
Advances in the Synthesis of Biologically Active Quaternary Ammonium Compounds


Abstract
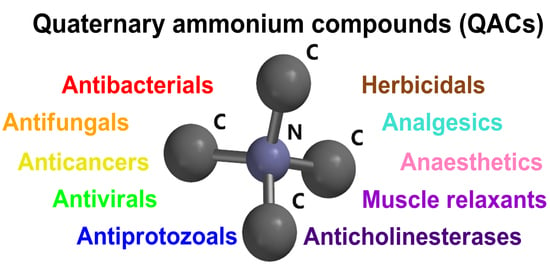
:
1. Introduction
2. Antibacterial Activity

| Compound | S. aureus | MRSA | VRE | E. coli | K. pneumoniae | P. aeruginosa | A. baumannii | S. epidermidis | M. luteus | Pseudomonas sp. | B. subtilis | B. cereus | E. faecalis | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1b | 1.47 | 1.95 | 1.95 | 5.86 | 7.81 | >500 | 3.91 | - | - | - | - | - | - | [35] |
| 2c | 0.5 | 4 | 8 | 32 | 32 | >1024 | - | 2 | - | - | - | - | - | [36] |
| 4d | 50 | - | - | 230 | - | - | - | - | 70 | 1010 | - | - | - | [37] |
| 5c | 80 | - | - | 230 | - | - | - | - | 80 | 2380 | - | - | - | [38] |
| 6c | - | - | - | - | - | 12.7 | - | - | - | - | 12.7 | - | - | [31] |
| 10 | 8 | - | - | 16 | - | 63 | - | - | - | - | - | - | - | [39] |
| 11c | 15.6 | - | - | >125 | - | - | - | - | - | - | - | - | - | [40] |
| 14c | 2.6 | - | - | 27.9 | - | 231.1 | - | - | - | - | - | - | - | [41] |
| 15 | 10 | - | - | 40 | 4 | 30 | - | 80 | - | - | - | 2 | - | [42] |
| 16a | 2.44 | - | - | 19.5 | 19.5 | 78 | - | 1.83 | - | - | - | 1.22 | - | [43] |
| 19 | 39.1 | - | - | - | - | 625 | - | - | - | - | - | - | - | [44] |
| 20 | 3.55 | 7.09 | - | 454 | 227 | - | - | - | - | - | - | - | - | [45] |
| 22 | 1.71 | 1.71 | - | 874 | 874 | >874 | - | - | - | - | - | - | - | [46] |
| 23 | 8.76 | 8.76 | - | 70.1 | 35.1 | 280 | - | - | - | - | - | - | - | [46] |
| 34c | 0.6 | - | - | 5.5 | - | - | - | - | - | - | - | - | - | [47] |
| 38 | 0.25 | 0.5 | - | 2 | - | 8 | - | - | - | - | - | - | 4 | [48] |
| 48 | 17 | - | - | 74.7 | - | - | - | - | - | - | - | - | - | [49] |
| 50d | 2 | 1 | - | 1 | - | 4 | - | - | - | - | - | - | 4 | [50] |
| 51c | 1 | 1 | - | 2 | - | 8 | - | - | - | - | - | - | 8 | [51] |
| 52a–c | >250 | >250 | - | >250 | - | >250 | - | - | - | - | - | - | >250 | [52] |
| 60d | 1 | - | - | 1 | - | - | - | - | - | - | - | - | - | [53] |
| 62d | 6.1 | - | - | 24.4 | - | 97.6 | - | - | - | - | 24.4 | - | - | [54] |
| 63e | 3.7 | - | - | 7.3 | - | - | - | - | - | - | - | - | - | [54] |
| 64l | 0.6 | - | - | 5 | - | 5 | - | - | - | - | 0.6 | - | - | [55] |
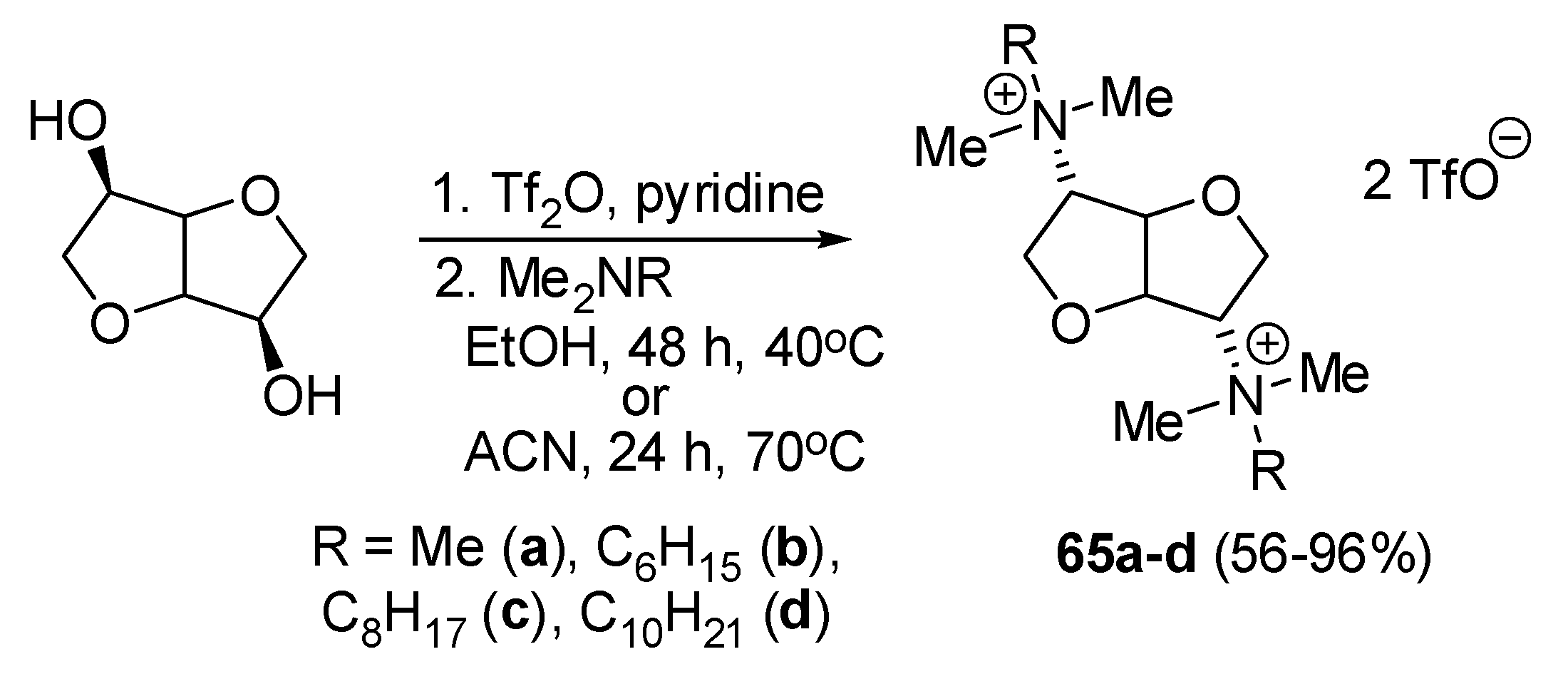
| 65d | 5–10 | - | - | 40 | - | - | - | - | - | - | - | - | - | [56] |
| 66d | 0.8 | - | - | 1.6 | - | - | - | - | - | - | - | - | - | [57] |
| 76e,g | 6.25 | - | - | - | - | 6.25 | - | - | - | - | - | - | - | [58] |
| 79 | 30 | - | - | 16 | - | 250 | - | - | - | - | - | - | - | [59] |
| 80a | 160 | - | - | 80 | - | 50 | - | - | - | - | 120 | - | - | [60] |
| 83c | 0.98 | 0.98 | 1.95 | 3.91 | 7.81 | - | 1.95 | - | - | - | - | - | - | [61] |
| 103a–c | - | - | - | >200 | - | - | - | - | - | - | >200 | - | - | [62] |
| Compound | S. aureus | MRSA | VRE | E. coli | K. pneumoniae | P. aeruginosa | A. baumannii | S. epidermidis | M. luteus | Pseudomonas sp. | B. subtilis | B. cereus | S. typhimurum | E. faecium | E. faecalis | S. pneumoniae | S. pyogenes | S. mutans | P. vulgaris | α-H-tococcus | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3c | 250 | 500 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [63] |
| 4d | 20 | - | - | 100 | - | - | - | - | 30 | 450 | - | - | - | - | - | - | - | - | - | - | [37] |
| 5c | 30 | - | - | 90 | - | - | - | - | 30 | 950 | - | - | - | - | - | - | - | - | - | - | [38] |
| 9 | 2.5 | - | - | 12.5 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [64] |
| 13c | 1 | - | - | 8 | - | 64 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [65] |
| 14c | 2 | - | - | 16 | - | 128 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [41] |
| 17 | - | >63 | - | 2 | - | 1 | 8 | - | - | - | - | - | - | - | - | - | - | - | - | - | [66] |
| 18a | 0.5 | 1 | - | - | - | - | - | - | - | - | - | - | - | 63 | 4 | - | - | - | - | - | [67] |
| 18b | 0.15 | 0.3 | - | - | - | - | - | - | - | - | - | - | - | 1.2 | 0.15 | - | - | - | - | - | [67] |
| 24e | - | - | - | - | - | - | - | - | - | - | - | - | - | - | >64 | 0.5 | 1 | - | - | - | [68] |
| 25g | 25 | - | - | >200 | - | - | - | - | - | - | 50 | - | - | - | 100 | - | 100 | 25 | - | - | [69] |
| 26 | - | 4 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [70] |
| 27 | 1 | - | - | 4 | - | 32 | - | - | 2 | - | 2 | - | - | - | - | - | - | - | - | - | [71] |
| 28 | 1 | - | - | 8 | - | >64 | - | - | 2 | - | 1 | - | - | - | - | - | - | - | - | - | [71] |
| 29b | 0.5 | 2–16 | - | 2 | - | >64 | - | 0.5 | 0.5 | - | - | - | 0.5 | - | - | - | - | - | - | - | [72] |
| 30 | 1 | - | - | 16 | 4 | 32 | - | 2 | - | - | 4 | - | - | - | - | - | - | - | - | - | [73] |
| 33b | 4 | - | - | 16 | >64 | 64 | - | 4 | - | - | 4 | - | - | - | - | - | - | - | - | - | [74] |
| 35g | 0.97 | - | - | 3.9 | 15.6 | 31.25 | 7.8 | - | - | - | - | - | 31.25 | - | 0.97 | - | 0.06 | - | - | - | [75] |
| 36b | 0.4 | - | - | 15.6 | - | 500 | - | - | - | - | - | 0.9 | - | - | - | - | - | - | - | - | [76] |
| 37a | 15.6 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [77] |
| 43 | 32 | - | - | 16 | - | - | - | - | - | - | 8 | - | - | - | - | - | - | - | - | - | [78] |
| 44 | 0.48 | - | - | 15.6 | - | - | - | - | - | - | - | 0.98 | - | - | - | - | - | - | - | - | [79] |
| 45a–c | 32 | - | - | 64 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [80] |
| 45d,e | <2 | - | - | 256 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [80] |
| 53b | - | 0.98 | - | 50 | 31 | 10.7 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [81] |
| 54 | - | 0.25 | - | 0.5 | 2 | 4 | 2 | - | - | - | - | - | - | - | - | - | - | - | - | - | [82] |
| 55d | - | - | - | 16 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [83] |
| 58 | - | - | - | 4 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [84] |
| 59b | - | - | - | 2 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [85] |
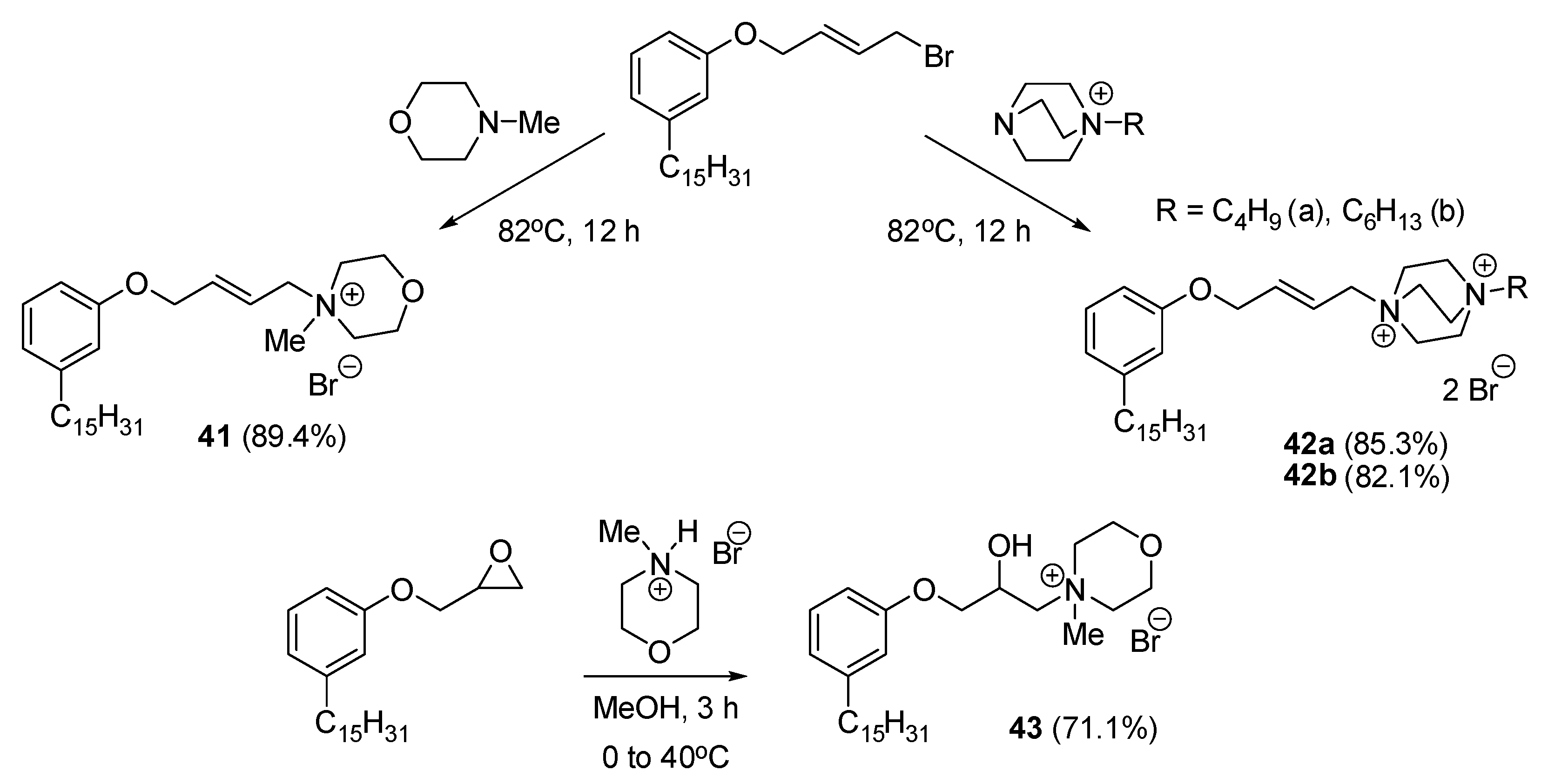
| 65d | 4–8 | - | - | 32 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [56] |
| 69b | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 3.12 | - | - | [86] |
| 70g | 2.5 | - | - | 2.5 | - | - | - | - | - | - | - | - | - | - | - | - | - | 2.5 | - | - | [87] |
| 71d | 61.3 | - | - | - | - | - | - | - | - | - | - | 15.6 | - | - | - | - | - | - | - | - | [88] |
| 73 | 1 | - | - | 8 | 32 | 16 | - | 1 | - | - | 2 | - | - | - | - | - | - | - | - | - | [89] |
| 75d | 2 | - | - | 0.25 | - | 1 | - | 1–4 | - | - | 0.5 | - | - | - | - | 512 | - | - | 0.125 | - | [90] |
| 77a | 6.25 | - | - | 25 | - | 12.5 | - | - | - | - | - | - | - | - | - | - | - | - | 25 | 6.25 | [91] |
| 77b | 6.25 | - | - | 12.5 | - | 25 | - | - | - | - | - | - | - | - | - | - | - | - | 12.5 | 6.25 | [92] |
| 81 | <90 | - | - | <90 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [93] |
| 86d | 64 | - | - | 512 | 512 | 1024 | - | - | - | - | - | - | - | - | - | - | 64 | - | - | - | [94] |
| 93a | 1.25 | - | - | 2.5 | - | 0.625 | - | - | - | - | 1.25 | - | - | - | - | - | - | - | - | - | [95] |
| 94 | - | - | - | 64 | - | - | - | >16 | - | - | - | - | - | - | - | - | - | - | - | - | [96] |
2.1. Benzalkonium Analogues
2.2. QA Peptidomimetics: Amino Acids, and Peptides
| Compound | C. albicans | C. tropicalis | A. niger | P. chrysogenum | Ref. |
|---|---|---|---|---|---|
| 4d | - | 90 | 1240 | - | [37] |
| 5c | - | 50 | 880 | - | [38] |
| 6c | - | - | 12.7 | - | [31] |
| 14c | 13.9 | - | - | - | [41] |
| 34c | 0.6 | - | - | - | [47] |
| 60e | 0.2 | - | - | - | [53] |
| 62e | 12.2 | - | 97.6 | 48.8 | [54] |
| 63e | 14.6 | - | 117 | 58 | [54] |
