
Herpes Simplex Virus ICP27 Protein Inhibits AIM 2-Dependent Inflammasome Influencing Pro-Inflammatory Cytokines Release in Human Pigment Epithelial Cells (hTert-RPE 1)
 , ,
, ,  , , and
, , and 
Abstract
:1. Introduction
2. Results
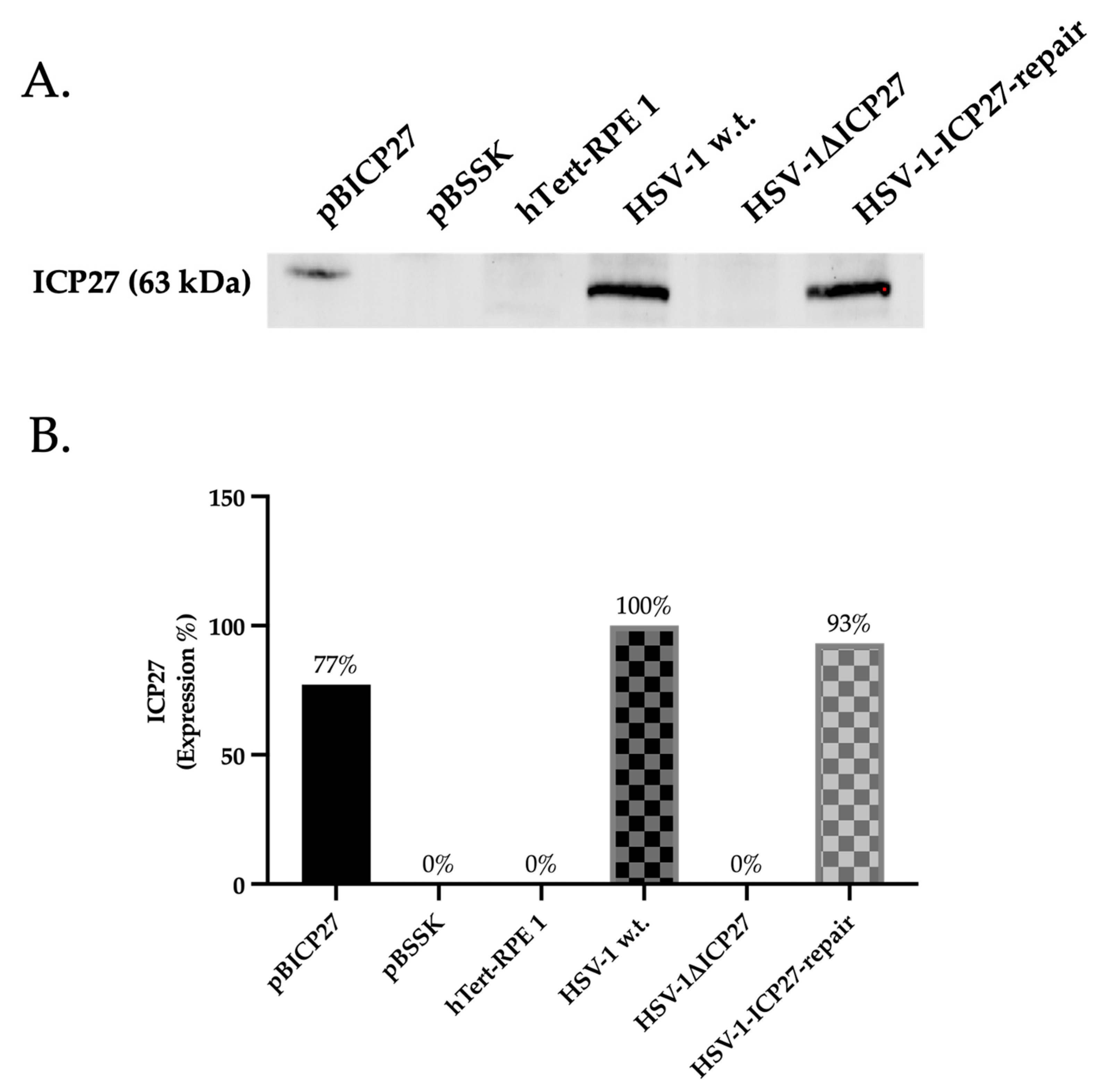
2.1. HSV-1ΔICP27 Mutant Virus Does Not Express the ICP27 Protein
2.2. HSV-1 ICP27 Protein Inhibits the Inflammasome’s Sensor Protein AIM 2
2.3. HSV-1’s ICP27 Protein Inhibits the Inflammasome’s Adaptor Protein ASC
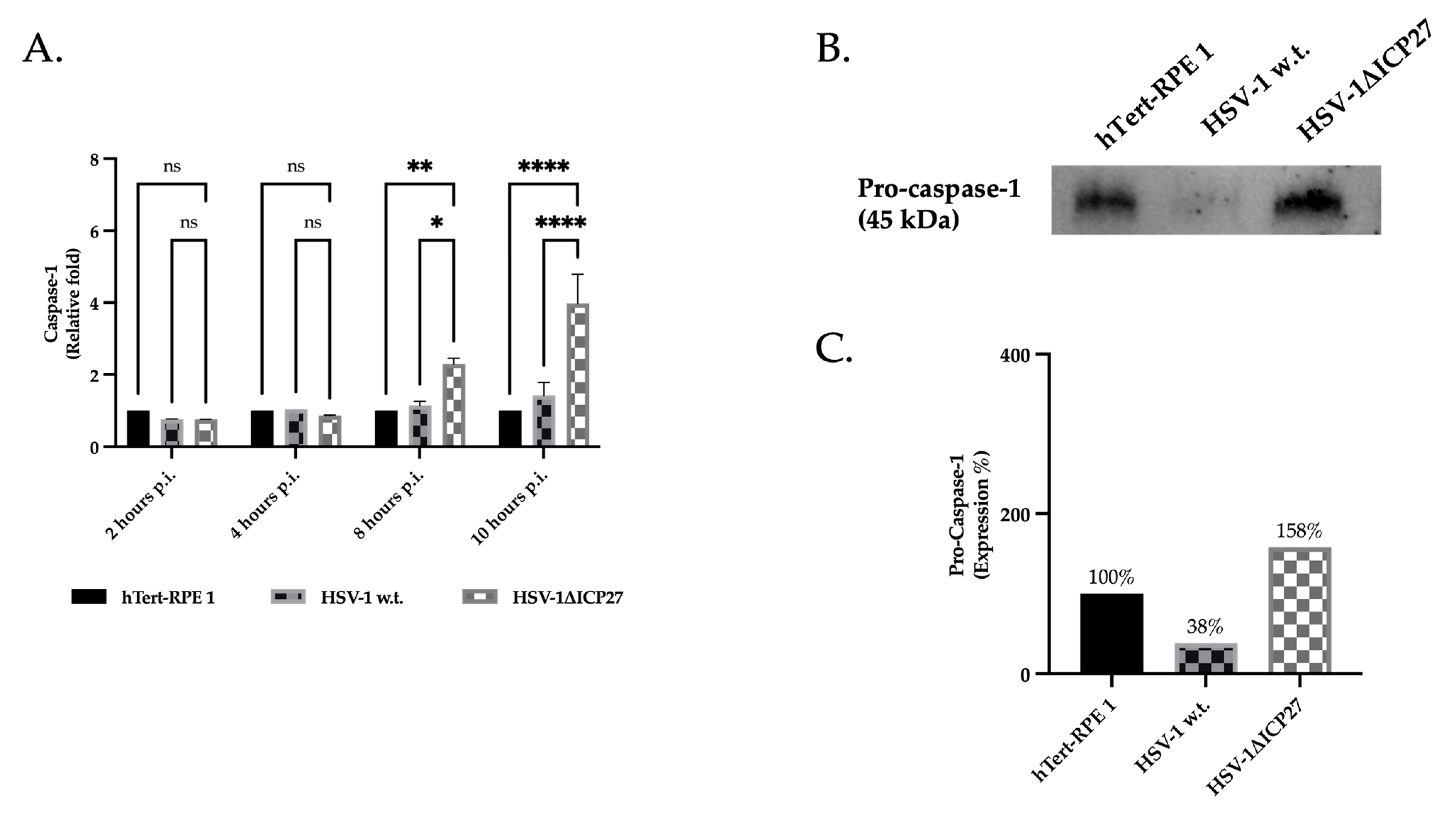
2.4. HSV-1 ICP27 Protein Inhibits the Inflammasome’s Effector Protein Pro-Caspase-1
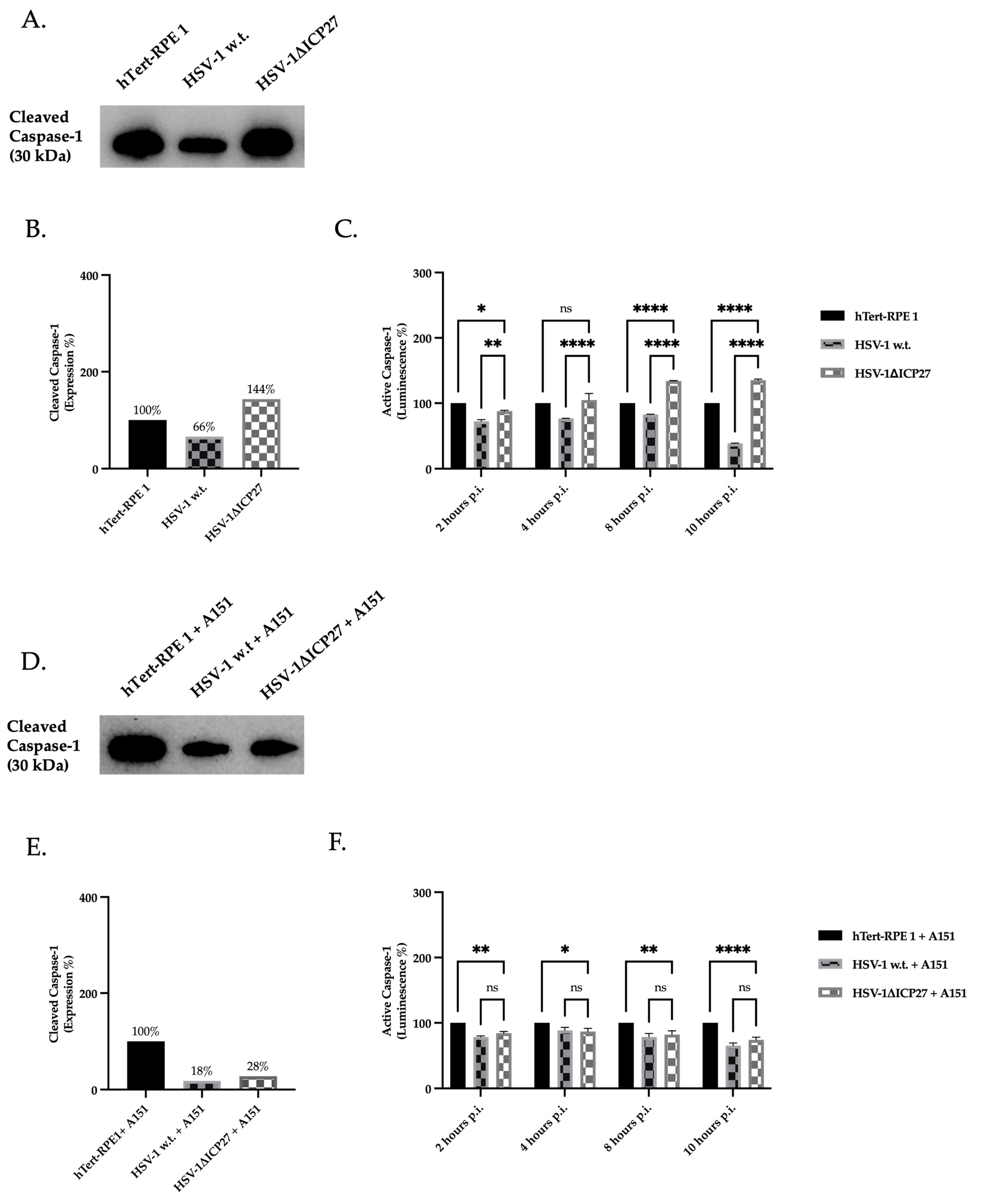
2.5. Inhibition of AIM 2 Inflammasome Constituent Proteins by ICP27 Results in Downregulation of Active Caspase-1
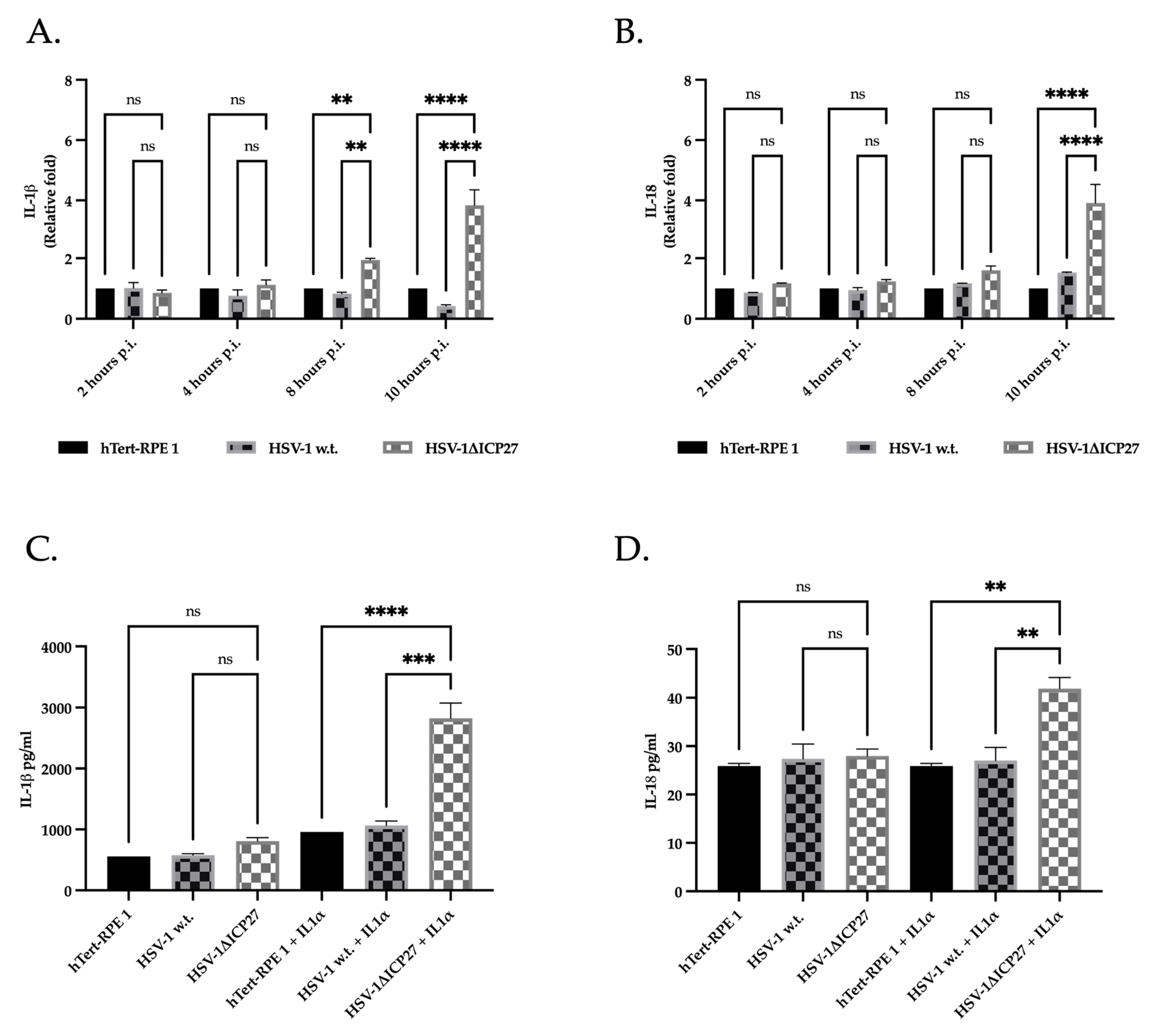
2.6. The Inhibition of Caspase-1 by ICP27 Affects the Expression and Release of Pro-Inflammatory Cytokines IL-1β and IL-18
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Plasmid Construction
4.3. Viruses Construction
4.4. Viruses: Propagation and In Vitro Titration
4.5. Transfection
4.6. Infection
4.7. AIM 2 Inhibition
4.8. RNA Extraction and Real-Time PCR
4.9. Protein Extraction and Western Blot Assay
4.10. Caspase-1 Bioluminescent Inflammasome Assay
4.11. ELISA and IL-1α Treatment
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thellman, N.M.; Triezenberg, S.J. Herpes simplex virus establishment, maintenance, and reactivation: In vitro modeling of latency. Pathogens 2017, 6, 28. [Google Scholar] [CrossRef]
- Menendez, C.M.; Carr, D.J.J. Defining nervous system susceptibility during acute and latent herpes simplex virus-1 infection. J. Neuroimmunol. 2017, 308, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Viejo-Borbolla, A. Pathogenesis and virulence of herpes simplex virus. Virulence 2021, 12, 2670–2702. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.; Illzam, E.M.; Muniandy, R.K.; Sharifah, A.M.; Nang, M.K.; Ramesh, B. Herpes simplex virus infections, Pathophysiology and Management. IOSR J. Dent. Med. Sci. 2016, 15, 85–91. [Google Scholar] [CrossRef]
- Kumar, S.P.; Chandy, M.L.; Shanavas, M.; Khan, S.; Suresh, K.V. Pathogenesis and life cycle of herpes simplex virus infection-stages of primary, latency and recurrence. J. Oral. Maxillofac. Surg. Med. Pathol. 2016, 28, 350–353. [Google Scholar] [CrossRef]
- Smith, J.B.; Herbert, J.J.; Truong, N.R.; Cunningham, A.L. Cytokines and chemokines: The vital role they play in herpes simplex virus mucosal immunology. Front. Immunol. 2022, 13, 936235. [Google Scholar] [CrossRef] [PubMed]
