
The Impact of the Aryl Hydrocarbon Receptor on Antenatal Chemical Exposure-Induced Cardiovascular–Kidney–Metabolic Programming


Abstract
:1. Introduction
2. Aryl Hydrocarbon Receptor
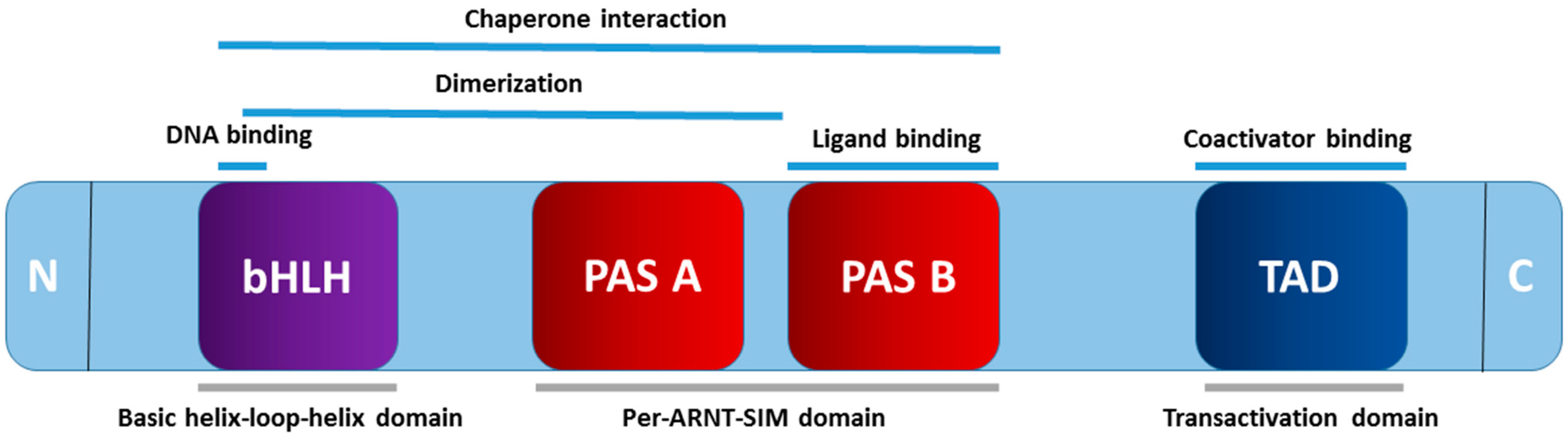
2.1. The Structure of AHR
2.2. AHR Ligands
2.3. AHR Signaling
3. AHR and CKM Syndrome
3.1. Cardiovascular Disease and Hypertension
3.2. Kidney Disease
3.3. Diabetes, Obesity, and NAFLD
4. Epidemiological Evidence: The Link between Chemical Exposure and CKM Syndrome
4.1. Dioxins
4.2. Plastic Chemicals
4.3. Per- and Polyfluoroalkyl Substances (PFAS)
4.4. Polycyclic Aromatic Hydrocarbon
4.5. Air Pollution
4.6. Heavy Metals
5. Evidence from Animal Models: The Role of AHR in CKM Programming
6. Reprogramming Strategies Targeting AHR Signaling
6.1. Tryptophan Metabolites
6.2. Resveratrol
6.3. Butyrate
6.4. Others
7. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hanson, M.; Gluckman, P. Developmental origins of noncommunicable disease: Population and public health implications. Am. J. Clin. Nutr. 2011, 94, 1754S–1758S. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Gluckman, P.D. Early developmental conditioning of later health and disease: Physiology or pathophysiology? Physiol. Rev. 2014, 94, 1027–1076. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.P.; Velazquez, M.A.; Eckert, J.J. Embryos, DOHaD and David Barker. J. Dev. Orig. Health Dis. 2015, 6, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.G.B.; Alves, L.V. Early-life nutrition and adult-life outcomes. J. Pediatr. 2024, 100, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Lapehn, S.; Paquette, A.G. The Placental Epigenome as a Molecular Link between Prenatal Exposures and Fetal Health Outcomes through the DOHaD Hypothesis. Curr. Environ. Health Rep. 2022, 9, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Fukuoka, H. Developmental origins of health and disease theory in cardiology. J. Cardiol. 2020, 76, 14–17. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecologists. Exposure to toxic environmental agents. Obstet. Gynecol. 2013, 122, 931–935. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (US) Committee on Improving Risk Analysis Approaches Used by the U.S. EPA. Science and Decisions: Advancing Risk Assessment; National Academies Press (US): Washington, DC, USA, 2009. [Google Scholar]
- Centers For Disease Control and Prevention. Fourth National Report on Human Exposure to Environmental Chemicals, Updated Tables, January 2019; US Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019.
- Jaradat, J.H.; Nashwan, A.J. Cardiovascular-kidney-metabolic syndrome: Understanding the interconnections and the need for holistic intervention. J. Med. Surg. Public Health 2023, 1, 100028. [Google Scholar] [CrossRef]
- Ndumele, C.E.; Rangaswami, J.; Chow, S.L.; Neeland, I.J.; Tuttle, K.R.; Khan, S.S.; Coresh, J.; Mathew, R.O.; Baker-Smith, C.M.; Carnethon, M.R.; et al. American Heart Association. Cardiovascular-Kidney-Metabolic Health: A Presidential Advisory from the American Heart Association. Circulation 2023, 148, 1606–1635. [Google Scholar] [CrossRef]
- Hoffman, D.J.; Powell, T.L.; Barrett, E.S.; Hardy, D.B. Developmental origins of metabolic diseases. Physiol. Rev. 2021, 101, 739–795. [Google Scholar] [CrossRef]
- Iturzaeta, A.; Sáenz Tejeira, M.M. Early programming of hypertension. Arch. Argent. Pediatr. 2022, 120, e8–e16. [Google Scholar] [PubMed]
- Chevalier, R.L. Evolution, kidney development, and chronic kidney disease. Semin. Cell Dev. Biol. 2019, 91, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, L.P.J.; Piovan, S.; Moreira, V.M.; Gonçalves, G.D.; Ferreira, A.R.O.; Ribeiro, M.V.G.; Peres, M.N.C.; Almeida, D.L.; Raposo, S.R.; da Silva, M.C.; et al. Epigenetic programming for obesity and noncommunicable disease: From womb to tomb. Rev. Endocr. Metab. Disord. 2023, 25, 309–324. [Google Scholar] [CrossRef]
- Paauw, N.D.; van Rijn, B.B.; Lely, A.T.; Joles, J.A. Pregnancy as a critical window for blood pressure regulation in mother and child: Programming and reprogramming. Acta Physiol. 2017, 219, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Interplay between oxidative stress and nutrient sensing signaling in the developmental origins of cardiovascular disease. Int. J. Mol. Sci. 2017, 18, 841. [Google Scholar] [CrossRef]
- Kett, M.M.; Denton, K.M. Renal programming: Cause for concern? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R791–R803. [Google Scholar] [CrossRef] [PubMed]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Yoo, J.Y.; Valeria Ozorio Dutra, S.; Morgan, K.H.; Groer, M. The Association between Early-Life Gut Microbiota and Long-Term Health and Diseases. J. Clin. Med. 2021, 10, 459. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. The Renin Angiotensin System and the Cardiovascular-Kidney-Metabolic Syndrome: Focus on Early-Life Programming. Int. J. Mol. Sci. 2024, 25, 3298. [Google Scholar] [CrossRef] [PubMed]
- Kou, Z.; Dai, W. Aryl hydrocarbon receptor: Its roles in physiology. Biochem. Pharmacol. 2021, 185, 114428. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef]
- Zablon, H.A.; Ko, C.I.; Puga, A. Converging Roles of the Aryl Hydrocarbon Receptor in Early Embryonic Development, Maintenance of Stemness, and Tissue Repair. Toxicol. Sci. 2021, 182, 1–9. [Google Scholar] [CrossRef]
- Yi, T.; Wang, J.; Zhu, K.; Tang, Y.; Huang, S.; Shui, X.; Ding, Y.; Chen, C.; Lei, W. Aryl Hydrocarbon Receptor: A New Player of Pathogenesis and Therapy in Cardiovascular Diseases. BioMed Res. Int. 2018, 2018, 6058784. [Google Scholar] [CrossRef]
- Brito, J.S.; Borges, N.A.; Esgalhado, M.; Magliano, D.C.; Soulage, C.O.; Mafra, D. Aryl Hydrocarbon Receptor Activation in Chronic Kidney Disease: Role of Uremic Toxins. Nephron 2017, 137, 1–7. [Google Scholar] [CrossRef]
- Sayed, T.S.; Maayah, Z.H.; Zeidan, H.A.; Agouni, A.; Korashy, H.M. Insight into the physiological and pathological roles of the aryl hydrocarbon receptor pathway in glucose homeostasis, insulin resistance, and diabetes development. Cell. Mol. Biol. Lett. 2022, 27, 103. [Google Scholar] [CrossRef]