| 64t | 8.2 | - | 33.2 | 16.5 | [55] |
| 65d | 40 | - | - | - | [56] |
| 66d | 1.6 | - | - | - | [57] |
| 83c | - | - | - | 100 | [61] |
| Compound | C. albicans | C. glabrata | C. tropicalis | A. niger | Ref. |
|---|---|---|---|---|---|
| 4d | - | - | 40 | 550 | [37] |
| 5c | - | - | 20 | 350 | [38] |
| 13c | 2 | - | - | - | [65] |
| 14c | 8 | - | - | - | [41] |
| 33b | 0.38 | - | - | - | [74] |
| 35g | 0.06 | - | - | 0.48 | [75] |
| 36c | 2 | - | - | 500 | [76] |
| 55d | 4 | - | - | - | [83] |
| 65d | 32 | 16 | - | - | [56] |
| 66d | 1.6 | - | - | - | [57,99] |
| 77a | 6.25 | - | - | - | [91,92] |
| 77b | 6.25 | - | - | 6.25 | [91,92] |
| 86d | 64 | - | - | - | [94] |
| 87 | 625 | >5000 | 2500 | - | [100] |
| 88c | 1.6 | - | - | - | [101] |
| 90a | 1 | 1 | 2 | - | [102] |
2.3. Carbohydrate Derivatives
2.4. Compounds Derived from Alkaloids and Natural Compounds
2.5. DABCO Derivatives
2.6. Gemini Surfactants and Polycationic QACs
2.7. Methacrylate Monomers for Dental Applications
2.8. Other Antibacterials
3. Antifungal Activity
| Compound | A549 | MDA-MB-231 | HCT-116 | HepG2 | MCF-7 | HEK-293 | Ref. |
|---|---|---|---|---|---|---|---|
| 93c | - | - | 1.1 | - | 2.46 | 5.08 | [95] |
| 96a | - | - | 1.04 | - | 1.53 | - | [139] |
| 97a | 46 | - | - | - | - | 107 | [140] |
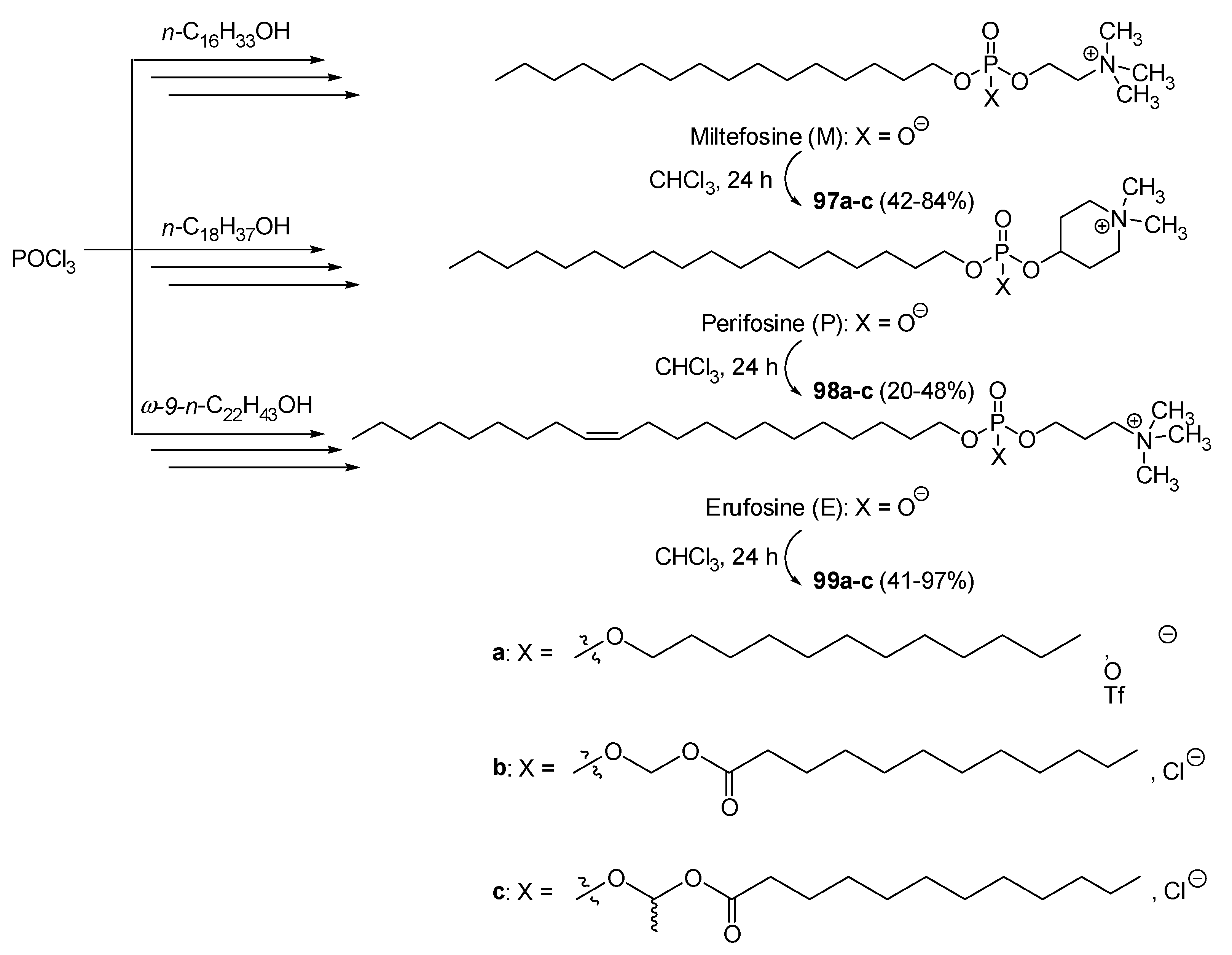
| 100 | 2.42 | - | 1.03 | - | - | 10.11 | [13] |
| 102d | - | 3.93 | - | 8.86 | 4.67 | - | [15] |
| 103a | - | - | - | 130.5 | - | - | [62] |
| 104 | - | 4.4 | - | - | 10.20 | - | [14] |
| 105c | - | - | - | - | 5.5–10.5 | - | [141] |
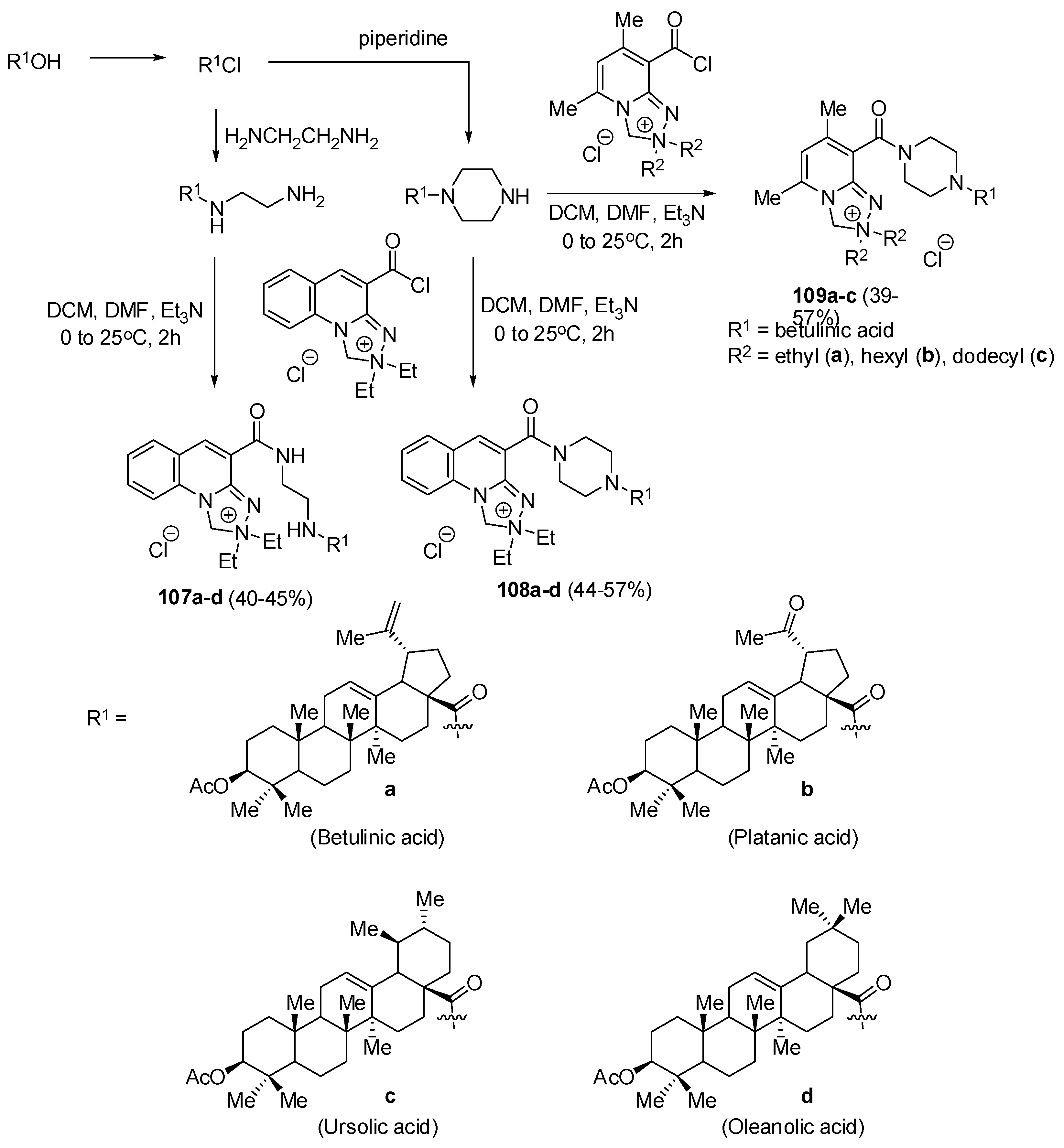
| 109b | - | - | - | - | 4.9 | 3.0 | [142] |
4. Anticancer Activity
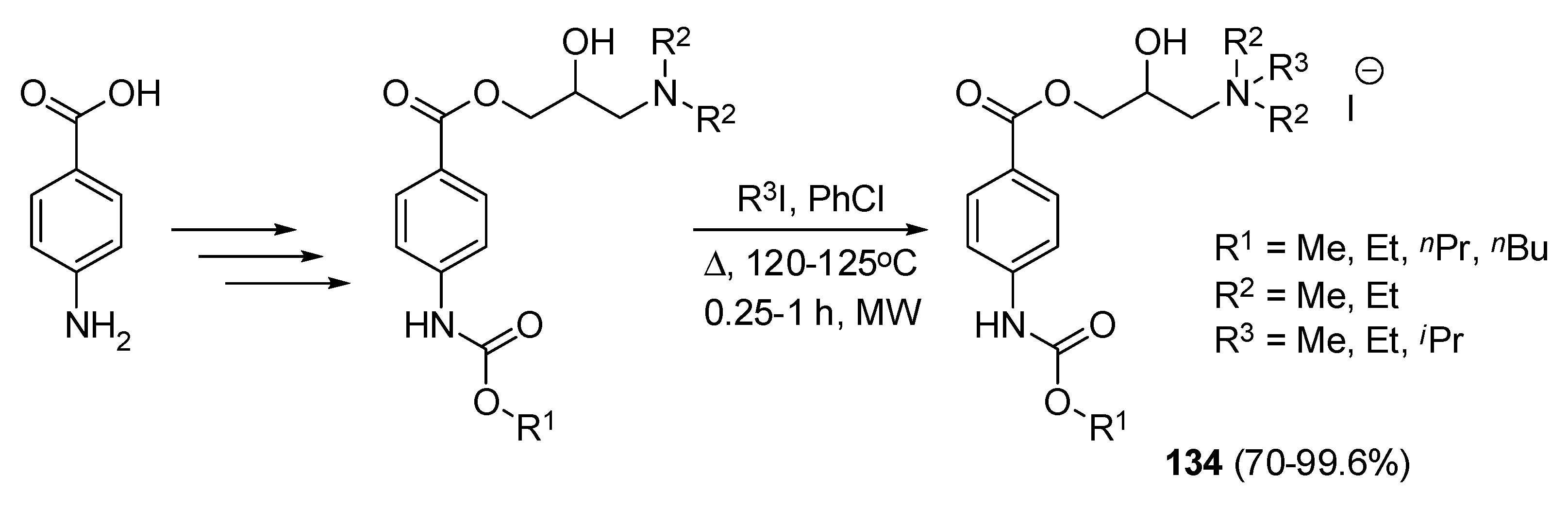
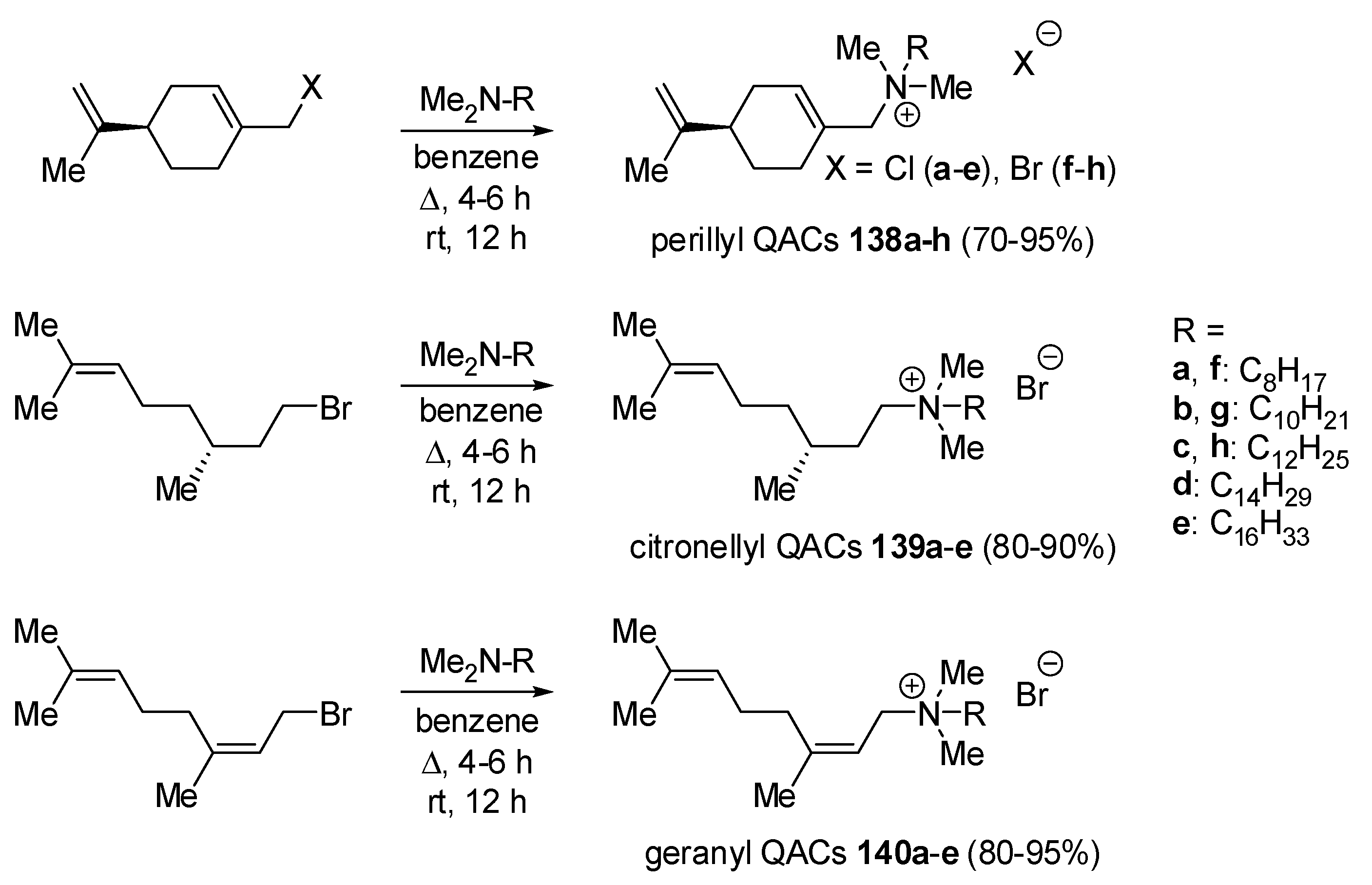
5. Various Applications
5.1. Antivirals
5.2. Antiprotozoals
5.3. Analgesics, Anesthetics, and Muscle Relaxants
5.4. Anticholinesterases
5.5. Herbicidals
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AChE | acetylcholinesterasese |
| AMP | antimicrobial peptides |
| BAC | benzalkonium chlorides |
| BBB | blood–brain barrier |
| BChE | butyrylcholinesterase |
| CFU | colony-forming unit |
| CNS | central nervous system |
| CPC | cetylpyridinium chloride |
| GS | gemini surfactants |
| G+ | Gram-positive |
| G− | Gram-negative |
| HAP | hypoxia-activated prodrugs |
| IC50 | half-maximal inhibitory concentration |
| LPS | lipopolysaccharides |
| MBC | minimum bactericidal concentration |
| MIC | minimum inhibitory concentration |
| mAChR | muscarinic acetylcholine receptor |
| MMP | matrix metalloproteinases |
| MRSA | methicillin-resistant Staphylococcus aureus |
| nAChR | nicotinic acetylcholine receptor |
| NMBAs | neuromuscular blocking agents |
| PAM | positive allosteric modulator |
| PDT | photodynamic therapy |
| PARP-1 | poly(ADP-ribose) polymerase |
| QA | auaternary ammonium |
| QACs | auaternary ammonium compounds |
| TMV | tobacco mosaic virus |
| VRE | vancomycin-resistant enterococci |
References
- McDonnell, G.; Russell, A.D. Antiseptics and Disinfectants: Activity, Action, and Resistance. Clin. Microbiol. Rev. 1999, 12, 147–179. [Google Scholar] [CrossRef] [PubMed]
- Tuo, X.; Yang, J.; Zhang, Y.; Wang, P. Synthesis of n-methylmorpholinium derivatives possessing a 1,3,4-oxadiazole core as feasible antibacterial agents against plant bacterial diseases. J. Chem. 2021, 2021, 5415950. [Google Scholar] [CrossRef]
- Hosseini, P.; Osipitan, O.; Mesgaran, M. Seed germination responses of broomrape species (Phelipanche ramosa and Phelipanche aegyptiaca) to various sanitation chemicals. Weed Technol. 2022, 36, 723–728. [Google Scholar] [CrossRef]
- Rajkowska, K.; Koziróg, A.; Otlewska, A.; Piotrowska, M.; Nowicka-Krawczyk, P.; Brycki, B.; Gutarowska, B. Quaternary ammonium biocides as antimicrobial agents protecting historical wood and brick. Acta Biochim. Pol. 2015, 63, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Thorsteinsson, T.; Másson, M.; Kristinsson, K.; Hjálmarsdóttir, M.; Hilmarsson, H.; Loftsson, T. Soft antimicrobial agents: synthesis and activity of labile environmentally friendly long chain quaternary ammonium compounds. J. Med. Chem. 2003, 46, 4173–4181. [Google Scholar] [CrossRef]
- Zhang, C.; Cui, F.; Zeng, G.; Jiang, M.; Yang, Z.; Yu, Z.; Shen, L. Quaternary ammonium compounds (QACs): A review on occurrence, fate and toxicity in the environment. Sci. Total Environ. 2015, 518–519, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Hora, P.; Pati, S.; McNamara, P.; Arnold, W. Increased use of quaternary ammonium compounds during the SARS-CoV-2 pandemic and beyond: Consideration of environmental implications. Environ. Sci. Technol. Lett. 2020, 7, 622–631. [Google Scholar] [CrossRef]
- Li, X.; Brownawell, B. Quaternary ammonium compounds in urban estuarine sediment environments—A class of contaminants in need of increased attention? Environ. Sci. Technol. 2010, 44, 7561–7568. [Google Scholar] [CrossRef]
- Ruan, T.; Song, S.; Wang, T.; Liu, R.; Lin, Y.; Jiang, G. Identification and composition of emerging quaternary ammonium compounds in municipal sewage sludge in China. Environ. Sci. Technol. 2014, 48, 4289–4297. [Google Scholar] [CrossRef]
- Vincent, G.; Kopferschmitt-Kubler, M.; Mirabel, P.; Pauli, G.; Millet, M. Sampling and analysis of quaternary ammonium compounds (QACs) traces in indoor atmosphere. Environ. Monit. Assess. 2006, 133, 25–30. [Google Scholar] [CrossRef]
- Arnold, W. Quaternary ammonium compounds: A chemical class of emerging concern. Environ. Sci. Technol. 2023, 57, 7645–7665. [Google Scholar] [CrossRef] [PubMed]
- Osimitz, T.; Droege, W. Quaternary ammonium compounds: Perspectives on benefits, hazards, and risk. Toxicol. Res. Appl. 2021, 5, 1–16. [Google Scholar] [CrossRef]
- Xia, X.; Chen, Y.; Wang, L.; Yang, Z.-G.; Ma, X.-D.; Zhao, Z.-G.; Yang, H.-J. Synthesis of Diosgenyl Quaternary Ammonium Derivatives and Their Antitumor Activity. Steroids 2021, 166, 108774. [Google Scholar] [CrossRef] [PubMed]
- Saturnino, C.; Caruso, A.; Iacopetta, D.; Rosano, C.; Ceramella, J.; Muià, N.; Mariconda, A.; Bonomo, M.G.; Ponassi, M.; Rosace, G.; et al. Inhibition of Human Topoisomerase II by N,N,N-Trimethylethanammonium Iodide Alkylcarbazole Derivatives. ChemMedChem 2018, 13, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Skrzypczak, N.; Pyta, K.; Ruszkowski, P.; Mikołajczak, P.Ł.; Kucińska, M.; Murias, M.; Gdaniec, M.; Bartl, F.; Przybylski, P. Anticancer Activity and Toxicity of New Quaternary Ammonium Geldanamycin Derivative Salts and Their Mixtures with Potentiators. J. Enzym. Inhib. Med. Chem. 2021, 36, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Fraise, A.P.; Lambert, P.A.; Maillard, J.-Y. Russell, Hugo & Ayliffe’s Principles and Practice of Disinfection, Preservation & Sterilization; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2004. [Google Scholar] [CrossRef]
- Vieira, D.B. Cationic Lipids and Surfactants as Antifungal Agents: Mode of Action. J. Antimicrob. Chemother. 2006, 58, 760–767. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, Y.; Jia, X.; Wang, Y.; Chen, T. Research progress on typical quaternary ammonium salt polymers. Molecules 2022, 27, 1267. [Google Scholar] [CrossRef]
- Tischer, M.; Pradel, G.; Ohlsen, K.; Holzgrabe, U. Quaternary Ammonium Salts and Their Antimicrobial Potential: Targets or Nonspecific Interactions? ChemMedChem 2011, 7, 22–31. [Google Scholar] [CrossRef]
