, Z., Qin, Y., Li, X., Sun, L., Wang, Z. -Q., & Zhou, Z. -W. (2024). Microcephaly Gene Mcph1 Deficiency Induces p19ARF-Dependent Cell Cycle Arrest and Senescence. International Journal of Molecular Sciences, 25(9), 4597. https://doi.org/10.3390/ijms25094597