- Jones, S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004, 5, 226. [Google Scholar] [CrossRef]
- Busbee, P.B.; Rouse, M.; Nagarkatti, M.; Nagarkatti, P.S. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutr. Rev. 2013, 71, 353–369. [Google Scholar] [CrossRef]
- Fukunaga, B.N.; Probst, M.R.; Reisz Porszasz, S.; Hankinson, O. Identification of functional domains of the aryl hydrocarbon receptor. J. Biol. Chem. 1995, 270, 29270–29278. [Google Scholar] [CrossRef]
- Ho, P.P.; Steinman, L. The aryl hydrocarbon receptor: A regulator of Th17 and Treg cell development in disease. Cell Res. 2008, 18, 605–608. [Google Scholar] [CrossRef]
- Kumar, M.B.; Ramadoss, P.; Reen, R.K.; Perdew, G.H. The Q-rich subdomain of the human Ah receptor transactivation domain is required for dioxin-mediated transcriptional activity. J. Biol. Chem. 2001, 276, 42302–42310. [Google Scholar] [CrossRef]
- Hankinson, O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 2005, 433, 379–386. [Google Scholar] [CrossRef]
- Wright, E.J.; Pereira De Castro, J.; Joshi, A.D.; Elferink, C.J. Canonical and non-canonical aryl hydrocarbon receptor signaling pathways. Curr. Opin. Toxicol. 2017, 2, 87–92. [Google Scholar] [CrossRef]
- Avilla, M.N.; Malecki, K.M.C.; Hahn, M.E.; Wilson, R.H.; Bradfield, C.A. The Ah receptor: Adaptive metabolism, ligand diversity, and the xenokine model. Chem. Res. Toxicol. 2020, 33, 860–879. [Google Scholar] [CrossRef]
- Meyer, B.K.; Petrulis, J.R.; Perdew, G.H. Aryl hydrocarbon (Ah) receptor levels are selectively modulated by hsp90-associated immunophilin homolog XAP2. Cell Stress Chaperones 2000, 5, 243–254. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Jackson, D.P.; Joshi, A.D.; Elferink, C.J. Ah receptor pathway intricacies; signaling through diverse protein partners and DNA-motifs. Toxicol. Res. 2015, 4, 1143–1158. [Google Scholar] [CrossRef]
- Vogel, C.F.; Matsumura, F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family. Biochem. Pharmacol. 2009, 77, 734–745. [Google Scholar] [CrossRef]
- Huang, G.; Elferink, C.J. A novel nonconsensus xenobiotic response element capable of mediating aryl hydrocarbon receptor-dependent gene expression. Mol. Pharmacol. 2012, 81, 338–347. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Wang, K.; Fang, R.; Zheng, J. Expression of aryl hydrocarbon receptor in human placentas and fetal tissues. J. Histochem. Cytochem. 2010, 58, 679–685. [Google Scholar] [CrossRef]
- Lund, A.K.; Goens, M.B.; Nuñez, B.A.; Walker, M.K. Characterizing the role of endothelin-1 in the progression of cardiac hypertrophy in aryl hydrocarbon receptor (AhR) null mice. Toxicol. Appl. Pharmacol. 2006, 212, 127–135. [Google Scholar] [CrossRef]
- Thackaberry, E.A.; Gabaldon, D.M.; Walker, M.K.; Smith, S.M. Aryl hydrocarbon receptor null mice develop cardiac hypertrophy and increased hypoxia-inducible factor-1alpha in the absence of cardiac hypoxia. Cardiovasc. Toxicol. 2002, 2, 263–274. [Google Scholar] [CrossRef]
- Ko, C.I.; Fan, Y.; de Gannes, M.; Wang, Q.; Xia, Y.; Puga, A. Repression of the Aryl Hydrocarbon Receptor Is Required to Maintain Mitotic Progression and Prevent Loss of Pluripotency of Embryonic Stem Cells. Stem Cells 2016, 34, 2825–2839. [Google Scholar] [CrossRef]
- Lahvis, G.P.; Lindell, S.L.; Thomas, R.S.; McCuskey, R.S.; Murphy, C.; Glover, E.; Bentz, M.; Southard, J.; Bradfield, C.A. Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 10442–10447. [Google Scholar] [CrossRef]
- Ichihara, S.; Yamada, Y.; Ichihara, G.; Nakajima, T.; Li, P.; Kondo, T.; Gonzalez, F.J.; Murohara, T. A role for the aryl hydrocarbon receptor in regulation of ischemia-induced angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1297–1304. [Google Scholar] [CrossRef]
- Cuartero, M.I.; Ballesteros, I.; de la Parra, J.; Harkin, A.L.; Abautret-Daly, A.; Sherwin, E.; Fernández-Salguero, P.; Corbí, A.L.; Lizasoain, I.; Moro, M.A. L-kynurenine/aryl hydrocarbon receptor pathway mediates brain damage after experimental stroke. Circulation 2014, 130, 2040–2051. [Google Scholar] [CrossRef]
- Nakagawa, K.; Kobayashi, F.; Kamei, Y.; Tawa, M.; Ohkita, M. Acute Kynurenine Exposure of Rat Thoracic Aorta Induces Vascular Dysfunction via Superoxide Anion Production. Biol. Pharm. Bull. 2022, 45, 522–527. [Google Scholar] [CrossRef]
- Lund, A.K.; Agbor, L.N.; Zhang, N.; Baker, A.; Zhao, H.; Fink, G.D.; Kanagy, N.L.; Walker, M.K. Loss of the Aryl Hydrocarbon Receptor Induces Hypoxemia, Endothelin-1, and Systemic Hypertension at Modest Altitude. Hypertension 2008, 51, 803–809. [Google Scholar] [CrossRef]
- Zhang, N.; Agbor, L.N.; Scott, J.A.; Zalobowski, T.; Elased, K.M.; Trujillo, A.; Duke, M.S.; Wolf, V.; Walsh, M.T.; Born, J.L.; et al. An Activated Renin-Angiotensin System Maintains Normal Blood Pressure in Aryl Hydrocarbon Receptor Heterozygous Mice but Not in Null Mice. Biochem. Pharmacol. 2010, 80, 197–204. [Google Scholar] [CrossRef]
- Stevens, E.A.; Mezrich, J.D.; Bradfield, C.A. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 2009, 127, 299–311. [Google Scholar] [CrossRef]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef]
- Curran, C.S.; Kopp, J.B. Aryl Hydrocarbon Receptor Mechanisms Affecting Chronic Kidney Disease. Front. Pharmacol. 2022, 13, 782199. [Google Scholar] [CrossRef]
- Ding, M.; Coward, R.J.; Jeansson, M.; Kim, W.; Quaggin, S.E. Regulation of Hypoxia-Inducible Factor 2-a Is Essential for Integrity of the Glomerular Barrier. Am. J. Physiol. Ren. Physiol. 2013, 304, F120–F126. [Google Scholar] [CrossRef]
- Nakano, N.; Sakata, N.; Katsu, Y.; Nochise, D.; Sato, E.; Takahashi, Y.; Yamaguchi, S.; Haga, Y.; Ikeno, S.; Motizuki, M.; et al. Dissociation of the AhR/ARNT complex by TGF-β/Smad signaling represses CYP1A1 gene expression and inhibits benze[a]pyrene-mediated cytotoxicity. J. Biol. Chem. 2020, 295, 9033–9051. [Google Scholar] [CrossRef]