- Gilbert, P.; Moore, L.E. Cationic Antiseptics: Diversity of Action under a Common Epithet. J. Appl. Microbiol. 2005, 99, 703–715. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, F.; Chen, H.; Li, M.; Qiao, F.; Liu, Z.; Hou, Y.; Wu, C.; Fan, Y.; Liu, L.; et al. Selective Antimicrobial Activities and Action Mechanism of Micelles Self-Assembled by Cationic Oligomeric Surfactants. ACS Appl. Mater. Interfaces 2016, 8, 4242–4249. [Google Scholar] [CrossRef]
- Zhao, X.; Li, Y.; Yuan, H.; Yin, J.; Hu, M. Antibacterial Mechanism of Octamethylene-1,8-Bis(Dodecyldimethylammonium Bromide) against E. coli. J. Surfact. Deterg. 2017, 20, 717–723. [Google Scholar] [CrossRef]
- Ioannou, C.J.; Hanlon, G.W.; Denyer, S.P. Action of Disinfectant Quaternary Ammonium Compounds against Staphylococcus aureus. Antimicrob. Agents Chemother. 2007, 51, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ding, S.; Yu, J.; Chen, X.; Lei, Q.; Fang, W. Antibacterial Activity, In Vitro Cytotoxicity, and Cell Cycle Arrest of Gemini Quaternary Ammonium Surfactants. Langmuir 2015, 31, 12161–12169. [Google Scholar] [CrossRef] [PubMed]
- Inácio, Â.S.; Domingues, N.; Nunes, B.; Martins, T.; Moreno, M.J.; Estronca, L.M.B.B.; Fernandes, R.; Moreno, A.J.; Borrego, M.J.; Gomes, J.P.; et al. Quaternary Ammonium Surfactant Structure Determines Selective Toxicity towards Bacteria: Mechanisms of Action and Clinical Implications in Antibacterial Prophylaxis. J. Antimicrob. Chemother. 2015, 71, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.C.; Minbiole, K.P.C.; Wuest, W.M. Quaternary Ammonium Compounds: An Antimicrobial Mainstay and Platform for Innovation to Address Bacterial Resistance. ACS Infect. Dis. 2015, 1, 288–303. [Google Scholar] [CrossRef]
- Brycki, B.; Szulc, A. Gemini Alkyldeoxy-D-Glucitolammonium Salts as Modern Surfactants and Microbiocides: Synthesis, Antimicrobial and Surface Activity, Biodegradation. PLoS ONE 2014, 9, e84936. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.D.; Gould, G.W. Resistance of Enterobacteriaceae to Preservatives and Disinfectants. J. Appl. Bacteriol. 1988, 65, 167S–195S. [Google Scholar] [CrossRef]
- Verkleij, A.J.; Zwaal, R.F.A.; Roelofsen, B.; Comfurius, P.; Kastelijn, D.; van Deenen, L.L.M. The Asymmetric Distribution of Phospholipids in the Human Red Cell Membrane. A Combined Study Using Phospholipases and Freeze-Etch Electron Microscopy. Biochim. Biophys. Acta–Biomembr. 1973, 323, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Scherer, C.; Müller, K.-D.; Rath, P.-M.; Ansorg, R.A. Influence of Culture Conditions on the Fatty Acid Profiles of Laboratory-Adapted and Freshly Isolated Strains of Helicobacter pylori. J. Clin. Microbiol. 2003, 41, 1114–1117. [Google Scholar] [CrossRef]
- Mechken, K.A.; Menouar, M.; Belkhodja, M.; Saïdi-Besbes, S. Synthesis, Surface Properties and Bioactivity of Novel 4-Substituted 1,2,3-Triazole Quaternary Ammonium Surfactants. J. Mol. Liq. 2021, 338, 116775. [Google Scholar] [CrossRef]
- Liao, Y.; Ye, Z.; Qian, M.; Wang, X.; Guo, Y.; Han, G.; Song, Y.; Hou, J.; Liu, Y. Photoactive NO Hybrids with Pseudo-Zero-Order Release Kinetics for Antimicrobial Applications. Org. Biomol. Chem. 2020, 18, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Tian, R.; Li, G.; Qiu, X.; Xu, L.; Guo, M.; Chigan, D.; Zhang, Y.; Chen, X.; He, G. Cationic Chalcogenoviologen Derivatives for Photodynamic Antimicrobial Therapy and Skin Regeneration. Chem. Eur. J. 2019, 25, 13472–13478. [Google Scholar] [CrossRef] [PubMed]
- Baysal, G.; Aydın, H.; Uzan, S.; Hoşgören, H. Investigation of Antimicrobial Properties of QASs+ (Novel Synthesis). Russ. J. Phys. Chem. B 2018, 12, 695–700. [Google Scholar] [CrossRef]
- Benková, M.; Soukup, O.; Prchal, L.; Sleha, R.; Eleršek, T.; Novák, M.; Sepčić, K.; Gunde-Cimerman, N.; Doležal, R.; Boštíková, V.; et al. Synthesis, Antimicrobial Effect and Lipophilicity-Activity Dependence of Three Series of Dichained N-Alkylammonium Salts. ChemistrySelect 2019, 4, 12076–12084. [Google Scholar] [CrossRef]
- Markova, A.; Hympanova, M.; Matula, M.; Prchal, L.; Sleha, R.; Benkova, M.; Pulkrabkova, L.; Soukup, O.; Krocova, Z.; Jun, D.; et al. Synthesis and Decontamination Effect on Chemical and Biological Agents of Benzoxonium-like Salts. Toxics 2021, 9, 222. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, E.A.; Guastavino, J.F.; Gutierrez, C.G.; Lancelle, M.V.; Russell-White, K.; Murguía, M.C. Synthesis and Properties of New Cleavable Cationic Surfactants Containing Carbonate Groups. J. Surfact. Deterg. 2021, 24, 411–419. [Google Scholar] [CrossRef]
- Gilbert, E.A.; Guastavino, J.F.; Nicollier, R.A.; Lancelle, M.V.; Russell-White, K.; Murguía, M.C. Synthesis and Properties of Cleavable Quaternary Ammonium Compounds. J. Oleo Sci. 2021, 70, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.T.; Kuppusamy, R.; Yasir, M.; Hassan, M.M.; Alghalayini, A.; Gadde, S.; Deplazes, E.; Cranfield, C.; Willcox, M.D.P.; Black, D.S.; et al. Design, Synthesis and Biological Evaluation of Biphenylglyoxamide-Based Small Molecular Antimicrobial Peptide Mimics as Antibacterial Agents. Int. J. Mol. Sci. 2020, 21, 6789. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, R.; Yasir, M.; Yee, E.; Willcox, M.; Black, D.S.C.; Kumar, N. Guanidine Functionalized Anthranilamides as Effective Antibacterials with Biofilm Disruption Activity. Org. Biomol. Chem. 2018, 16, 5871–5888. [Google Scholar] [CrossRef]
- Perinelli, D.R.; Petrelli, D.; Vitali, L.A.; Bonacucina, G.; Cespi, M.; Vllasaliu, D.; Giorgioni, G.; Palmieri, G.F. Quaternary Ammonium Leucine-Based Surfactants: The Effect of a Benzyl Group on Physicochemical Properties and Antimicrobial Activity. Pharmaceutics 2019, 11, 287. [Google Scholar] [CrossRef]
- Laulloo, S.J.; Caumul, P.; Joondan, N.; Jawaheer, S.; Parboteeah, S.; Dyall, S.D.; Bhowon, M.G. A Study of the Antibacterial Activities and the Mode of Action of L-Methionine and L-Cystine Based Surfactants and Their Interaction with Bovine Serum Albumin Using Fluorescence Spectroscopy and In Silico Modelling. Biointerface Res. Appl. Chem. 2021, 12, 7356–7375. [Google Scholar] [CrossRef]
- Joondan, N.; Caumul, P.; Jackson, G.; Laulloo, S.J. Novel Quaternary Ammonium Compounds Derived from Aromatic and Cyclic Amino Acids: Synthesis, Physicochemical Studies and Biological Evaluation. Chem. Phys. Lipids 2021, 235, 105051. [Google Scholar] [CrossRef]
- Peng, G.; Hou, X.; Zhang, W.; Song, M.; Yin, M.; Wang, J.; Li, J.; Liu, Y.; Zhang, Y.; Zhou, W.; et al. Alkyl Rhamnosides, a Series of Amphiphilic Materials Exerting Broad-Spectrum Anti-Biofilm Activity against Pathogenic Bacteria via Multiple Mechanisms. Artif. Cells Nanomed. Biotechnol. 2018, 46, S217–S232. [Google Scholar] [CrossRef]
- Delbeke, E.I.P.; Everaert, J.; Lozach, O.; Le Gall, T.; Berchel, M.; Montier, T.; Jaffrès, P.-A.; Rigole, P.; Coenye, T.; Brennich, M.; et al. Synthesis and Biological Evaluation of Bolaamphiphilic Sophorolipids. ACS Sustain. Chem. Eng. 2018, 6, 8992–9005. [Google Scholar] [CrossRef]
- Delbeke, E.I.P.; Everaert, J.; Lozach, O.; Le Gall, T.; Berchel, M.; Montier, T.; Jaffrès, P.-A.; Rigole, P.; Coenye, T.; Brennich, M.; et al. Lipid-Based Quaternary Ammonium Sophorolipid Amphiphiles with Antimicrobial and Transfection Activities. ChemSusChem 2019, 12, 3642–3653. [Google Scholar] [CrossRef] [PubMed]
- Mikláš, R.; Miklášová, N.; Bukovský, M. Synthesis and Correlation of Aggregation and Antimicrobial Properties of Homochiral Quaternary Ammonium Bromides Derived from Camphoric Acid. Eur. Pharm. J. 2021, 68, 10–16. [Google Scholar] [CrossRef]