- Chentoufi, A.A.; Benmohamed, L. Mucosal herpes immunity and immunopathology to ocular and genital herpes simplex virus infections. Clin. Dev. Immunol. 2012, 2012, 149135. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.S.; Guarda, G.; D’Ombrain, M.C.; Drexler, S.K. Inflammatory caspases in innate immunity and inflammation. J. Innate Immun. 2010, 2, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhou, J.; Shi, C. Inflammasome: Structure, biological functions, and therapeutic targets. MedComm 2023, 4, e391. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Pradeu, T.; Masters, S.L.; Mogensen, T.H. Constitutive immune mechanisms: Mediators of host defence and immune regulation. Nat. Rev. Immunol. 2021, 21, 137–150. [Google Scholar] [CrossRef]
- Muruve, D.A.; Pétrilli, D.A.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A. A virological view of innate immune recognition. Annu. Rev. Microbiol. 2012, 66, 177–196. [Google Scholar] [CrossRef]
- Malik, A.; Kanneganti, T.D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Wu, H. Structural mechanisms of inflammasome assembly. FEBS J. 2015, 282, 435–444. [Google Scholar] [CrossRef]
- Kihara, T.; Toriuchi, K.; Aoki, H.; Kakita, H.; Yamada, Y.; Aoyama, M. Interleukin-1β enhances cell adhesion in human endothelial cells via microRNA-1914–5p suppression. Biochem. Biophys. Rep. 2021, 27, 101046. [Google Scholar] [CrossRef] [PubMed]
- Ihim, S.A.; Abubakar, S.D.; Zian, Z.; Sasaki, T.; Saffarioun, M.; Maleknia, S.; Azizi, G. Interleukin-18 cytokine in immunity, inflammation, and autoimmunity: Biological role in induction, regulation, and treatment. Front. Immunol. 2022, 13, 919973. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.; Taylor, K.E.; Mossman, K.L. Innate and adaptive immune responses to herpes simplex virus. Viruses 2009, 1, 979–1002. [Google Scholar] [CrossRef]
- O’connor, C.M.; Sen, G.C. Innate immune responses to herpesvirus infection. Cells 2021, 10, 2122. [Google Scholar] [CrossRef] [PubMed]
- Gram, A.M.; Frenkel, J.; Ressing, M.E. Inflammasomes and viruses: Cellular defence versus viral offence. J. Gen. Virol. 2012, 93, 2063–2075. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Qin, C.; Liu, Y.; Rao, Y.; Feng, P. Herpes Simplex Virus and Pattern Recognition Receptors: An Arms Race. Front. Immunol. 2021, 11, 613799. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Goulet, M.-L.; Sze, A.; Hadj, S.B.; Belgnaoui, S.M.; Lababidi, R.R.; Zheng, C.; Fritz, J.H.; Olagnier, D.; Lin, R. RIG-I-Mediated STING Upregulation Restricts Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 9406–9419. [Google Scholar]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes Simplex Virus 1 Infection Induces Activation and Subsequent Inhibition of the IFI16 and NLRP3 Inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar]