- Thackaberry, E.A.; Bedrick, E.J.; Goens, M.B.; Danielson, L.; Lund, A.K.; Gabaldon, D.; Smith, S.M.; Walker, M.K. Insulin regulation in AhR-null Mice: Embryonic cardiac enlargement, neonatal macrosomia, and altered insulin regulation and response in pregnant and aging AhR-null females. Toxicol. Sci. 2003, 76, 407–417. [Google Scholar] [CrossRef]
- Xia, H.; Zhu, X.; Zhang, X.; Jiang, H.; Li, B.; Wang, Z.; Li, D.; Jin, Y. Alpha-naphthoflavone attenuates non-alcoholic fatty liver disease in oleic acid-treated HepG2 hepatocytes and in high fat diet-fed mice. Biomed. Pharmacother. 2019, 118, 109287. [Google Scholar] [CrossRef]
- Tseng, H.L.; Yang, S.C.; Yang, S.H.; Shieh, K.R. Hepatic circadian-clock system altered by insulin resistance, diabetes and insulin sensitizer in mice. PLoS ONE 2015, 10, e0120380. [Google Scholar] [CrossRef]
- Dou, H.; Duan, Y.; Zhang, X.; Yu, Q.; Di, Q.; Song, Y.; Li, P.; Gong, Y. Aryl hydrocarbon receptor (AhR) regulates adipocyte differentiation by assembling CRL4B ubiquitin ligase to target PPARγ for proteasomal degradation. J. Biol. Chem. 2019, 294, 18504–18515. [Google Scholar] [CrossRef]
- Kern, P.A.; Dicker-Brown, A.; Said, S.T.; Kennedy, R.; Fonseca, V.A. The stimulation of tumor necrosis factor and inhibition of glucose transport and lipoprotein lipase in adipose cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Metabolism 2002, 51, 65–68. [Google Scholar] [CrossRef]
- Ha, M.H.; Lee, D.H.; Jacobs, D.R., Jr. Association between serum concentrations of persistent organic pollutants and self-reported cardiovascular disease prevalence: Results from the National Health and Nutrition Examination Survey, 1999–2002. Environ. Health Perspect. 2007, 115, 1204–1209. [Google Scholar] [CrossRef]
- Magliano, D.J.; Loh, V.H.Y.; Harding, J.L.; Botton, J.; Shaw, J.E. Persistent organic pollutants and diabetes: A review of the epidemiological evidence. Diabetes Metab. 2014, 40, 1–14. [Google Scholar] [CrossRef]
- Gao, J.; Xu, Y.; Zhong, T.; Yu, X.; Wang, L.; Xiao, Y.; Peng, Y.; Sun, Q. A review of food contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin and its toxicity associated with metabolic disorders. Curr. Res. Food Sci. 2023, 7, 100617. [Google Scholar] [CrossRef]
- Kataria, A.; Trasande, L.; Trachtmanm, H. The effects of environmental chemicals on renal function. Nat. Rev. Nephrol. 2015, 11, 610–625. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Adverse Impact of Environmental Chemicals on Developmental Origins of Kidney Disease and Hypertension. Front. Endocrinol. 2021, 12, 745716. [Google Scholar] [CrossRef]
- Chen, M.; Yang, Y.; Baral, K.; Fu, Y.; Meng, Y.; Zhang, Y.; Sun, F.; Zhao, M. Relationship between bisphenol A and the cardiovascular disease metabolic risk factors in American adults: A population-based study. Chemosphere 2023, 324, 138289. [Google Scholar] [CrossRef]
- Pérez-Bermejo, M.; Mas-Pérez, I.; Murillo-Llorente, M.T. The Role of the Bisphenol A in Diabetes and Obesity. Biomedicines 2021, 9, 666. [Google Scholar] [CrossRef]
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of bisphenol A as environmental factor in the promotion of non-alcoholic fatty liver disease: In vitro and clinical study. Aliment. Pharmacol. Ther. 2018, 47, 826–837. [Google Scholar] [CrossRef]
- Mariana, M.; Cairrao, E. Phthalates Implications in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2020, 7, 26. [Google Scholar] [CrossRef]
- Mariana, M.; Cairrao, E. The Relationship between Phthalates and Diabetes: A Review. Metabolites 2023, 13, 746. [Google Scholar] [CrossRef]
- Mérida, D.M.; Moreno-Franco, B.; Marquès, M.; León-Latre, M.; Laclaustra, M.; Guallar-Castillón, P. Phthalate exposure and the metabolic syndrome: A systematic review and meta-analysis. Environ. Pollut. 2023, 333, 121957. [Google Scholar] [CrossRef]
- Cai, S.; Fan, J.; Ye, J.; Rao, X.; Li, Y. Phthalates exposure is associated with non-alcoholic fatty liver disease among US adults. Ecotoxicol. Environ. Saf. 2021, 224, 112665. [Google Scholar] [CrossRef]
- Guo, X.; Wu, B.; Xia, W.; Gao, J.; Xie, P.; Feng, L.; Sun, C.; Liang, M.; Ding, X.; Zhao, D.; et al. Association of organophosphate ester exposure with cardiovascular disease among US adults: Cross-sectional findings from the 2011–2018 National Health and Nutrition Examination Survey. Chemosphere 2022, 308, 136428. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Zhang, R.; Aimuzi, R.; Wang, Y.; Nian, M.; Zhang, J. Exposure to Organophosphate esters and metabolic syndrome in adults. Environ. Int. 2020, 143, 105941. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.F.; Cheng, F.J.; Huang, W.T.; Kung, C.T.; Lee, C.T.; Cheng, B.C.; Chen, J.B.; Li, S.H.; Wang, C.C.; Wang, L.J.; et al. The associations between renal disease severity and exposure to organophosphate flame retardants in patients with chronic kidney disease. Environ. Int. 2022, 170, 107573. [Google Scholar] [CrossRef]
- Guo, X.; Ke, Y.; Wu, B.; Song, Q.; Sun, C.; Li, Y.; Wang, H.; Su, W.; Liang, Q.; Lowe, S.; et al. Exploratory analysis of the association between organophosphate ester mixtures with high blood pressure of children and adolescents aged 8–17 years: Cross-sectional findings from the National Health and Nutrition Examination Survey. Environ. Sci. Pollut. Res. Int. 2023, 30, 22900–22912. [Google Scholar] [CrossRef]
- Marfella, R.; Prattichizzo, F.; Sardu, C.; Fulgenzi, G.; Graciotti, L.; Spadoni, T.; D’Onofrio, N.; Scisciola, L.; La Grotta, R.; Frigé, C.; et al. Microplastics and Nanoplastics in Atheromas and Cardiovascular Events. N. Engl. J. Med. 2024, 390, 900–910. [Google Scholar] [CrossRef]
- Qi, W.; Clark, J.M.; Timme-Laragy, A.R.; Park, Y. Per- and Polyfluoroalkyl Substances and Obesity, Type 2 Diabetes and Non-alcoholic Fatty Liver Disease: A Review of Epidemiologic Findings. Toxicol. Environ. Chem. 2020, 102, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Mallah, M.A.; Changxing, L.; Mallah, M.A.; Naveed, M.; Liu, Y.; Noreen, S.; Xi, H.; Wang, W.; Feng, F.; Zhang, Q. Association of urinary polycyclic aromatic hydrocarbon metabolites and cardiovascular disease among US population: A cross-sectional study. Environ. Res. 2022, 209, 112775. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xue, Q.; Wen, Y.; Huang, Y.; Wang, Y.; Mahai, G.; Yan, T.; Liu, Y.; Rong, T.; Wang, Y.; et al. Environmental polycyclic aromatic hydrocarbon exposure in relation to metabolic syndrome in US adults. Sci. Total Environ. 2022, 840, 156673. [Google Scholar] [CrossRef]