- Kontos, R.C.; Schallenhammer, S.A.; Bentley, B.S.; Morrison, K.R.; Feliciano, J.A.; Tasca, J.A.; Kaplan, A.R.; Bezpalko, M.W.; Kassel, W.S.; Wuest, W.M.; et al. An Investigation into Rigidity–Activity Relationships in BisQAC Amphiphilic Antiseptics. ChemMedChem 2018, 14, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Taleb, K.; Mohamed-Benkada, M.; Benhamed, N.; Saidi-Besbes, S.; Grohens, Y.; Derdour, A. Benzene Ring Containing Cationic Gemini Surfactants: Synthesis, Surface Properties and Antibacterial Activity. J. Mol. Liq. 2017, 241, 81–90. [Google Scholar] [CrossRef]
- Schallenhammer, S.A.; Duggan, S.M.; Morrison, K.R.; Bentley, B.S.; Wuest, W.M.; Minbiole, K.P.C. Hybrid BisQACs: Potent Biscationic Quaternary Ammonium Compounds Merging the Structures of Two Commercial Antiseptics. ChemMedChem 2017, 12, 1931–1934. [Google Scholar] [CrossRef]
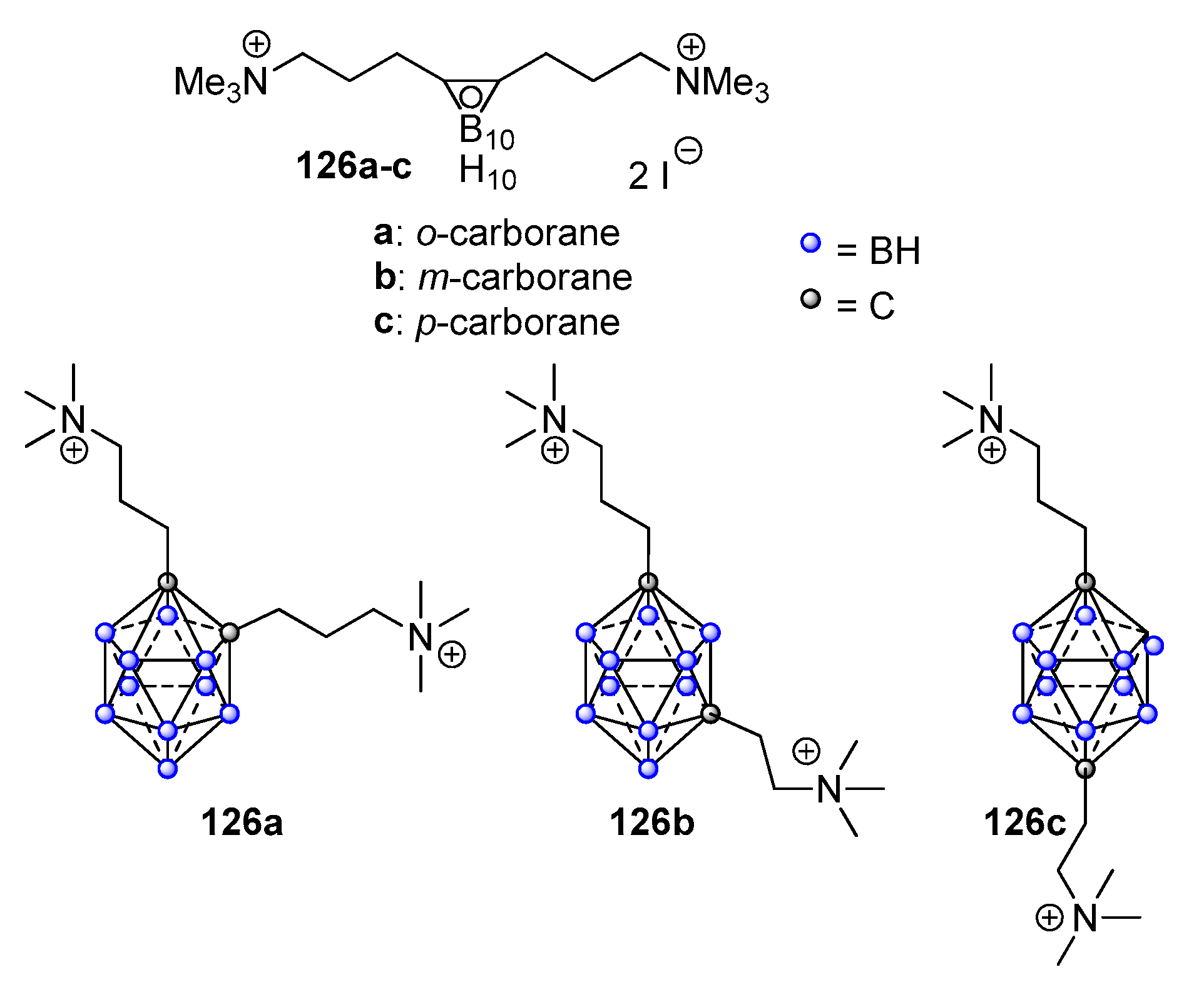
- Sommers, K.J.; Bentley, B.S.; Carden, R.G.; Post, S.J.; Allen, R.A.; Kontos, R.C.; Black, J.W.; Wuest, W.M.; Minbiole, K.P.C. Metallocene QACs: The Incorporation of Ferrocene Moieties into MonoQAC and BisQAC Structures. ChemMedChem 2020, 16, 467–471. [Google Scholar] [CrossRef]
- Feliciano, J.A.; Leitgeb, A.J.; Schrank, C.L.; Allen, R.A.; Minbiole, K.P.C.; Wuest, W.M.; Carden, R.G. Trivalent Sulfonium Compounds (TSCs): Tetrahydrothiophene-Based Amphiphiles Exhibit Similar Antimicrobial Activity to Analogous Ammonium-Based Amphiphiles. Bioorg. Med. Chem. Lett. 2021, 37, 127809. [Google Scholar] [CrossRef]
- Pisárčik, M.; Pupák, M.; Lukáč, M.; Devínsky, F.; Hubčík, L.; Bukovský, M.; Horváth, B. The Synthesis, Self-Assembled Structures, and Microbicidal Activity of Cationic Gemini Surfactants with Branched Tridecyl Chains. Molecules 2019, 24, 4380. [Google Scholar] [CrossRef] [PubMed]
- Brycki, B.; Koziróg, A.; Kowalczyk, I.; Pospieszny, T.; Materna, P.; Marciniak, J. Synthesis, Structure, Surface and Antimicrobial Properties of New Oligomeric Quaternary Ammonium Salts with Aromatic Spacers. Molecules 2017, 22, 1810. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, I.; Pakiet, M.; Szulc, A.; Koziróg, A. Antimicrobial Activity of Gemini Surfactants with Azapolymethylene Spacer. Molecules 2020, 25, 4054. [Google Scholar] [CrossRef] [PubMed]
- Sikora, K.; Nowacki, A.; Szweda, P.; Woziwodzka, A.; Bartoszewska, S.; Piosik, J.; Dmochowska, B. Antimicrobial, Cytotoxic and Mutagenic Activity of Gemini QAS Derivatives of 1,4:3,6-Dianhydro-l-Iditol. Molecules 2022, 27, 757. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Liu, C.; Song, S.; Qiao, Y.; Hu, Y.; Li, P.; Li, Z.; Sun, S. Construction of a Quaternary Ammonium Salt Platform with Different Alkyl Groups for Antibacterial and Biosensor Applications. RSC Adv. 2018, 8, 2941–2949. [Google Scholar] [CrossRef] [PubMed]
- Fedorowicz, J.; Cruz, C.D.; Morawska, M.; Ciura, K.; Gilbert-Girard, S.; Mazur, L.; Mäkkylä, H.; Ilina, P.; Savijoki, K.; Fallarero, A.; et al. Antibacterial and Antibiofilm Activity of Permanently Ionized Quaternary Ammonium Fluoroquinolones. Eur. J. Med. Chem. 2023, 254, 115373. [Google Scholar] [CrossRef]
- Hafidi, Z.; Yakkou, L.; Guouguaou, F.-E.; Amghar, S.; Achouri, M.E. Aminoalcohol-Based Surfactants (N-(Hydroxyalkyl)-N,N-Dimethyl N-Alkylammonium Bromide): Evaluation of Antibacterial Activity and Molecular Docking Studies against Dehydrosqualene Synthase Enzyme (CrtM). J. Dispers. Sci. Technol. 2019, 42, 514–525. [Google Scholar] [CrossRef]
- Zheng, J.; Li, Y.; Yang, X.; Wei, T.; Li, T. Aggregation Behavior and Reactivity of N-Alkyl-N,N-Dimethyl-N-(2,3-Epoxy Propyl) Ammonium Chloride. J. Dispers. Sci. Technol. 2019, 41, 168–178. [Google Scholar] [CrossRef]
- Soukup, O.; Benkova, M.; Dolezal, R.; Sleha, R.; Malinak, D.; Salajkova, S.; Markova, A.; Hympanova, M.; Prchal, L.; Ryskova, L.; et al. The Wide-Spectrum Antimicrobial Effect of Novel N-Alkyl Monoquaternary Ammonium Salts and Their Mixtures; the QSAR Study against Bacteria. Eur. J. Med. Chem. 2020, 206, 112584. [Google Scholar] [CrossRef]
- Karataş, M.O.; Noma, A.; Gürses, C.; Balcıoğlu, S.; Ateş, B.; Alıcı, B.; Çakır, Ü. Water Soluble Coumarin Quaternary Ammonium Chlorides: Synthesis and Biological Evaluation. Chem. Biodivers. 2020, 17, e2000258. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, M.; Kalhapure, R.S.; Rambharose, S.; Mocktar, C.; Govender, T. Synthesis, Characterization and Antibacterial Activity of Novel Heterocyclic Quaternary Ammonium Surfactants. J. Ind. Eng. Chem. 2017, 47, 405–414. [Google Scholar] [CrossRef]
- Shen, S.; Huang, Y.; Yuan, A.; Fengting, L.; Liu, L.; Wang, S. Electrochemical Regulation of Antibacterial Activity Using Ferrocene-Containing Antibiotics. CCS Chem. 2021, 3, 129–135. [Google Scholar] [CrossRef]
- Perinelli, D.R.; Petrelli, D.; Vitali, L.A.; Vllasaliu, D.; Cespi, M.; Giorgioni, G.; Elmowafy, E.; Bonacucina, G.; Palmieri, G.F. Quaternary Ammonium Surfactants Derived from Leucine and Methionine: Novel Challenging Surface Active Molecules with Antimicrobial Activity. J. Mol. Liq. 2019, 283, 249–256. [Google Scholar] [CrossRef]
- Ongwae, G.M.; Morrison, K.R.; Allen, R.A.; Kim, S.; Im, W.; Wuest, W.M.; Pires, M.M. Broadening Activity of Polymyxin by Quaternary Ammonium Grafting. ACS Infect. Dis. 2020, 6, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-C.; Isley, N.A.; Boger, D.L. N-Terminus Alkylation of Vancomycin: Ligand Binding Affinity, Antimicrobial Activity, and Site-Specific Nature of Quaternary Trimethylammonium Salt Modification. ACS Infect. Dis. 2018, 4, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Janas, A.; Pecyna, P.; Gajecka, M.; Bartl, F.; Przybylski, P. Synthesis and Antibacterial Activity of New N-Alkylammonium and Carbonate-Triazole Derivatives within Desosamine of 14- and 15-Membered Lactone Macrolides. ChemMedChem 2020, 15, 1529–1551. [Google Scholar] [CrossRef]
- Bielawski, K.; Leszczyńska, K.; Kałuża, Z.; Bielawska, A.; Michalak, O.; Daniluk, T.; Staszewska-Krajewska, O.; Czajkowska, A.; Pawłowska, N.; Gornowicz, A. Synthesis and Antimicrobial Activity of Chiral Quaternary N-Spiro Ammonium Bromides with 3′,4′-Dihydro-1′H-Spiro[Isoindoline-2,2′-Isoquinoline] Skeleton. Drug Des. Dev. Ther. 2017, 11, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Chalothorn, T.; Rukachaisirikul, V.; Phongpaichit, S.; Pannara, S.; Tansakul, C. Synthesis and Antibacterial Activity of Emodin and Its Derivatives against Methicillin-Resistant Staphylococcus aureus. Tetrahedron Lett. 2019, 60, 151004. [Google Scholar] [CrossRef]
- Sapozhnikov, S.V.; Sabirova, A.E.; Shtyrlin, N.V.; Druk, A.Y.; Agafonova, M.N.; Chirkova, M.N.; Kazakova, P.P.; Grishaev, D.Y.; Nikishova, T.B.; Krylova, E.S.; et al. Design, Synthesis, Antibacterial Activity and Toxicity of Novel Quaternary Ammonium Compounds Based on Pyridoxine and Fatty Acids. Eur. J. Med. Chem. 2021, 211, 113100. [Google Scholar] [CrossRef]
- Sapozhnikov, S.V.; Shtyrlin, N.V.; Kayumov, A.R.; Zamaldinova, A.E.; Iksanova, A.G.; Nikitina, E.V.; Krylova, E.S.; Grishaev, D.Y.; Balakin, K.V.; Shtyrlin, Y.G. New Quaternary Ammonium Pyridoxine Derivatives: Synthesis and Antibacterial Activity. Med. Chem. Res. 2017, 26, 3188–3202. [Google Scholar] [CrossRef]
- Shtyrlin, N.V.; Pugachev, M.V.; Sapozhnikov, S.V.; Garipov, M.R.; Vafina, R.M.; Grishaev, D.Y.; Pavelyev, R.S.; Kazakova, R.R.; Agafonova, M.N.; Iksanova, A.G.; et al. Novel Bis-Ammonium Salts of Pyridoxine: Synthesis and Antimicrobial Properties. Molecules 2020, 25, 4341. [Google Scholar] [CrossRef]