- Søby, S.; Laursen, R.R.; Østergaard, L.; Melchjorsen, J. HSV-1-induced chemokine expression via IFI16-dependent and IFI16-independent pathways in human monocyte-derived macrophages. Herpesviridae 2012, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.R.; Lum, K.K.; Kennedy, M.A.; Cristea, I.M. The Nuclear DNA Sensor IFI16 Indiscriminately Binds to and Diminishes Accessibility of the HSV-1 Genome to Suppress Infection. mSystems 2022, 7, e0019822. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Stavrakis, G.; Karaba, A.H. Herpesviruses and Inflammasomes: One Sensor Does Not Fit All. mBio 2022, 13, e0173721. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Karki, R.; Kanneganti, T.D. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, E.L.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016, 46, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Bürckstümmer, T.; Baumann, C.; Blüml, S.; Dixit, E.; Dürnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, A.; Frera, G.; Lugrin, J.; Jamilloux, Y.; Hsu, E.-T.; Tardivel, A.; De Gassart, A.; Zaffalon, L.; Bujisic, B.; Siegert, S.; et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc. Natl. Acad. Sci. USA 2016, 113, E4671–E4680. [Google Scholar] [CrossRef] [PubMed]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Søby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal Degradation of Herpes Simplex Virus Capsids in Macrophages Releases DNA to the Cytosol for Recognition by DNA Sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Karaba, A.H.; Figueroa, A.; Massaccesi, G.; Botto, S.; DeFilippis, V.R.; Cox, A.L. Herpes simplex virus type 1 inflammasome activation in proinflammatory human macrophages is dependent on NLRP3, ASC, and caspase-1. PLoS ONE 2020, 15, e0229570. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, G.E.; Sand, J.; Sauter, M.; Seyffert, M.; Steigerwald, R.; Fraefel, C.; Smola, S.; French, L.E.; Beer, H.-D. IFN-γ Primes Keratinocytes for HSV-1–Induced Inflammasome Activation. J. Investig. Dermatol. 2016, 136, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Maruzuru, Y.; Ichinohe, T.; Sato, R.; Miyake, K.; Okano, T.; Suzuki, T.; Koshiba, T.; Koyanagi, N.; Tsuda, S.; Watanabe, M.; et al. Herpes Simplex Virus 1 VP22 Inhibits AIM2-Dependent Inflammasome Activation to Enable Efficient Viral Replication. Cell Host Microbe 2018, 23, 254–265.e7. [Google Scholar] [CrossRef] [PubMed]
- Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. Role of the DNA Binding Activity of Herpes Simplex Virus 1 VP22 in Evading AIM2-Dependent Inflammasome Activation Induced by the Virus. J. Virol. 2021, 95, 10–1128. [Google Scholar]
- Deng, C.H.; Li, T.Q.; Zhang, W.; Zhao, Q.; Wang, Y. Targeting Inflammasome Activation in Viral Infection: A Therapeutic Solution? Viruses 2023, 15, 1451. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Patel, A.; Krause, P.R. Herpes simplex virus ICP27 regulates alternative pre-mRNA polyadenylation and splicing in a sequence-dependent manner. Proc. Natl. Acad. Sci. USA 2016, 113, 12256–12561. [Google Scholar] [CrossRef]
- Sandri-Goldin, R.M. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol. 2011, 6, 1261–1277. [Google Scholar] [CrossRef] [PubMed]
- Hargett, D.; McLean, T.; Bachenheimer, S.L. Herpes Simplex Virus ICP27 Activation of Stress Kinases JNK and p38. J. Virol. 2005, 79, 8348–8360. [Google Scholar]
- Gillis, P.A.; Okagaki, L.H.; Rice, S.A. Herpes Simplex Virus Type 1 ICP27 Induces p38 Mitogen-Activated Protein Kinase Signaling and Apoptosis in HeLa Cells. J. Virol. 2009, 83, 1767–1777. [Google Scholar]
- Kim, J.A.; Kim, J.C.; Min, J.S.; Kang, I.; Oh, J.; Ahn, J.K. HSV-1 ICP27 induces apoptosis by promoting Bax translocation to mitochondria through interacting with 14-3-3θ. BMB Rep. 2017, 50, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Sandri-Goldin, R.M. Properties of an HSV-1 regulatory protein that appears to impair host cell splicing. Infect. Agents Dis. 1994, 3, 59–67. [Google Scholar] [PubMed]
- Smith, R.W.P.; Malik, P.; Clements, J.B. The herpes simplex virus ICP27 protein: A multifunctional post-transcriptional regulator of gene expression. Biochem. Soc. Trans. 2005, 33, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hennig, T.; Whisnant, A.W.; Erhard, F.; Prusty, B.K.; Friedel, C.C.; Forouzmand, E.; Hu, W.; Erber, L.; Chen, Y.; et al. Herpes simplex virus blocks host transcription termination via the bimodal activities of ICP27. Nat. Commun. 2020, 11, 293. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Choi, M.S.; Min, J.S.; Kang, I.; Oh, J.; Kim, J.C.; Ahn, J.K. HSV-1 ICP27 represses NF-κB activity by regulating Daxx sumoylation. BMB Rep. 2017, 50, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Melchjorsen, J.; Sirén, J.; Julkunen, I.; Paludan, S.R.; Matikainen, S. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-κB and IRF-3. J. Gen. Virol. 2006, 87, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Song, B.; Knipe, D.M. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 2008, 374, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. HSV-1 ICP 27 targets the TBK 1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Brandstetter, C.; Patt, J.; Holz, F.G.; Krohne, T.U. Inflammasome priming increases retinal pigment epithelial cell susceptibility to lipofuscin phototoxicity by changing the cell death mechanism from apoptosis to pyroptosis. J. Photochem. Photobiol. B 2016, 161, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H.; Melchjorsen, J.; Malmgaard, L.; Casola, A.; Paludan, S.R. Suppression of Proinflammatory Cytokine Expression by Herpes Simplex Virus Type 1. J. Virol. 2004, 78, 5883–5890. [Google Scholar]
- Tognarelli, E.I.; Palomino, T.F.; Corrales, N.; Bueno, S.M.; Kalergis, A.M.; González, P.A. Herpes simplex virus evasion of early host antiviral responses. Front. Cell. Infect. Microbiol. 2019, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhan, G.; Zheng, C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol. J. 2016, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Stuart, P.M. Developments in Vaccination for Herpes Simplex Virus. Front. Microbiol. 2021, 12, 798927. [Google Scholar] [CrossRef] [PubMed]
- Cuddy, S.R.; Cliffe, A.R. The Intersection of Innate Immune Pathways with the Latent Herpes Simplex Virus Genome. J. Virol. 2023, 97, e0135222. [Google Scholar]
- Zhu, H.; Zheng, C. The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus 1. Microbiol. Mol. Biol. Rev. 2020, 84, 10–1128. [Google Scholar]
- Ike, A.C.; Onu, C.J.; Ononugbo, C.M.; Reward, E.E.; Muo, S.O. Immune response to herpes simplex virus infection and vaccine development. Vaccines 2020, 8, 302. [Google Scholar] [CrossRef] [PubMed]
- de Paiva, C.S.; Leger, A.J.S.; Caspi, R.R. Mucosal immunology of the ocular surface. Mucosal Immunol. 2022, 15, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Antony, F.; Rouse, B.T.; Suryawanshi, A. Role of Innate Interferon Responses at the Ocular Surface in Herpes Simplex Virus-1-Induced Herpetic Stromal Keratitis. Pathogens 2023, 12, 437. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.W.; Hsu, S.; Ng, T.F. The Role of Retinal Pigment Epithelial Cells in Regulation of Macrophages/Microglial Cells in Retinal Immunobiology. Front. Immunol. 2021, 12, 724601. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, G.M.; Kijlstra, A.; Peek, R.; De Vos, A.F. Retinal Pigment Epithelium-immune System Interactions: Cytokine Production and Cytokine-induced Changes. Prog. Retin. Eye Res. 2001, 20, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Du, F.; Chen, S.-W.; Nakasaki, M.; Rana, I.; Shih, V.F.; Hoffmann, A.; Jamora, C. Regulation and Function of the Caspase-1 in an Inflammatory Microenvironment. J. Investig. Dermatol. 