- Choi, Y.H.; Lee, J.Y.; Moon, K.W. Exposure to volatile organic compounds and polycyclic aromatic hydrocarbons is associated with the risk of non-alcoholic fatty liver disease in Korean adolescents: Korea National Environmental Health Survey (KoNEHS) 2015–2017. Ecotoxicol. Environ. Saf. 2023, 251, 114508. [Google Scholar] [CrossRef] [PubMed]
- Krittanawong, C.; Qadeer, Y.K.; Hayes, R.B.; Wang, Z.; Virani, S.; Thurston, G.D.; Lavie, C.J. PM2.5 and Cardiovascular Health Risks. Curr. Probl. Cardiol. 2023, 48, 101670. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, M.; Song, Y.; Ma, H.; Zhou, T.; Liang, Z.; Qi, L. Obesity and the relation between joint exposure to ambient air pollutants and incident type 2 diabetes: A cohort study in UK Biobank. PLoS Med. 2021, 18, e1003767. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, L.; Yang, G.; Zhang, C.; Liu, X.; Sun, X.; Chen, X.; Wang, N. The influence of PM2.5 exposure on non-alcoholic fatty liver disease. Life Sci. 2021, 270, 119135. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Xu, C.; Liu, Q.; Xu, J.; Weng, Z.; Zhang, X.; Basnet, T.B.; Dahal, M.; Gu, A. Levels of a mixture of heavy metals in blood and urine and all-cause, cardiovascular disease and cancer mortality: A population-based cohort study. Environ. Pollut. 2020, 263, 114630. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.J.; Hung, C.H.; Wang, C.W.; Tu, H.P.; Li, C.H.; Tsai, C.C.; Lin, W.Y.; Chen, S.C.; Kuo, C.H. Associations among Heavy Metals and Proteinuria and Chronic Kidney Disease. Diagnostics 2021, 11, 282. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mukherjee, B.; Park, S.K. Associations of cumulative exposure to heavy metal mixtures with obesity and its comorbidities among U.S. adults in NHANES 2003–2014. Environ. Int. 2018, 121, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Dopico, M.; Gόmez, A. Review of the Current State and Main Sources of Dioxins around the World. J. Air Waste Manag. Assoc. 2015, 65, 1033–1049. [Google Scholar] [CrossRef] [PubMed]
- Milbrath, M.O.; Wenger, Y.; Chang, C.W.; Emond, C.; Garabrant, D.; Gillespie, B.W.; Jolliet, O. Apparent half-lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast-feeding. Environ. Health Perspect. 2009, 117, 417–425. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Ropero, A.B.; Soriano, S.; García-Arévalo, M.; Ripoll, C.; Fuentes, E.; Quesada, I.; Nadal, A. Bisphenol-A acts as a potent estrogen via non-classical estrogen triggered pathways. Mol. Cell. Endocrinol. 2012, 355, 201–207. [Google Scholar] [CrossRef]
- Agay-Shay, K.; Martinez, D.; Valvi, D.; Garcia-Esteban, R.; Basagaña, X.; Robinson, O.; Casas, M.; Sunyer, J.; Vrijheid, M. Exposure to Endocrine-Disrupting Chemicals during Pregnancy and Weight at 7 Years of Age: A Multi-pollutant Approach. Environ. Health Perspect. 2015, 123, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Lim, Y.H.; Lee, Y.A.; Shin, C.H.; Oh, S.Y.; Hong, Y.C. Maternal Urinary Bisphenol a Concentration during Midterm Pregnancy and Children’s Blood Pressure at Age 4. Hypertension 2017, 69, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.C.; Hsieh, Y.K.; Huang, B.W.; Cheruiyot, N.K.; Chang-Chien, G.P. An Overview: Organophosphate Flame Retardants in the Atmosphere. Aerosol Air Qual. Res. 2022, 22, 220148. [Google Scholar] [CrossRef]
- Cox, K.D.; Covernton, G.A.; Davies, H.L.; Dower, J.F.; Juanes, F.; Dudas, S.E. Human Consumption of Microplastics. Environ. Sci. Technol. 2019, 53, 7068–7074. [Google Scholar] [CrossRef] [PubMed]
- Kutralam-Muniasamy, G.; Shruti, V.C.; Pérez-Guevara, F.; Roy, P.D. Microplastic diagnostics in humans: “The 3Ps” Progress, problems, and prospects. Sci. Total Environ. 2023, 856, 159164. [Google Scholar] [CrossRef] [PubMed]
- Sunderland, E.M.; Hu, X.C.; Dassuncao, C.; Tokranov, A.K.; Wagner, C.C.; Allen, J.G. A Review of the Pathways of Human Exposure to Poly- and Perfluoroalkyl Substances (Pfass) and Present Understanding of Health Effects. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Xiao, J.; Ducatman, A. Perfluoroalkyl Chemicals and Chronic Kidney Disease in US Adults. Am. J. Epidemiol. 2011, 174, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Starling, A.P.; Adgate, J.L.; Hamman, R.F.; Kechris, K.; Calafat, A.M.; Dabelea, D. Prenatal exposure to per- and polyfluoroalkyl substances and infant growth and adiposity: The Healthy Start Study. Environ. Int. 2019, 131, 104983. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Eliot, M.N.; Papandonatos, G.D.; Kelsey, K.T.; Fore, R.; Langevin, S.; Buckley, J.; Chen, A.; Lanphear, B.P.; Cecil, K.M.; et al. Gestational Perfluoroalkyl Substance Exposure and DNA Methylation at Birth and 12 Years of Age: A Longitudinal Epigenome-Wide Association Study. Environ. Health Perspect. 2022, 130, 37005. [Google Scholar] [CrossRef]
- Patel, A.B.; Shaikh, S.; Jain, K.R.; Desai, C.; Madamwar, D. Polycyclic Aromatic Hydrocarbons: Sources, Toxicity, and Remediation Approaches. Front. Microbiol. 2020, 11, 562813. [Google Scholar] [CrossRef]
- Rahman, H.H.; Niemann, D.; Munson-McGee, S.H. Association of chronic kidney disease with exposure to polycyclic aromatic hydrocarbons in the US population. Environ. Sci. Pollut. Res. Int. 2022, 29, 24024–24034. [Google Scholar] [CrossRef]
- Drwal, E.; Rak, A.; Gregoraszczuk, E.L. Review: Polycyclic Aromatic Hydrocarbons (Pahs)-Action on Placental Function and Health Risks in Future Life of Newborns. Toxicology 2019, 411, 133–142. [Google Scholar] [CrossRef]
- Bukowska, B.; Sicińska, P. Influence of Benzo(a)pyrene on Different Epigenetic Processes. Int. J. Mol. Sci. 2021, 22, 13453. [Google Scholar] [CrossRef]
- Bianco-Miotto, T.; Craig, J.M.; Gasser, Y.P.; van Dijk, S.J.; Ozanne, S.E. Epigenetics and DOHaD: From basics to birth and beyond. J. Dev. Orig. Health Dis. 2017, 8, 513–519. [Google Scholar] [CrossRef]