- Garipov, M.R.; Sabirova, A.E.; Pavelyev, R.S.; Shtyrlin, N.V.; Lisovskaya, S.A.; Bondar, O.V.; Laikov, A.V.; Romanova, J.G.; Bogachev, M.I.; Kayumov, A.R.; et al. Targeting Pathogenic Fungi, Bacteria and Fungal-Bacterial Biofilms by Newly Synthesized Quaternary Ammonium Derivative of Pyridoxine and Terbinafine with Dual Action Profile. Bioorg. Chem. 2020, 104, 104306. [Google Scholar] [CrossRef]
- Bazina, L.; Maravić, A.; Krce, L.; Soldo, B.; Odžak, R.; Popovic, V.; Aviani, I.; Primožič, I.; Šprung, M. Discovery of Novel Quaternary Ammonium Compounds Based on Quinuclidine-3-Ol as New Potential Antimicrobial Candidates. Eur. J. Med. Chem. 2019, 163, 626–635. [Google Scholar] [CrossRef]
- Burilova, E.A.; Pashirova, T.N.; Lukashenko, S.S.; Sapunova, A.S.; Voloshina, A.D.; Zhiltsova, E.P.; Campos, J.R.; Souto, E.B.; Zakharova, L.Y. Synthesis, Biological Evaluation and Structure-Activity Relationships of Self-Assembled and Solubilization Properties of Amphiphilic Quaternary Ammonium Derivatives of Quinuclidine. J. Mol. Liq. 2018, 272, 722–730. [Google Scholar] [CrossRef]
- Bogdanov, A.V.; Kadomtseva, M.E.; Bukharov, S.V.; Voloshina, A.D.; Mironov, V.F. Effect of the Cationic Moiety on the Antimicrobial Activity of Sterically Hindered Isatin 3-Hydrazone Derivatives. Russ. J. Org. Chem. 2020, 56, 555–558. [Google Scholar] [CrossRef]
- Ma, J.; Liu, N.; Huang, M.; Wang, L.; Han, J.; Qian, H.; Che, F. Synthesis, Physicochemical and Antimicrobial Properties of Cardanol-Derived Quaternary Ammonium Compounds (QACs) with Heterocyclic Polar Head. J. Mol. Liq. 2019, 294, 111669. [Google Scholar] [CrossRef]
- Pashirova, T.N.; Sapunova, A.S.; Lukashenko, S.S.; Burilova, E.A.; Lubina, A.P.; Shaihutdinova, Z.M.; Gerasimova, T.P.; Kovalenko, V.I.; Voloshina, A.D.; Souto, E.; et al. Synthesis, Structure-Activity Relationship and Biological Evaluation of Tetracationic Gemini Dabco-Surfactants for Transdermal Liposomal Formulations. Int. J. Pharm. 2020, 575, 118953. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, H.; Shen, Y.; Chai, S.; Zhang, J.; Zou, Q. Application of Gemini Quaternary Ammonium with Ester Groups in Cationic P(St-Co-BA) Nanolatex and Study on Its Antibacterial Properties. Mater. Sci. Eng. C 2017, 80, 417–424. [Google Scholar] [CrossRef]
- Rohand, T.; Ramli, Y.; Baruah, M.; Budka, J.; Das, A.M. Synthesis, Structure Elucidation and Antimicrobial Properties of New Bis-1,3,4-Oxadiazole Derivatives. Pharm. Chem. J. 2019, 53, 150–154. [Google Scholar] [CrossRef]
- Dey, R.; De, K.; Mukherjee, R.; Ghosh, S.; Haldar, J. Small Antibacterial Molecules Highly Active against Drug-Resistant Staphylococcus aureus. MedChemComm 2019, 10, 1907–1915. [Google Scholar] [CrossRef]
- Wan, M.; Hua, L.; Zeng, Y.; Jiao, P.; Xie, D.; Tong, Z.; Wu, G.; Zhou, Y.; Tang, Q.; Mo, F. Synthesis and Properties of Novel Stilbene-Twelve Alkyl Quaternary Ammonium Salts as Antibacterial Optical Whitening Agents. Cellulose 2017, 24, 3209–3218. [Google Scholar] [CrossRef]
- Limwongyut, J.; Nie, C.; Moreland, A.S.; Bazan, G.C. Molecular Design of Antimicrobial Conjugated Oligoelectrolytes with Enhanced Selectivity toward Bacterial Cells. Chem. Sci. 2020, 11, 8138–8144. [Google Scholar] [CrossRef]
- Limwongyut, J.; Moreland, A.S.; Nie, C.; Read de Alaniz, J.; Bazan, G.C. Amide Moieties Modulate the Antimicrobial Activities of Conjugated Oligoelectrolytes against Gram-Negative Bacteria. ChemistryOpen 2022, 11, e202100260. [Google Scholar] [CrossRef] [PubMed]
- Manouchehri, F.; Sadeghi, B.; Najafi, F.; Mosslemin, M.H.; Niakan, M. Synthesis and Characterization of Novel Polymerizable Bis-Quaternary Ammonium Dimethacrylate Monomers with Antibacterial Activity as an Efficient Adhesive System for Dental Restoration. Polym. Bull. 2018, 76, 1295–1315. [Google Scholar] [CrossRef]
- Fanfoni, L.; Marsich, E.; Turco, G.; Breschi, L.; Cadenaro, M. Development of Di-Methacrylate Quaternary Ammonium Monomers with Antibacterial Activity. Acta Biomater. 2021, 129, 138–147. [Google Scholar] [CrossRef]
- Chugunova, E.A.; Akylbekov, N.I.; Mahrous, E.M.; Voloshina, A.D.; Kulik, N.V.; Zobov, V.V.; Strelnik, A.G.; Gerasimova, T.P.; Dobrynin, A.B.; Burilov, A.R. Synthesis and Study of Antimicrobial Activity of Quaternary Ammonium Benzofuroxan Salts. Monatsh. Chem. 2017, 149, 119–126. [Google Scholar] [CrossRef]
- Padnya, P.L.; Terenteva, O.S.; Akhmedov, A.A.; Iksanova, A.G.; Shtyrlin, N.V.; Nikitina, E.V.; Krylova, E.S.; Shtyrlin, Y.G.; Stoikov, I.I. Thiacalixarene Based Quaternary Ammonium Salts as Promising Antibacterial Agents. Bioorg. Med. Chem. 2021, 29, 115905. [Google Scholar] [CrossRef]
- Fedorowicz, J.; Sączewski, J.; Konopacka, A.; Waleron, K.; Lejnowski, D.; Ciura, K.; Tomašič, T.; Skok, Ž.; Savijoki, K.; Morawska, M.; et al. Synthesis and Biological Evaluation of Hybrid Quinolone-Based Quaternary Ammonium Antibacterial Agents. Eur. J. Med. Chem. 2019, 179, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xie, X.; Liu, W.; Hou, G.-G.; Sun, J.-F.; Zhao, F.; Cong, W.; Li, H.; Xin, W. Quaternary Ammonium Salts Substituted by 5-Phenyl-1,3,4-Oxadiazole-2-Thiol as Novel Antibacterial Agents with Low Cytotoxicity. Chem. Biol. Drug Des. 2017, 90, 943–952. [Google Scholar] [CrossRef]
- Xie, X.; Cong, W.; Zhao, F.; Li, H.; Xin, W.; Hou, G.; Wang, C. Synthesis, Physiochemical Property and Antimicrobial Activity of Novel Quaternary Ammonium Salts. J. Enzym. Inhib. Med. Chem. 2017, 33, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Li, Q.; Geng, T.; Jiang, Y. Properties of the Quaternary Ammonium Salts with Novel Counterions. J. Surfact. Deterg. 2012, 15, 593–599. [Google Scholar] [CrossRef]
- Mancuso, R.; Amuso, R.; Armentano, B.; Grasso, G.; Rago, V.; Cappello, A.R.; Galiano, F.; Figoli, A.; De Luca, G.; Hoinkis, J.; et al. Synthesis and Antibacterial Activity of Polymerizable Acryloyloxyalkyltriethyl Ammonium Salts. ChemPlusChem 2017, 82, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Feng, X.-Z.; Xiao, Z.-Q.; Fan, G.-R.; Chen, S.-X.; Liao, S.-L.; Luo, H.; Wang, Z.-D. Design, Synthesis, Antibacterial, Antifungal and Anticancer Evaluations of Novel β-Pinene Quaternary Ammonium Salts. Int. J. Mol. Sci. 2021, 22, 11299. [Google Scholar] [CrossRef] [PubMed]
- Miladi, I.; Vivier, M.; Dauplat, M.-M.; Chatard, M.; Besse, S.; Vidal, A.; Chassain, K.; Jean, B.; Forestier, C.; Chezal, J.-M.; et al. Doxycycline and Its Quaternary Ammonium Derivative for Adjuvant Therapies of Chondrosarcoma. Cancer Chemother. Pharmacol. 2017, 80, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.T.; Nizalapur, S.; Ho, K.K.K.; Yee, E.; Berry, T.; Cranfield, C.G.; Willcox, M.; Black, D.S.; Kumar, N. Design, Synthesis and Biological Evaluation of N-Sulfonylphenyl Glyoxamide-Based Antimicrobial Peptide Mimics as Novel Antimicrobial Agents. ChemistrySelect 2017, 2, 3452–3461. [Google Scholar] [CrossRef]
- Mohamed, M.; Abdelkhalek, A.; Abdelkhalek, M. Evaluation of Short Synthetic Antimicrobial Peptides for Treatment of Drug-Resistant and Intracellular Staphylococcus aureus. Sci. Rep. 2016, 6, 29707. [Google Scholar] [CrossRef] [PubMed]
- Cherchali, F.Z.; Mouzali, M.; Tommasino, J.B.; Decoret, D.; Attik, N.; Aboulleil, H.; Seux, D.; Grosgogeat, B. Effectiveness of the DHMAI Monomer in the Development of an Antibacterial Dental Composite. Dent. Mater. 2017, 33, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Dmochowska, B.; Pellowska-Januszek, L.; Samaszko-Fiertek, J.; Slusarz, R.; Wakiec, R.; Madaj, J. Efficient Synthesis and Antifungal Investigation of Nucleosides’ Quaternaryammonium Salt Derivatives. Turk. J. Chem. 2019, 43, 157–171. [Google Scholar] [CrossRef]