2015, 135, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Unlu, S.; Kumar, A.; Waterman, W.R.; Tsukada, J.; Wang, K.Z.; Galson, D.L.; Auron, P.E. Phosphorylation of IRF8 in a pre-associated complex with Spi-1/PU.1 and non-phosphorylated Stat1 is critical for LPS induction of the IL1B gene. Mol. Immunol. 2007, 44, 3364–3379. [Google Scholar] [CrossRef] [PubMed]
- Cornut, M.; Bourdonnay, E.; Henry, T. Transcriptional regulation of inflammasomes. Int. J. Mol. Sci. 2020, 21, 8087. [Google Scholar] [CrossRef]
- Doody, G.M.; Care, M.A.; Burgoyne, N.J.; Bradford, J.R.; Bota, M.; Bonifer, C.; Westhead, D.R.; Tooze, R.M. An extended set of PRDM1/BLIMP1 target genes links binding motif type to dynamic repression. Nucleic Acids Res. 2010, 38, 5336–5350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, J.; Tang, Y.; Jin, T.; Tao, J. Inflammasomes cross-talk with lymphocytes to connect the innate and adaptive immune response. J. Adv. Res. 2023, 54, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Caucheteux, S.M.; Hu-Li, J.; Guo, L.; Bhattacharyya, N.; Crank, M.; Collins, M.T.; Paul, W.E. IL-1β enhances inflammatory TH2 differentiation. J. Allergy Clin. Immunol. 2016, 138, 898–901.e4. [Google Scholar] [CrossRef] [PubMed]
- Ciraci, C.; Janczy, J.R.; Sutterwala, F.S.; Cassel, S.L. Control of innate and adaptive immunity by the inflammasome. Microbes Infect. 2012, 14, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sasson, S.Z.; Wang, K.; Cohen, J.; Paul, W.E. IL-1β strikingly enhances antigen-driven CD4 and CD8 T-cell responses. Cold Spring Harb. Symp. Quant. Biol. 2013, 78, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sasson, S.Z.; Hogg, A.; Hu-Li, J.; Wingfield, P.; Chen, X.; Crank, M.; Caucheteux, S.; Ratner-Hurevich, M.; Berzofsky, J.A.; Nir-Paz, R.; et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J. Exp. Med. 2013, 210, 491–502. [Google Scholar] [CrossRef]
- Ben-Sasson, S.Z.; Hu-Li, J.; Quiel, J.; Cauchetaux, S.; Ratner, M.; Shapira, I.; Dinarello, C.A.; Paul, W.E. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. 2009, 106, 7119–7124. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Chan, W.L.; Leung, B.P.; Hunter, D.; Schulz, K.; Carter, R.W.; McInnes, I.B.; Robinson, J.H.; Liew, F.Y. Selective Expression and Functions of Interleukin 18 Receptor on T Helper (Th) Type 1 but not Th2 Cells. J. Exp. Med. 1998, 188, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Van Den Eeckhout, B.; Tavernier, J.; Gerlo, S. Interleukin-1 as Innate Mediator of T Cell Immunity. Front. Immunol. 2020, 11, 621931. [Google Scholar] [CrossRef] [PubMed]