- Kampa, M.; Castanas, E. Human health effects of air pollution. Environ. Pollut. 2008, 151, 362–367. [Google Scholar] [CrossRef]
- Zhang, S.; Qian, Z.M.; Chen, L.; Zhao, X.; Cai, M.; Wang, C.; Zou, H.; Wu, Y.; Zhang, Z.; Li, H.; et al. Exposure to Air Pollution during Pre-Hypertension and Subsequent Hypertension, Cardiovascular Disease, and Death: A Trajectory Analysis of the UK Biobank Cohort. Environ. Health Perspect. 2023, 131, 17008. [Google Scholar] [CrossRef]
- An, Y.; Liu, Z.H. Air Pollution and Kidney Diseases: PM2.5 as an Emerging Culprit. Contrib. Nephrol. 2021, 199, 274–284. [Google Scholar]
- Zhang, M.; Mueller, N.T.; Wang, H.; Hong, X.; Appel, L.J.; Wang, X. Maternal Exposure to Ambient Particulate Matter ≤ 2.5 μm During Pregnancy and the Risk for High Blood Pressure in Childhood. Hypertension 2018, 72, 194–201. [Google Scholar] [CrossRef]
- Elten, M.; Donelle, J.; Lima, I.; Burnett, R.T.; Weichenthal, S.; Stieb, D.M.; Hystad, P.; van Donkelaar, A.; Chen, H.; Paul, L.A.; et al. Ambient air pollution and incidence of early-onset paediatric type 1 diabetes: A retrospective population-based cohort study. Environ. Res. 2020, 184, 109291. [Google Scholar] [CrossRef]
- Rehman, K.; Fatima, F.; Waheed, I.; Akash, M.S.H. Prevalence of exposure of heavy metals and their impact on health consequences. J. Cell. Biochem. 2018, 119, 157–184. [Google Scholar] [CrossRef]
- Li, S.; Wang, Q.; Luo, W.; Jia, S.; Liu, D.; Ma, W.; Gu, H.; Wei, X.; He, Y.; Cao, S.; et al. Relationship between maternal heavy metal exposure and congenital heart defects: A systematic review and meta-analysis. Environ. Sci. Pollut. Res. Int. 2022, 29, 55348–55366. [Google Scholar] [CrossRef]
- Iwaya, Y.; Sanefuji, M.; Nishiyama, K.; Sonoda, Y.; Hamada, N.; Suga, R.; Ochiai, M.; Shimono, M.; Kusuhara, K.; Ohga, S.; et al. Prenatal metal levels and congenital anomalies of the kidney and urinary tract: The Japan Environment and Children’s Study. Sci. Total Environ. 2023, 890, 164356. [Google Scholar] [CrossRef]
- Saylor, C.; Tamayo-Ortiz, M.; Pantic, I.; Amarasiriwardena, C.; McRae, N.; Estrada-Gutierrez, G.; Parra-Hernandez, S.; Tolentino, M.C.; Baccarelli, A.A.; Fadrowski, J.J.; et al. Prenatal blood lead levels and reduced preadolescent glomerular filtration rate: Modification by body mass index. Environ. Int. 2021, 154, 106414. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, T.; Wang, G.; Buckley, J.P.; Guallar, E.; Hong, X.; Wang, M.C.; Wills-Karp, M.; Wang, X.; Mueller, N.T. In Utero Exposure to Heavy Metals and Trace Elements and Childhood Blood Pressure in a U.S. Urban, Low-Income, Minority Birth Cohort. Environ. Health Perspect. 2021, 129, 67005. [Google Scholar] [CrossRef]
- Hsu, C.N.; Chan, J.Y.H.; Yu, H.R.; Lee, W.C.; Wu, K.L.H.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on Gut Microbiota-Derived Metabolite Trimethylamine to Protect Adult Male Rat Offspring against Hypertension Programmed by Combined Maternal High-Fructose Intake and Dioxin Exposure. Int. J. Mol. Sci. 2020, 21, 5488. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hung, C.H.; Hou, C.Y.; Chang, C.I.; Tain, Y.L. Perinatal Resveratrol Therapy to Dioxin-Exposed Dams Prevents the Programming of Hypertension in Adult Rat Offspring. Antioxidants 2021, 10, 1393. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Lee, C.T.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal 3,3-Dimethyl-1-Butanol Therapy Protects Adult Male Rat Offspring against Hypertension Programmed by Perinatal TCDD Exposure. Nutrients 2021, 13, 3041. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, Y.J.; Lu, P.C.; Tain, Y.L. Maternal Resveratrol Therapy Protects Male Rat Offspring Against Programmed Hypertension Induced by TCDD and Dexamethasone Exposures: Is it Relevant to Aryl Hydrocarbon Receptor? Int. J. Mol. Sci. 2018, 19, 2459. [Google Scholar] [CrossRef]
- Aragon, A.C.; Kopf, P.G.; Campen, M.J.; Huwe, J.K.; Walker, M.K. In Utero and Lactational 2,3,7,8-Tetrachlorodibenzo-P-Dioxin Exposure: Effects on Fetal and Adult Cardiac Gene Expression and Adult Cardiac and Renal Morphology. Toxicol. Sci. 2008, 101, 321–330. [Google Scholar] [CrossRef]
- Nuñez, P.; Fernandez, T.; García-Arévalo, M.; Alonso-Magdalena, P.; Nadal, A.; Perillan, C. Effects of Bisphenol a Treatment During Pregnancy on Kidney Development in Mice: A Stereological and Histopathological Study. J. Dev. Orig. Health Dis. 2018, 9, 208–214. [Google Scholar] [CrossRef]
- Shih, M.K.; Tain, Y.L.; Chen, Y.W.; Hsu, W.H.; Yeh, Y.T.; Chang, S.K.C.; Liao, J.X.; Hou, C.Y. Resveratrol Butyrate Esters Inhibit Obesity Caused by Perinatal Exposure to Bisphenol A in Female Offspring Rats. Molecules 2021, 26, 4010. [Google Scholar] [CrossRef]
- Liao, J.X.; Chen, Y.W.; Shih, M.K.; Tain, Y.L.; Yeh, Y.T.; Chiu, M.H.; Chang, S.K.C.; Hou, C.Y. Resveratrol Butyrate Esters Inhibit BPA-Induced Liver Damage in Male Offspring Rats by Modulating Antioxidant Capacity and Gut Microbiota. Int. J. Mol. Sci. 2021, 22, 5273. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, Y.J.; Tain, Y.L. Maternal Exposure to Bisphenol a Combined with High-Fat Diet-Induced Programmed Hypertension in Adult Male Rat Offspring: Effects of Resveratrol. Int. J. Mol. Sci. 2019, 20, 4382. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Vieira, E.; Soriano, S.; Menes, L.; Burks, D.; Quesada, I.; Nadal, A. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ. Health Perspect. 2010, 118, 1243–1250. [Google Scholar] [CrossRef]
- Wei, Z.; Song, L.; Wei, J.; Chen, T.; Chen, J.; Lin, Y.; Xia, W.; Xu, B.; Li, X.; Chen, X.; et al. Maternal Exposure to Di-(2-Ethylhexyl)Phthalate Alters Kidney Development through the Renin-Angiotensin System in Offspring. Toxicol. Lett. 2012, 212, 212–221. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Hsu, C.N. Resveratrol Butyrate Ester Supplementation Blunts the Development of Offspring Hypertension in a Maternal Di-2-ethylhexyl Phthalate Exposure Rat Model. Nutrients 2023, 15, 697. [Google Scholar] [CrossRef]