- Jain, T.; Muktapuram, P.R.; Pochampalli, S.; Sharma, K.; Pant, G.; Mitra, K.; Bathula, S.R.; Banerjee, D. Chain-Length-Specific Anti-Candida Activity of Cationic Lipo-Oxazoles: A New Class of Quaternary Ammonium Compounds. J. Med. Microbiol. 2017, 66, 1706–1714. [Google Scholar] [CrossRef]
- Hsu, L.-H.; Kwaśniewska, D.; Wang, S.-C.; Shen, T.-L.; Wieczorek, D.; Chen, Y.-L. Gemini Quaternary Ammonium Compound PMT12-BF4 Inhibits Candida Albicans via Regulating Iron Homeostasis. Sci. Rep. 2020, 10, 2911. [Google Scholar] [CrossRef] [PubMed]
- Okano, A.; Isley, N.A.; Boger, D.L. Peripheral Modifications of [Ψ[CH2NH]Tpg4]Vancomycin with Added Synergistic Mechanisms of Action Provide Durable and Potent Antibiotics. Proc. Natl. Acad. Sci. USA 2017, 114, E5052–E5061. [Google Scholar] [CrossRef]
- Zeraik, A.E.; Nitschke, M. Biosurfactants as Agents to Reduce Adhesion of Pathogenic Bacteria to Polystyrene Surfaces: Effect of Temperature and Hydrophobicity. Curr. Microbiol. 2010, 61, 554–559. [Google Scholar] [CrossRef]
- Brackman, G.; Cos, P.; Maes, L.; Nelis, H.J.; Coenye, T. Quorum Sensing Inhibitors Increase the Susceptibility of Bacterial Biofilms to Antibiotics In Vitro and In Vivo. Antimicrob. Agents Chemother. 2011, 55, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Xu, W.F. Facile Synthesis of Emodin Derivatives as Potential MMPIs. Bull. Korean Chem. Soc. 2005, 26, 1923–1924. [Google Scholar] [CrossRef]
- Sharma, S.; Verma, A.; Singh, J.; Teja, B.V.; Mittapelly, N.; Pandey, G.; Urandur, S.; Shukla, R.P.; Konwar, R.; Mishra, P.R. Vitamin B6 Tethered Endosomal PH Responsive Lipid Nanoparticles for Triggered Intracellular Release of Doxorubicin. ACS App. Mater. Interfaces 2016, 8, 30407–30421. [Google Scholar] [CrossRef]
- Bogdanov, A.; Voloshina, A.D.; Sapunova, A.S.; Kulik, N.V.; Bukharov, S.V.; Dobrynin, A.B.; Voronina, J.K.; Terekhova, N.V.; Samorodov, A.V.; Pavlov, V.N.; et al. Isatin-3-Acylhydrazones with Enhanced Lipophilicity: Synthesis, Antimicrobial Activity Evaluation and the Influence on Hemostasis System. Chem. Biodivers. 2022, 19, e202100496. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, A.V.; Zaripova, I.F.; Voloshina, A.D.; Sapunova, A.S.; Kulik, N.V.; Bukharov, S.V.; Voronina, J.K.; Vandyukov, A.E.; Mironov, V.F. Synthesis and Biological Evaluation of New Isatin-Based QACs with High Antimicrobial Potency. ChemistrySelect 2019, 4, 6162–6166. [Google Scholar] [CrossRef]
- Bogdanov, A.V.; Voloshina, A.D.; Sapunova, A.S.; Kulik, N.V.; Mironov, V.F. Effect of Structure of 1-Substituted Isatins on Direction of Their Reactions with Some Acetohydrazide Ammonium Derivatives. Russ. J. Gen. Chem. 2020, 90, 1591–1600. [Google Scholar] [CrossRef]
- Bogdanov, A.V.; Iskhakova, K.R.; Voloshina, A.D.; Sapunova, A.S.; Kulik, N.V.; Terekhova, N.V.; Arsenyev, M.V.; Ziyatdinova, G.K.; Bukharov, S.V. Ammonium-Charged Sterically Hindered Phenols with Antioxidant and Selective Anti-Gram-Positive Bacterial Activity. Chem. Biodivers. 2020, 17, e2000147. [Google Scholar] [CrossRef]
- Hu, S.; Fu, D.; Chen, H.; Liu, H.; Xu, B. Surface Activities, Antibacterial Activity and Corrosion Inhibition Properties of Gemini Quaternary Ammonium Surfactants with Amido Group and Carboxylic Counterions. J. Oleo Sci. 2020, 69, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhou, X.; Zhang, X.; Shi, G.; Hu, J.; Liu, C.; Xu, B. Green Synthesis, Surface Activity, Micellar Aggregation, and Corrosion Inhibition Properties of New Gemini Quaternary Ammonium Surfactants. J. Chem. Eng. Data 2018, 63, 1304–1315. [Google Scholar] [CrossRef]
- Zhou, X.; Hu, S.; Wang, Y.; Ullah, S.; Hu, J.; Liu, H.; Xu, B. The Surface Adsorption, Aggregate Structure and Antibacterial Activity of Gemini Quaternary Ammonium Surfactants with Carboxylic Counterions. R. Soc. Open Sci. 2019, 6, 190378. [Google Scholar] [CrossRef] [PubMed]
- Akram, M.; Lal, H.; Osama, M.; Ansari, F.; Anwar, S.; Kabir-ud-Din; Ahmad, A.; Samreen; Azum, N.; Marwani, H.M.; et al. An Insight View on Synthetic Protocol, Surface Activity, and Biological Aspects of Novel Biocompatible Quaternary Ammonium Cationic Gemini Surfactants. J. Surfact. Deterg. 2020, 24, 35–49. [Google Scholar] [CrossRef]
- Arnitz, R.; Sarg, B.; Ott, H.W.; Neher, A.; Lindner, H.; Nagl, M. Protein Sites of Attack of N-Chlorotaurine in Escherichia coli. Proteomics 2006, 6, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Jekle, A.; Najafi, R.; Ruado, F.S.; Zuck, M.; Khosrovi, B.; Memarzadeh, B.; Debabov, D.; Wang, L.; Anderson, M.E. Virucidal Mechanism of Action of NVC-422, a Novel Antimicrobial Drug for the Treatment of Adenoviral Conjunctivitis. Antivir. Res. 2011, 92, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Ghanbar, S.; Kazemian, M.R.; Liu, S. New Generation of N-Chloramine/QAC Composite Biocides: Efficient Antimicrobial Agents to Target Antibiotic-Resistant Bacteria in the Presence of Organic Load. ACS Omega 2018, 3, 9699–9709. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H.; Jia, D.; Wang, P. Synthesis of Gemini-QA N-Chloramine Biocides for Antibacterial Applications. ChemistrySelect 2019, 4, 13198–13203. [Google Scholar] [CrossRef]
- Li, L.; Jin, Y.; Zhou, H.; Wang, H. Synthesis of Zwitterionic N-Chlorohydantoins for Antibacterial Applications. Bioorg. Med. Chem. Lett. 2018, 28, 3665–3669. [Google Scholar] [CrossRef]
- Li, L.; Pu, T.; Zhanel, G.; Zhao, N.; Ens, W.; Liu, S. New Biocide with BothN-Chloramine and Quaternary Ammonium Moieties Exerts Enhanced Bactericidal Activity. Adv. Healthc. Mater. 2012, 1, 609–620. [Google Scholar] [CrossRef]
- Wan, M.; Luo, Y.; Tong, Z.; Geng, Q.; Hua, L. Novel Amino Acid-Stilbene Quaternary Ammonium Salt Fluorescent Whitening Agents: Synthesis, Optical Properties, Acid Resistance and Anti-Bacterial Activity. Color. Technol. 2021, 138, 201–209. [Google Scholar] [CrossRef]
- Kim, M.; Linstadt, R.T.H.; Ando, K.A.; Ahn, J. Gemini-Mediated Self-Disinfecting Surfaces to Address the Contact Transmission of Infectious Diseases. Langmuir 2022, 38, 2162–2173. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shi, Y.; Chen, X.; Liu, F.; Zhao, W. A Simple Method of Simultaneously Endowing Paper or Fluff Pulp with Both High Softness or Appropriate Fluffing Properties and Antimicrobial Properties. Cellulose 2021, 28, 7327–7339. [Google Scholar] [CrossRef]
- Zhou, M.; Zhao, J.; Hu, X. Synthesis of Bis[N,N′-(Alkylamideethyl)Ethyl] Triethylenediamine Bromide Surfactants and Their Oilfield Application Investigation. J. Surfact. Deterg. 2011, 15, 309–315. [Google Scholar] [CrossRef]
- Brycki, B.E.; Szulc, A.; Kowalczyk, I.; Koziróg, A.; Sobolewska, E. Antimicrobial Activity of Gemini Surfactants with Ether Group in the Spacer Part. Molecules 2021, 26, 5759. [Google Scholar] [CrossRef]
- Xu, X.M.; Wang, Y.; Liao, S.; Wen, Z.T.; Fan, Y. Synthesis and Characterization of Antibacterial Dental Monomers and Composites. J. Biomed. Mater. Res. Part B 2012, 100, 1151–1162. [Google Scholar] [CrossRef]
- Wang, Y.; Costin, S.; Zhang, J.-F.; Liao, S.; Wen, Z.T.; Lallier, T.; Yu, Q.; Xu, X. Synthesis, Antibacterial Activity, and Biocompatibility of New Antibacterial Dental Monomers. Am. J. Dent. 2018, 31, 17B–23B. [Google Scholar] [PubMed]