- Boyaka, P.N.; Mcghee, J.R. Cytokines as adjuvants for the induction of mucosal immunity. Adv. Drug Deliv. Rev. 2001, 51, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.; Garanina, E.; Rizvanov, A.; Khaiboullina, S. Inflammasomes as targets for adjuvants. Pathogens 2020, 9, 252. [Google Scholar] [CrossRef] [PubMed]
- Martynova, E.; Rizvanov, A.; Urbanowicz, R.A.; Khaiboullina, S. Inflammasome Contribution to the Activation of Th1, Th2, and Th17 Immune Responses. Front. Microbiol. 2022, 13, 851835. [Google Scholar] [CrossRef] [PubMed]
- Krisky, D.M.; Marconi, P.C.; Oligino, T.; Rouse, R.J.D.; Fink, D.J.; Glorioso, J.C. Rapid method for construction of recombinant HSV gene transfer vectors. Gene Ther. 1997, 4, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Marconi, P.; Krisky, D.; Oligino, T.; Poliani, P.L.; Ramakrishnan, R.; Goins, W.F.; Fink, D.J.; Glorioso, J.C. Replication-defective herpes simplex virus vectors for gene transfer in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 11319–11320. [Google Scholar] [CrossRef] [PubMed]
- Warden, C.; Tang, Q.; Zhu, H. Herpesvirus BACs: Past, present, and future. J. Biomed. Biotechnol. 2011, 2011, 124595. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Yajima, M.; Ahsan, N.; Tanaka, M.; Takada, K. Production of High-Titer Epstein-Barr Virus Recombinants Derived from Akata Cells by Using a Bacterial Artificial Chromosome System. J. Virol. 2004, 78, 7004–7015. [Google Scholar]
- Fraefel, C.; Marconi, P.; Epstein, A.L. Herpes Simplex Virus Type 1-Derived Recombinant and Amplicon Vectors. In Viral Vectors for Gene Therapy. Methods in Molecular Biology; Marten, O., Al-Rubeai, M., Eds.; Humana Press: Totowa, NJ, USA, 2011; Volume 737, pp. 303–343. [Google Scholar] [CrossRef]
- Marconi, P.; Manservigi, R. Herpes Simplex Virus Growth, Preparation, and Assay. In Herpes Simplex Virus; Humana Press: Totowa, NJ, USA, 2014; Volume 1144, pp. 19–29. [Google Scholar] [CrossRef]
- Juarez, D.; Long, K.C.; Aguilar, P.; Kochel, T.J.; Halsey, E.S. Assessment of plaque assay methods for alphaviruses. J. Virol. Methods 2013, 187, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Maloy, A.; Alexander, S.; Andreas, A.; Nyunoya, T.; Chandra, D. Stain-Free total-protein normalization enhances the reproducibility of Western blot data. Anal. Biochem. 2022, 654, 114840. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Moehring, D.; Muñoz-Planillo, R.; Núñez, G.; Callaway, J.; Ting, J.; Scurria, M.; Ugo, T.; Bernad, L.; Cali, J.; et al. A bioluminescent caspase-1 activity assay rapidly monitors inflammasome activation in cells. J. Immunol. Methods 2017, 447, 1–13. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Forward | Primer Reverse |