- Fan, Y.; Qin, Y.; Chen, M.; Li, X.; Wang, R.; Huang, Z.; Xu, Q.; Yu, M.; Zhang, Y.; Han, X.; et al. Prenatal low-dose DEHP exposure induces metabolic adaptation and obesity: Role of hepatic thiamine metabolism. J. Hazard. Mater. 2020, 385, 121534. [Google Scholar] [CrossRef]
- Ye, Q.; Zhao, S.; Zhang, Y.; Su, Y.M.; Chen, M.; Zhao, J.; Jia, G.Z.; Han, B.M.; Jiang, J.T. Activation of the Rhoa/ROCK Pathway Contributes to Renal Fibrosis in Offspring Rats Induced by Maternal Exposure to Di-N-Butyl Phthalate. Toxicology 2020, 443, 152573. [Google Scholar] [CrossRef]
- Zhou, K.; Cheng, R.; Zhu, M.; Yang, M.; Shen, X.; Luo, X.; Ma, L.; Xu, L.; Zhang, J. The influence of perinatal maternal exposure to dibutyl phthalate on glucolipid metabolism in adult female offspring. Obes. Res. Clin. Pract. 2022, 16, 500–506. [Google Scholar] [CrossRef]
- Dangudubiyyam, S.V.; Mishra, J.S.; Zhao, H.; Kumar, S. Perfluorooctane sulfonic acid (PFOS) exposure during pregnancy increases blood pressure and impairs vascular relaxation mechanisms in the adult offspring. Reprod. Toxicol. 2020, 98, 165–173. [Google Scholar] [CrossRef]
- Jules, G.E.; Pratap, S.; Ramesh, A.; Hood, D.B. In Utero Exposure to Benzo(a)Pyrene Predisposes Offspring to Cardiovascular Dysfunction in Later-Life. Toxicology 2012, 295, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Saillenfait, A.M.; Payan, J.P.; Brondeau, M.T.; Zissu, D.; de Ceaurriz, J. Changes in Urinary Proximal Tubule Parameters in Neonatal Rats Exposed to Cadmium Chloride during Pregnancy. J. Appl. Toxicol. 1991, 11, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Jacquillet, G.; Barbier, O.; Rubera, I.; Tauc, M.; Borderie, A.; Namorado, M.C.; Martin, D.; Sierra, G.; Reyes, J.L.; Poujeol, P.; et al. Cadmium Causes Delayed Effects on Renal Function in the Offspring of Cadmium-Contaminated Pregnant Female Rats. Am. J. Physiol. Renal Physiol. 2007, 293, F1450–F1560. [Google Scholar] [CrossRef]
- Jackson, T.W.; Ryherd, G.L.; Scheibly, C.M.; Sasser, A.L.; Guillette, T.C.; Belcher, S.M. Gestational Cd Exposure in the CD-1 Mouse Induces Sex-Specific Hepatic Insulin Insensitivity, Obesity, and Metabolic Syndrome in Adult Female Offspring. Toxicol. Sci. 2020, 178, 264–280. [Google Scholar] [CrossRef]
- Pan, K.; Jiang, S.; Du, X.; Zeng, X.; Zhang, J.; Song, L.; Lei, L.; Zhou, J.; Kan, H.; Sun, Q. Parental PM2.5 Exposure Changes Th17/Treg Cells in Offspring, Is Associated with the Elevation of Blood Pressure. Environ. Toxicol. 2021, 36, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Pan, B.; Liu, L.; Zhao, W.; Zhu, J.; Huang, X.; Tian, J. In utero exposure to PM2.5 during gestation caused adult cardiac hypertrophy through histone acetylation modification. J. Cell. Biochem. 2019, 120, 4375–4384. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Lu, X.; Deng, Y.; Wang, X.; Zheng, S.; Ren, H.; Zhang, M.; Chen, T.; Jose, P.A.; Yang, J.; et al. In Utero Exposure to Fine Particulate Matter Causes Hypertension due to Impaired Renal Dopamine D1 Receptor in Offspring. Cell. Physiol. Biochem. 2018, 46, 148–159. [Google Scholar] [CrossRef]
- Chen, M.; Wang, X.; Hu, Z.; Zhou, H.; Xu, Y.; Qiu, L.; Qin, X.; Zhang, Y.; Ying, Z. Programming of mouse obesity by maternal exposure to concentrated ambient fine particles. Part. Fibre Toxicol. 2017, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liang, S.; Qin, X.; Zhang, L.; Qiu, L.; Chen, S.; Hu, Z.; Xu, Y.; Wang, W.; Zhang, Y.; et al. Prenatal exposure to diesel exhaust PM2.5 causes offspring β cell dysfunction in adulthood. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E72–E80. [Google Scholar] [CrossRef]
- Sengupta, P. The Laboratory Rat: Relating Its Age with Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar]
- Lin, L.; Dai, Y.; Xia, Y. An overview of aryl hydrocarbon receptor ligands in the Last two decades (2002–2022): A medicinal chemistry perspective. Eur. J. Med. Chem. 2022, 244, 114845. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Wu, X.; Huang, Q.; Liao, Y.; Liu, L.; Qiu, L.; Shen, H.; Dong, S. PFOS elicits transcriptional responses of the ER, AHR and PPAR pathways in Oryzias melastigma in a stage-specific manner. Aquat. Toxicol. 2012, 106–107, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Li, Y.; Xi, H.; Niu, Z.; Chen, N.; Wang, R.; Yan, Y.; Gan, X.; Wang, M.; Zhang, W.; et al. Benzo(a)pyrene and cardiovascular diseases: An overview of pre-clinical studies focused on the underlying molecular mechanism. Front. Nutr. 2022, 9, 978475. [Google Scholar] [CrossRef]
- Feng, S.; Duan, E.; Shi, X.; Zhang, H.; Li, H.; Zhao, Y.; Chao, L.; Zhong, X.; Zhang, W.; Li, R.; et al. Hydrogen ameliorates lung injury in a rat model of subacute exposure to concentrated ambient PM2.5 via Aryl hydrocarbon receptor. Int. Immunopharmacol. 2019, 77, 105939. [Google Scholar] [CrossRef]
- Safe, S.; Jin, U.H.; Park, H.; Chapkin, R.S.; Jayaraman, A. Aryl Hydrocarbon Receptor (AHR) Ligands as Selective AHR Modulators (SAhRMs). Int. J. Mol. Sci. 2020, 21, 6654. [Google Scholar] [CrossRef]
- Sládeková, L.; Mani, S.; Dvořák, Z. Ligands and agonists of the aryl hydrocarbon receptor AhR: Facts and myths. Biochem. Pharmacol. 2023, 213, 115626. [Google Scholar] [CrossRef]
- Pinto, C.J.G.; Ávila-Gálvez, M.Á.; Lian, Y.; Moura-Alves, P.; Nunes Dos Santos, C. Targeting the aryl hydrocarbon receptor by gut phenolic metabolites: A strategy towards gut inflammation. Redox Biol. 2023, 61, 102622. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, R.P. A modification of receptor theory. Br. J. Pharmacol. Chemother. 1956, 11, 379–393. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, I.C.; Yu, H.R.; Huang, L.T.; Tiao, M.M.; Tain, Y.L. Maternal Tryptophan Supplementation Protects Adult Rat Offspring against Hypertension Programmed by Maternal Chronic Kidney Disease: Implication of Tryptophan-Metabolizing Microbiome and Aryl Hydrocarbon Receptor. Int. J. Mol. Sci. 2020, 21, 4552. [Google Scholar] [CrossRef]
- Zakrocka, I.; Załuska, W. Kynurenine pathway in kidney diseases. Pharmacol. Rep. 2022, 74, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef] [PubMed]