- Arora, D.P.; Hossain, S.; Xu, Y.; Boon, E.M. Nitric Oxide Regulation of Bacterial Biofilms. Biochemistry 2015, 54, 3717–3728. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, R.V.; Gaynanova, G.A.; Kuznetsova, D.A.; Vasileva, L.A.; Zueva, I.V.; Sapunova, A.S.; Buzyurova, D.N.; Babaev, V.M.; Voloshina, A.D.; Lukashenko, S.S.; et al. Biomedical Potentialities of Cationic Geminis as Modulating Agents of Liposome in Drug Delivery across Biological Barriers and Cellular Uptake. Int. J. Pharm. 2020, 587, 119640. [Google Scholar] [CrossRef]
- Consoli, G.M.L.; Bari, I.D.; Blanco, A.R.; Nostro, A.; D’Arrigo, M.; Pistarà, V.; Sortino, S. Design, Synthesis, and Antibacterial Activity of a Multivalent Polycationic Calix[4]Arene–NO Photodonor Conjugate. ACS Med. Chem. Lett. 2017, 8, 881–885. [Google Scholar] [CrossRef]
- Fedorowicz, J.; Sączewski, J. Modifications of Quinolones and Fluoroquinolones: Hybrid Compounds and Dual-Action Molecules. Monatsh. Chem. 2018, 149, 1199–1245. [Google Scholar] [CrossRef] [PubMed]
- Sączewski, J.; Fedorowicz, J.; Korcz, M.; Sączewski, F.; Wicher, B.; Gdaniec, M.; Konopacka, A. Experimental and Theoretical Studies on the Tautomerism and Reactivity of Isoxazolo[3,4-b]Quinolin-3(1H)-Ones. Tetrahedron 2015, 71, 8975–8984. [Google Scholar] [CrossRef]
- Ciura, K.; Fedorowicz, J.; Kapica, H.; Adamkowska, A.; Sawicki, W.; Sączewski, J. Affinity of Fluoroquinolone–Safirinium Dye Hybrids to Phospholipids Estimated by IAM-HPLC. Processes 2020, 8, 1148. [Google Scholar] [CrossRef]
- Ciura, K.; Fedorowicz, J.; Andrić, F.; Greber, K.E.; Gurgielewicz, A.; Sawicki, W.; Sączewski, J. Lipophilicity Determination of Quaternary (Fluoro)Quinolones by Chromatographic and Theoretical Approaches. Int. J. Mol. Sci. 2019, 20, 5288. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Q.; Wang, Y.; Zhang, W.; Li, T. Synthesis and Properties of Organosilicon Quaternary Salts Surfactants. J. Surfact. Deterg. 2011, 15, 339–344. [Google Scholar] [CrossRef]
- Chen, Z.; Li, S.; Tian, B.; Liang, T.; Jin, Y. Synthesis of a Rosin Gemini Surfactant and Its Properties. Environ. Eng. Sci. 2012, 29, 606–610. [Google Scholar] [CrossRef]
- Feng, X.Z.; Xiao, Z.; Zhang, L.; Liao, S.; Chen, S.; Luo, H.; He, L.; Fan, G.; Wang, Z. Antifungal Activity of β-Pinene-Based Hydronopyl Quaternary Ammonium Salts against Phytopathogenic Fungi. Nat. Prod. Commun. 2020, 15, 1–6. [Google Scholar] [CrossRef]
- Omran, Z.; Guise, C.P.; Chen, L.; Rauch, C.; Abdalla, A.N.; Abdullah, O.; Sindi, I.A.; Fischer, P.M.; Smaill, J.B.; Patterson, A.V.; et al. Design, Synthesis and In-Vitro Biological Evaluation of Antofine and Tylophorine Prodrugs as Hypoxia-Targeted Anticancer Agents. Molecules 2021, 26, 3327. [Google Scholar] [CrossRef]
- Gaillard, B.; Remy, J.-S.; Pons, F.; Lebeau, L. Synthesis and Evaluation of Antitumor Alkylphospholipid Prodrugs. Pharm. Res. 2020, 37, 106. [Google Scholar] [CrossRef]
- Ma, D.-Y.; Wang, L.-L.; Lai, Q.; Peng, K.-J.; Li, X.; Li, Z.-X.; Liu, L.-J.; Luo, Z.-Y.; Liu, S.-Y. Synthesis and Antiproliferative Activities of Novel Quartenary Ammonium Spinosyn Derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 3346–3349. [Google Scholar] [CrossRef]
- Kraft, O.; Kozubek, M.; Hoenke, S.; Serbian, I.; Major, D.; Csuk, R. Cytotoxic Triterpenoid–Safirinium Conjugates Target the Endoplasmic Reticulum. Eur. J. Med. Chem. 2021, 209, 112920. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.; Hogendoorn, P.C.W.; Dijkstra, S.D.; van Rijswijk, C.S.; Krol, A.D.; Taminiau, A.H.M.; Bovée, J.V.M.G. The Clinical Approach towards Chondrosarcoma. Oncologist 2008, 13, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Söderström, M.; Aro, H.; Ahonen, M.; Johansson, N.; Aho, A.; Ekfors, T.; Böhling, T.; Kähäri, V.; Vuorio, E. Expression of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Human Chondrosarcomas. APMIS 2001, 109, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia 2015, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Ghedira, D.; Voissière, A.; Peyrode, C.; Kraiem, J.; Gerard, Y.; Maubert, E.; Vivier, M.; Miot-Noirault, E.; Chezal, J.-M.; Farhat, F.; et al. Structure-Activity Relationship Study of Hypoxia-Activated Prodrugs for Proteoglycan-Targeted Chemotherapy in Chondrosarcoma. Eur. J. Med. Chem. 2018, 158, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Gerard, Y.; Voissière, A.; Peyrode, C.; Galmier, M.-J.; Maubert, E.; Ghedira, D.; Tarrit, S.; Gaumet, V.; Canitrot, D.; Miot-Noirault, E.; et al. Design, Synthesis and Evaluation of Targeted Hypoxia-Activated Prodrugs Applied to Chondrosarcoma Chemotherapy. Bioorg. Chem. 2020, 98, 103747. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, B.; Seguin, C.; Remy, J.-S.; Pons, F.; Lebeau, L. Erufosine (ErPC3) Cationic Prodrugs as Dual Gene Delivery Reagents for Combined Antitumor Therapy. Chem. A Eur. J. 2019, 25, 15662–15679. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Aprille, J.R. Delocalized Lipophilic Cations Selectively Target the Mitochondria of Carcinoma Cells. Adv. Drug Deliv. Rev. 2001, 49, 63–70. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Roayapalley, P.K.; Dimmock, J.R.; Contreras, L.; Balderrama, K.S.; Aguilera, R.J.; Sakagami, H.; Amano, S.; Sharma, R.K.; Das, U. Design, Synthesis and Tumour-Selective Toxicity of Novel 1-[3-{3,5-Bis(Benzylidene)-4-Oxo-1-Piperidino}-3-Oxopropyl]-4-Piperidone Oximes and Related Quaternary Ammonium Salts. Molecules 2021, 26, 7132. [Google Scholar] [CrossRef]
- Fedorowicz, J.; Sączewski, J.; Drażba, Z.; Wiśniewska, P.; Gdaniec, M.; Wicher, B.; Suwiński, G.; Jalińska, A. Synthesis and Fluorescence of Dihydro-[1,2,4]Triazolo[4,3-a]Pyridin-2-Ium-Carboxylates: An Experimental and TD-DFT Comparative Study. Dye. Pigment. 2019, 161, 347–359. [Google Scholar] [CrossRef]
- Sączewski, J.; Hinc, K.; Obuchowski, M.; Gdaniec, M. The Tandem Mannich-Electrophilic Amination Reaction: A Versatile Platform for Fluorescent Probing and Labeling. Chem. Eur. J. 2013, 19, 11531–11535. [Google Scholar] [CrossRef] [PubMed]
- Fedorowicz, J.; Bazar, D.; Brankiewicz, W.; Kapica, H.; Ciura, K.; Zalewska-Piątek, B.; Piątek, R.; Cal, K.; Mojsiewicz-Pieńkowska, K.; Sączewski, J. Development of Safirinium Dyes for New Applications: Fluorescent Staining of Bacteria, Human Kidney Cells, and the Horny Layer of the Epidermis. Sci. Rep. 2022, 12, 15098. [Google Scholar] [CrossRef] [PubMed]
- Fedorowicz, J.; Cebrat, M.; Wierzbicka, M.; Wiśniewska, P.; Jalińska, A.; Dziomba, S.; Gdaniec, M.; Jaremko, M.; Jaremko, Ł.; Chandra, K.; et al. Synthesis and Evaluation of Dihydro-[1,2,4]Triazolo[4,3-a]Pyridin-2-Ium Carboxylates as Fixed Charge Fluorescent Derivatization Reagents for MEKC and MS Proteomic Analyses. J. Mol. Struct. 2020, 1217, 128426. [Google Scholar] [CrossRef]
- Fedorowicz, J.; Wierzbicka, M.; Cebrat, M.; Wiśniewska, P.; Piątek, R.; Zalewska-Piątek, B.; Szewczuk, Z.; Sączewski, J. Application of Safirinium N-Hydroxysuccinimide Esters to Derivatization of Peptides for High-Resolution Mass Spectrometry, Tandem Mass Spectrometry, and Fluorescent Labeling of Bacterial Cells. Int. J. Mol. Sci. 2020, 21, 9643. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lee, S.; Yoon, J. Supramolecular Photosensitizers Rejuvenate Photodynamic Therapy. Chem. Soc. Rev. 2018, 47, 1174–1188. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, H.; He, Z.; Fang, F.; Wang, C.; Zeng, K.; Gao, S.; Meng, F.; Luo, L.; Tang, B.Z. Multicationic AIEgens for Unimolecular Photodynamic Theranostics and Two-Photon Fluorescence Bioimaging. Mater. Chem. Front. 2020, 4, 1623–1633. [Google Scholar] [CrossRef]