|---|---|---|
| Caspase 1 | TGAATACCAAGAACTGCCCAAG | GCATCATCCTCAAACTCTTCTGTAG |
| ASC | CGCGAGGGTCACAAACGT | TGCTCATCCGTCAGGACCTT |
| AIM 2 | ATCTCCTGCTGCCTTCTTGG | AAGTCTCTCCTCATGTTAAGCCTG |
| IL-1β | AGATGATAAGCCCACTCTACAG | ACATTCAGCACAGACTCTC |
| IL-18 | CAGCCGCTTTAGCAGCCA | GCAAGGAATGTCTCCCAGTGC |
| GAPDH | GGTGTGAACCATGAGAAGTA | GAGTCCTTCCACGATACCAA |
| 18s | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| Antibodies | Source | Identifier |
|---|---|---|
| Mouse monoclonal anti-ICP27 antibody | Virusys | Cat#P1113 |
| Mouse monoclonal Human Caspase-1 Antibody | R&D System | Cat#MAB6215 |
| Rabbit polyclonal anti-caspase 1 (c-term) antibody | Biorad | Cat#AHP963 |
| Rabbit polyclonal anti-TMS1/ASC antibody | Abcam | Cat#EPR23978-28 |
| Rabbit monoclonal anti-AIM2 antibody | R&D System | Cat#MAB9965 |
| Polyclonal Goat Anti-Mouse Immunoglobulins/HRP | Dako | Cat#P0447 |
| Polyclonal Goat Anti-Mouse Immunoglobulins/HRP | Dako | Cat#P0448 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caproni, A.; Nordi, C.; Fontana, R.; Facchini, M.; Melija, S.; Pappadà, M.; Buratto, M.; Marconi, P. Herpes Simplex Virus ICP27 Protein Inhibits AIM 2-Dependent Inflammasome Influencing Pro-Inflammatory Cytokines Release in Human Pigment Epithelial Cells (hTert-RPE 1). Int. J. Mol. Sci. 2024, 25, 4608. https://doi.org/10.3390/ijms25094608
Caproni A, Nordi C, Fontana R, Facchini M, Melija S, Pappadà M, Buratto M, Marconi P. Herpes Simplex Virus ICP27 Protein Inhibits AIM 2-Dependent Inflammasome Influencing Pro-Inflammatory Cytokines Release in Human Pigment Epithelial Cells (hTert-RPE 1). International Journal of Molecular Sciences. 2024; 25(9):4608. https://doi.org/10.3390/ijms25094608
Chicago/Turabian StyleCaproni, Anna, Chiara Nordi, Riccardo Fontana, Martina Facchini, Sara Melija, Mariangela Pappadà, Mattia Buratto, and Peggy Marconi. 2024. "Herpes Simplex Virus ICP27 Protein Inhibits AIM 2-Dependent Inflammasome Influencing Pro-Inflammatory Cytokines Release in Human Pigment Epithelial Cells (hTert-RPE 1)" International Journal of Molecular Sciences 25, no. 9: 4608. https://doi.org/10.3390/ijms25094608
APA StyleCaproni, A., Nordi, C., Fontana, R., Facchini, M., Melija, S., Pappadà, M., Buratto, M., & Marconi, P. (2024). Herpes Simplex Virus ICP27 Protein Inhibits AIM 2-Dependent Inflammasome Influencing Pro-Inflammatory Cytokines Release in Human Pigment Epithelial Cells (hTert-RPE 1). International Journal of Molecular Sciences, 25(9), 4608. https://doi.org/10.3390/ijms25094608