- Hus, C.N.; Tain, Y.L. Developmental Programming and Reprogramming of Hypertension and Kidney Disease: Impact of Tryptophan Metabolism. Int. J. Mol. Sci. 2020, 21, 8705. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed]
- Kursvietiene, L.; Staneviciene, I.; Mongirdiene, A.; Bernatoniene, J. Multiplicity of effects and health benefits of resveratrol. Medicina 2016, 52, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Bardbori, A.; Bengtsson, J.; Rannug, U.; Rannug, A.; Wincent, E. Quercetin, resveratrol, and curcumin are indirect activators of the aryl hydrocarbon receptor (AHR). Chem. Res. Toxicol. 2012, 25, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Ciolino, H.P.; Yeh, G.C. Inhibition of aryl hydrocarbon-induced cytochrome P-450 1A1 enzyme activity and CYP1A1 expression by resveratrol. Mol. Pharmacol. 1999, 56, 760–767. [Google Scholar]
- Hsu, C.N.; Hou, C.Y.; Tain, Y.L. Preventive Aspects of Early Resveratrol Supplementation in Cardiovascular and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 4210. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Developmental Programming of the Metabolic Syndrome: Can We Reprogram with Resveratrol? Int. J. Mol. Sci. 2018, 19, 2584. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Chan, J.Y.H.; Lee, C.T.; Tain, Y.L. Maternal resveratrol therapy protected adult rat offspring against hypertension programmed by combined exposures to asymmetric dimethylarginine and trimethylamine-N oxide. J. Nutr. Biochem. 2021, 93, 108630. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Chang, S.K.C.; Liao, J.X.; Chen, Y.W.; Huang, H.T.; Li, Y.L.; Hou, C.Y. Synthesis of Short-Chain-Fatty-Acid Resveratrol Esters and Their Antioxidant Properties. Antioxidants 2021, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A Double-Edged Sword for Health? Adv. Nutr. 2018, 9, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L. Microbial short-chain fatty acids and blood pressure regulation. Curr. Hypertens. Rep. 2017, 19, 25. [Google Scholar] [CrossRef]
- Marinelli, L.; Martin-Gallausiaux, C.; Bourhis, J.M.; Béguet-Crespel, F.; Blottière, H.M.; Lapaque, N. Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci. Rep. 2019, 9, 643. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Yu, H.R.; Lin, I.C.; Tiao, M.M.; Huang, L.T.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Sodium butyrate modulates blood pressure and gut microbiota in maternal tryptophan-free diet-induced hypertension rat offspring. J. Nutr. Biochem. 2022, 108, 109090. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tzeng, H.T.; Lee, W.C.; Wu, K.L.H.; Yu, H.R.; Chan, J.Y.H.; Hsu, C.N. Reprogramming Effects of Postbiotic Butyrate and Propionate on Maternal High-Fructose Diet-Induced Offspring Hypertension. Nutrients 2023, 15, 1682. [Google Scholar] [CrossRef]
- Wu, K.L.H.; Liu, W.C.; Wu, C.W.; Fu, M.H.; Huang, H.M.; Tain, Y.L.; Liang, C.K.; Hung, C.Y.; Chen, I.C.; Hung, P.L.; et al. Butyrate reduction and HDAC4 increase underlie maternal high fructose-induced metabolic dysfunction in hippocampal astrocytes in female rats. J. Nutr. Biochem. 2024, 126, 109571. [Google Scholar] [CrossRef] [PubMed]
- Rejano-Gordillo, C.M.; Marín-Díaz, B.; Ordiales-Talavero, A.; Merino, J.M.; González-Rico, F.J.; Fernández-Salguero, P.M. From Nucleus to Organs: Insights of Aryl Hydrocarbon Receptor Molecular Mechanisms. Int. J. Mol. Sci. 2022, 23, 14919. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Interplay between maternal nutrition and epigenetic programming on offspring hypertension. J. Nutr. Biochem. 2024, 127, 109604. [Google Scholar] [CrossRef]
- Tain, Y.L.; Huang, L.T.; Chan, J.Y.; Lee, C.T. Transcriptome analysis in rat kidneys: Importance of genes involved in programmed hypertension. Int. J. Mol. Sci. 2015, 16, 4744–4758. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Runov, A.L.; Ponomartsev, S.V.; Vonsky, M.S.; Elmuratov, A.U.; Vinogradov, A.E. Long-Term Transcriptomic Changes and Cardiomyocyte Hyperpolyploidy after Lactose Intolerance in Neonatal Rats. Int. J. Mol. Sci. 2023, 24, 7063. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, C.S. Gut Dysbiosis in Animals due to Environmental Chemical Exposures. Front. Cell. Infect. Microbiol. 2017, 7, 396. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Metabolic Syndrome Programming and Reprogramming: Mechanistic Aspects of Oxidative Stress. Antioxidants 2022, 11, 2108. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Tain, Y.L. Targeting the Renin-Angiotensin-Aldosterone System to Prevent Hypertension and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 2298. [Google Scholar] [CrossRef] [PubMed]
- Dornbos, P.; Warren, M.; Crawford, R.B.; Kaminski, N.E.; Threadgill, D.W.; LaPres, J.J. Characterizing Serpinb2 as a Modulator of TCDD-Induced Suppression of the B Cell. Chem. Res. Toxicol. 2018, 31, 1248–1259. [Google Scholar] [CrossRef]
- Jurgelewicz, A.; Dornbos, P.; Warren, M.; Nault, R.; Arkatkar, A.; Lin, H.; Threadgill, D.W.; Zacharewski, T.; LaPres, J.J. Genetics-Based Approach to Identify Novel Genes Regulated by the Aryl Hydrocarbon Receptor in Mouse Liver. Toxicol. Sci. 2021, 181, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Faber, S.C. And Now for Something Completely Different: Diversity in Ligand-Dependent Activation of Ah Receptor Responses. Curr. Opin. Toxicol. 2017, 2, 124–131. [Google Scholar] [CrossRef]
- Dolciami, D.; Ballarotto, M.; Gargaro, M.; Lopez-Cara, L.C.; Fallarino, F.; Macchiarulo, A. Targeting Aryl hydrocarbon receptor for next-generation immunotherapies: Selective modulators (SAhRMs) versus rapidly metabolized ligands (RMAhRLs). Eur. J. Med. Chem. 2020, 185, 111842. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Environmental Chemicals | Common Substances or Derivatives | Major Sources | Exposure-Associated CKM Phenotypes |
|---|---|---|---|
| Dioxins | TCDD, PCDF, PCDD, PCB | Consumption of animal products rich in fat, pesticide production, wood pulp bleaching, and the process of waste incineration | Cardiovascular disease [63], diabetes [64], metabolic syndrome [65], hypertension, and kidney disease [66,67] |