- Hu, X.; Cao, Y.; Yin, X.; Zhu, L.; Chen, Y.; Wang, W.; Hu, J. Design and Synthesis of Various Quinizarin Derivatives as Potential Anticancer Agents in Acute T Lymphoblastic Leukemia. Bioorg. Med. Chem. 2019, 27, 1362–1369. [Google Scholar] [CrossRef]
- Karpova, Y.; Wu, C.; Divan, A.; McDonnell, M.E.; Hewlett, E.; Makhov, P.; Gordon, J.; Ye, M.; Reitz, A.B.; Childers, W.E.; et al. Non-NAD-like PARP-1 Inhibitors in Prostate Cancer Treatment. Biochem. Pharmacol. 2019, 167, 149–162. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Baranova, D.; Galochkina, A.V.; Shtro, A.A.; Kireeva, M.V.; Borisevich, S.S.; Gatilov, Y.V.; Zarubaev, V.V.; Salakhutdinov, N.F. Quaternary Ammonium Salts Based on (−)-Borneol as Effective Inhibitors of Influenza Virus. Arch. Virol. 2021, 166, 1965–1976. [Google Scholar] [CrossRef]
- Han, G.; Chen, L.; Wang, Q.; Wu, M.; Liu, Y.; Wang, Q. Design, Synthesis, and Antitobacco Mosaic Virus Activity of Water-Soluble Chiral Quaternary Ammonium Salts of Phenanthroindolizidines Alkaloids. J. Agric. Food Chem. 2018, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- López-Muñoz, M.; Gomez-Peña, J.J.; Ríos-Vásquez, L.A.; Ocampo-Cardona, R.; Jones, M.A.; Haynes, C.S.; Wallace, C.; Robledo, S.M. Novel Fluorinated Quaternary Ammonium Salts and Their In Vitro Activity as Trypanocidal Agents. Med. Chem. Res. 2019, 28, 300–319. [Google Scholar] [CrossRef]
- Mourot, A.; Herold, C.J.; Kienzler, M.A.; Kramer, R.H. Understanding and Improving Photo-Control of Ion Channels in Nociceptors with Azobenzene Photo-Switches. Br. J. Pharmacol. 2017, 175, 2296–2311. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Arozamena, C.; Estrada-Valencia, M.; Martí-Marí, O.; Pérez, C.; de la Fuente Revenga, M.; Villalba-Galea, C.A.; Rodríguez-Franco, M.I. Optical Control of Muscular Nicotinic Channels with Azocuroniums, Photoswitchable Azobenzenes Bearing Two N-Methyl-N-Carbocyclic Quaternary Ammonium Groups. Eur. J. Med. Chem. 2020, 200, 112403. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, M.; Sun, S.-L.; Wang, J.-J.; Zhong, Y.; Chen, H.-H.; Li, H.-M.; Xu, H.; Li, N.-G.; Ma, H.-Y.; et al. Bufotenine and Its Derivatives: Synthesis, Analgesic Effects Identification and Computational Target Prediction. Chin. J. Nat. Med. 2021, 19, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Quadri, M.; Matera, C.; Silnović, A.; Pismataro, M.C.; Horenstein, N.A.; Stokes, C.; Papke, R.L.; Dallanoce, C. Identification of α7 Nicotinic Acetylcholine Receptor Silent Agonists Based on the Spirocyclic Quinuclidine-Δ2-Isoxazoline Scaffold: Synthesis and Electrophysiological Evaluation. ChemMedChem 2017, 12, 1335–1348. [Google Scholar] [CrossRef]
- Del Bello, F.; Bonifazi, A.; Giorgioni, G.; Piergentili, A.; Sabbieti, M.G.; Agas, D.; Dell’Aera, M.; Matucci, R.; Górecki, M.; Pescitelli, G.; et al. Novel Potent Muscarinic Receptor Antagonists: Investigation on the Nature of Lipophilic Substituents in the 5- And/or 6-Positions of the 1,4-Dioxane Nucleus. J. Med. Chem. 2020, 63, 5763–5782. [Google Scholar] [CrossRef]
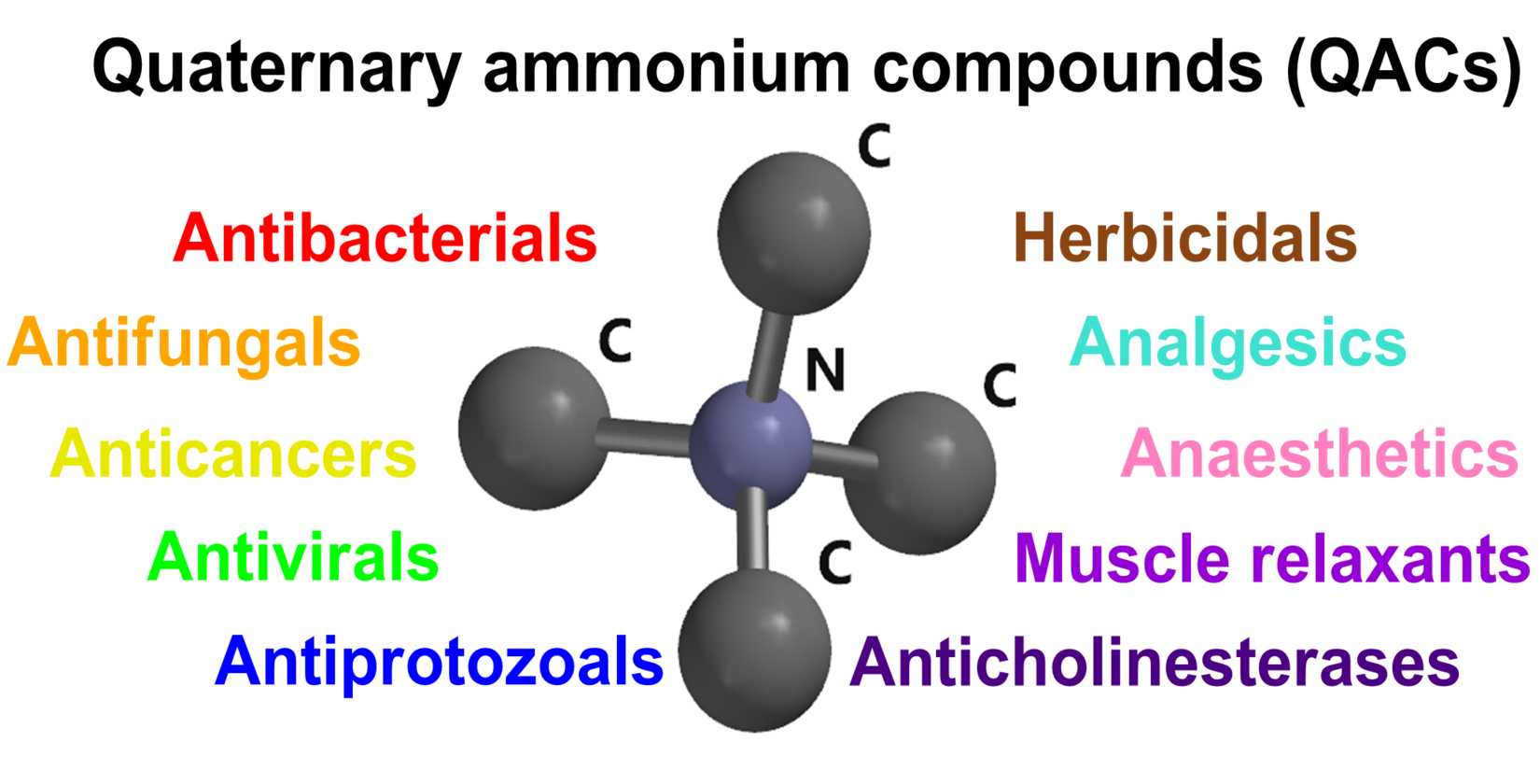
- Goswami, L.N.; Olds, T.J.; Monk, T.G.; Johnson, Q.L.; Dilger, J.P.; Shanawaz, M.A.; Jalisatgi, S.S.; Hawthorne, M.F.; Kracke, G.R. Isomeric Carborane Neuromuscular Blocking Agents. ChemMedChem 2019, 14, 1108–1114. [Google Scholar] [CrossRef]
- Pashirova, T.N.; Burilova, E.A.; Tagasheva, R.G.; Zueva, I.V.; Gibadullina, E.M.; Nizameev, I.R.; Sudakov, I.A.; Vyshtakalyuk, A.B.; Voloshina, A.D.; Kadirov, M.K.; et al. Delivery Nanosystems Based on Sterically Hindered Phenol Derivatives Containing a Quaternary Ammonium Moiety: Synthesis, Cholinesterase Inhibition and Antioxidant Activity. Chem.-Biol. Interact. 2019, 310, 108753. [Google Scholar] [CrossRef]
- Topuzyan, V.O.; Tosunyan, S.R.; Chshmarityan, S.G.; Paronikyan, R.V. Synthesis and Biological Activity of (Z)-Dialkylaminoalkylamides of N-Benzoyl-α,β-Dehydroamino Acids and Their Iodomethylates. Pharm. Chem. J. 2018, 51, 877–880. [Google Scholar] [CrossRef]
- Heise, N.; Friedrich, S.; Temml, V.; Schuster, D.; Siewert, B.; Csuk, R. N-Methylated Diazabicyclo[3.2.2]Nonane Substituted Triterpenoic Acids Are Excellent, Hyperbolic and Selective Inhibitors for Butyrylcholinesterase. Eur. J. Med. Chem. 2022, 227, 113947. [Google Scholar] [CrossRef] [PubMed]
- Padrtova, T.; Marvanova, P.; Odehnalova, K.; Kubinova, R.; Parravicini, O.; Garro, A.; Enriz, R.D.; Humpa, O.; Oravec, M.; Mokry, P. Synthesis, Analysis, Cholinesterase-Inhibiting Activity and Molecular Modelling Studies of 3-(Dialkylamino)-2-Hydroxypropyl 4-[(Alkoxy-Carbonyl)Amino]Benzoates and Their Quaternary Ammonium Salts. Molecules 2017, 22, 2048. [Google Scholar] [CrossRef] [PubMed]
- Godinez, J.; Lee, C.Y.; Schwans, J.P. Synthesis and Evaluation of Fmoc-Amino Esters and Amides Bearing a Substrate like Quaternary Ammonium Group as Selective Butyrylcholinesterase Inhibitors. Bioorg. Med. Chem. Lett. 2023, 92, 129392. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, C.; Yang, J.; Guan, A.; Ma, H. Design, Synthesis, and Herbicidal Activity of Novel Quaternary Ammonium Salt Derivatives. Pestic. Biochem. Physiol. 2017, 143, 246–251. [Google Scholar] [CrossRef]
- Żelechowski, K.; Gucma, M.; Golebiewski, W.M.; Krawczyk, M.; Michalczyk, A.K. Synthesis of New Quaternary Ammonium Salts with a Terpene Function and Evaluation of Their Fungicidal and Herbicidal Activities. Acta Chim. Slov. 2020, 67, 325–335. [Google Scholar] [CrossRef]
- Vallaro, M.; Ermondi, G.; Saame, J.; Leito, I.; Caron, G. Ionization and Lipophilicity in Nonpolar Media Mimicking the Cell Membrane Interior. Bioorg. Med. Chem. 2023, 81, 117203. [Google Scholar] [CrossRef]
- Shen, B.-Y.; Wang, M.-M.; Xu, S.-M.; Gao, C.; Wang, M.; Li, S.; Ampomah-Wireko, M.; Chen, S.-C.; Yan, D.-C.; Qin, S.; et al. Antibacterial Efficacy Evaluation and Mechanism Probe of Small Lysine Chalcone Peptide Mimics. Eur. J. Med. Chem. 2022, 244, 114885. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Baidya, A.T.K.; Mathew, A.T.; Yadav, A.K.; Kumar, R. Structural Modification Aimed for Improving Solubility of Lead Compounds in Early Phase Drug Discovery. Bioorg. Med. Chem. 2022, 56, 116614. [Google Scholar] [CrossRef]
- Ballari, M.S.; Porta, E.O.J.; Zalazar, E.A.; Etichetti, C.M.B.; Padrón, J.M.; Girardini, J.E.; Labadie, G.R. Lipophilic Modification of Salirasib Modulates the Antiproliferative and Antimigratory Activity. Bioorg. Med. Chem. 2023, 92, 117417. [Google Scholar] [CrossRef]




















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedorowicz, J.; Sączewski, J. Advances in the Synthesis of Biologically Active Quaternary Ammonium Compounds. Int. J. Mol. Sci. 2024, 25, 4649. https://doi.org/10.3390/ijms25094649
Fedorowicz J, Sączewski J. Advances in the Synthesis of Biologically Active Quaternary Ammonium Compounds. International Journal of Molecular Sciences. 2024; 25(9):4649. https://doi.org/10.3390/ijms25094649
Chicago/Turabian StyleFedorowicz, Joanna, and Jarosław Sączewski. 2024. "Advances in the Synthesis of Biologically Active Quaternary Ammonium Compounds" International Journal of Molecular Sciences 25, no. 9: 4649. https://doi.org/10.3390/ijms25094649
APA StyleFedorowicz, J., & Sączewski, J. (2024). Advances in the Synthesis of Biologically Active Quaternary Ammonium Compounds. International Journal of Molecular Sciences, 25(9), 4649. https://doi.org/10.3390/ijms25094649