| Bisphenol A | BPA | Plastic containers, lenses, medical tubing, and apparatus | Cardiovascular disease [68], diabetes [69], obesity [69], NAFLD [70], hypertension, and kidney disease [66,67] |
| Phthalates | DEHP, DBP | Vinyl plastics, cosmetics, shampoos, medical devices, and food packaging | Cardiovascular disease [71], diabetes [72], metabolic syndrome [73], NAFLD [74], hypertension [67], and kidney disease [66,67] |
| Organophosphate flame retardants | DPHP, TPHP, TDCPP | Plastics, rubbers, textiles, upholstered furniture, building materials, and electronic equipment | Cardiovascular disease [75], metabolic syndrome [76], kidney disease [77], and hypertension [78] |
| Microplastics | MPs | Various foods, including drinking water, seafood, milk, sugar, and salt | Cardiovascular disease [79] |
| Per- and polyfluoroalkyl substances | PFOA, PFOS | Firefighting foams, non-stick cookware, stain-resistant fabrics, water-repellent coatings, and food packaging materials | Obesity, diabetes, NAFLD [80], hypertension, and kidney disease [66,67] |
| Polycyclic aromatic hydrocarbon | BaP | Vehicle exhaust, industrial processes, tobacco smoke, and grilled food | Cardiovascular disease [81], metabolic syndrome [82], NAFLD [83], and kidney disease [66] |
| Air pollution | PM10, PM2.5 | Factories and manufacturing plants, transportation, agriculture, and waste management | Cardiovascular disease [84], diabetes [85], NAFLD [86], hypertension [67], and kidney disease [67] |
| Heavy metals | Pb, Cd, Hg | Manufacturing processes, emissions from vehicles, combustion of fossil fuels, improper disposal of electronic waste and batteries, and contaminating soil, water, and air | Cardiovascular disease [87], kidney disease [88], obesity [89], diabetes [89], and hypertension [89]. |
| Chemical | Exposure Dose and Period | Species | Age at Evaluation (Weeks) | CKM Phenotypes | Refs. |
|---|---|---|---|---|---|
| TCDD | 200 ng/kg orally on gestational days 14 and 21 and days 7 and 14 after birth | SD rats/M | 12 | Hypertension | [117] |
| TCDD | 200 ng/kg in four once-weekly oral doses throughout pregnancy and lactation | SD rats/M | 12 | Hypertension | [118,119] |
| TCDD | 200 ng/kg in four weekly oral doses throughout pregnancy and lactation | SD rats/M | 16 | Hypertension | [120] |
| TCDD | 6.0 µg/g orally on gestational day 14.5 | C57BL/6N mice/M | 12 | Cardiovascular dysfunction and kidney malformation | [121] |
| BPA | 10 or 100 mg/kg/day throughout gestational days 9–16 | OF1 mice/ M and F | 5 | Kidney dysfunction | [122] |
| BPA | 50 μg/kg/day throughout gestation and lactation | SD rats/F | 7 | Obesity | [123] |
| BPA | 50 μg/kg/day throughout gestation and lactation | SD rats/F | 7 | NAFLD | [124] |
| BPA | 50 mg/kg/day throughout gestation and lactation | SD rats/M | 16 | Hypertension | [125] |
| BPA | 10 or 100 μg/kg/day throughout gestational days 9–16 | SD rats/F | 16 | Insulin resistance and hyperlipidemia | [126] |
| DEHP | 0.25 or 6.25 mg/kg/day throughout pregnancy | Wistar rats/ M and F | 21 | Kidney dysfunction and hypertension | [127] |
| DEHP | 10 mg/kg/day throughout pregnancy and lactation | SD rats/M | 12 | Hypertension | [128] |
| DEHP | 0.2, 2, or 20 mg/kg/day throughout pregnancy and lactation | ICR mice/M | 12 | Abnormal adipogenesis and glucose metabolism | [129] |
| DBP | 850 mg/kg/day throughout gestational days 14–18 | SD rats/M | 8 | Kidney dysfunction and renal fibrosis | [130] |
| DBP | 33, 66, or 132 mg/kg/day from gestational day 7 throughout postnatal day 21 | SD rats/F | 12 | Obesity | [131] |
| PFOS | 50 μg/mL from gestational day 4 until delivery | SD rats/ M and F | 16 | Hypertension | [132] |
| BaP | 600 or 1200 mg/kg/day throughout gestational days 14–17 | LEH rats/ M and F | 8 | Hypertension | [133] |
| Cd | Cd chloride 2.0 or 2.5 mg/kg/day on gestational days 8, 10, 12, and 14 | SD rats/M | 7 | Kidney injury | [134] |
| Cd | Cd chloride 0.5 mg/kg/day throughout pregnancy | Wistar rats/ M and F | 8 | Kidney dysfunction | [135] |
| Cd | 500 ppb CdCl2 in drinking water throughout pregnancy to postnatal day 10 | CD-1 mice/ M and F | 17 | Obesity, hyperlipidemia, insulin resistance, and steatosis in F | [136] |
| PM2.5 | PM2.5 exposure for 16 weeks prior to delivery | C57BL/6N mice/M and F | 12 | Hypertension | [137] |
| PM2.5 | PM2.5 exposure 300 µg/m3 for 2 h/day throughout | C57BL/6N mice/M and F | 12 | Cardiac hypertrophy | [138] |
| PM2.5 | Oropharyngeal drip of PM2.5 (1.0 mg/kg) on gestational days 8, 10, and 12 | SD rats/M | 14 | Hypertension | [139] |
| PM2.5 | Concentrated ambient PM2.5 exposure throughout gestation and lactation | C57BL/6N mice/M and F | 22 | Obesity | [140] |
| PM2.5 | Diesel exhaust PM2.5 8.6 μg/day intratracheal instillation throughout pregnancy and lactation | C57BL/6N mice/M and F | 22 | Glucose intolerance and pancreatic islet dysfunction | [141] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tain, Y.-L.; Hsu, C.-N. The Impact of the Aryl Hydrocarbon Receptor on Antenatal Chemical Exposure-Induced Cardiovascular–Kidney–Metabolic Programming. Int. J. Mol. Sci. 2024, 25, 4599. https://doi.org/10.3390/ijms25094599
Tain Y-L, Hsu C-N. The Impact of the Aryl Hydrocarbon Receptor on Antenatal Chemical Exposure-Induced Cardiovascular–Kidney–Metabolic Programming. International Journal of Molecular Sciences. 2024; 25(9):4599. https://doi.org/10.3390/ijms25094599
Chicago/Turabian StyleTain, You-Lin, and Chien-Ning Hsu. 2024. "The Impact of the Aryl Hydrocarbon Receptor on Antenatal Chemical Exposure-Induced Cardiovascular–Kidney–Metabolic Programming" International Journal of Molecular Sciences 25, no. 9: 4599. https://doi.org/10.3390/ijms25094599
APA StyleTain, Y. -L., & Hsu, C. -N. (2024). The Impact of the Aryl Hydrocarbon Receptor on Antenatal Chemical Exposure-Induced Cardiovascular–Kidney–Metabolic Programming. International Journal of Molecular Sciences, 25(9), 4599. https://doi.org/10.3390/ijms25094599