Chemical Reactivity Dynamics and Quantum Chaos in Highly Excited Hydrogen Atoms in an External Field: A Quantum Potential Approach

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:I. Introduction
II. Theoretical Background
III. Numerical Solution
IV. Results and Discussions
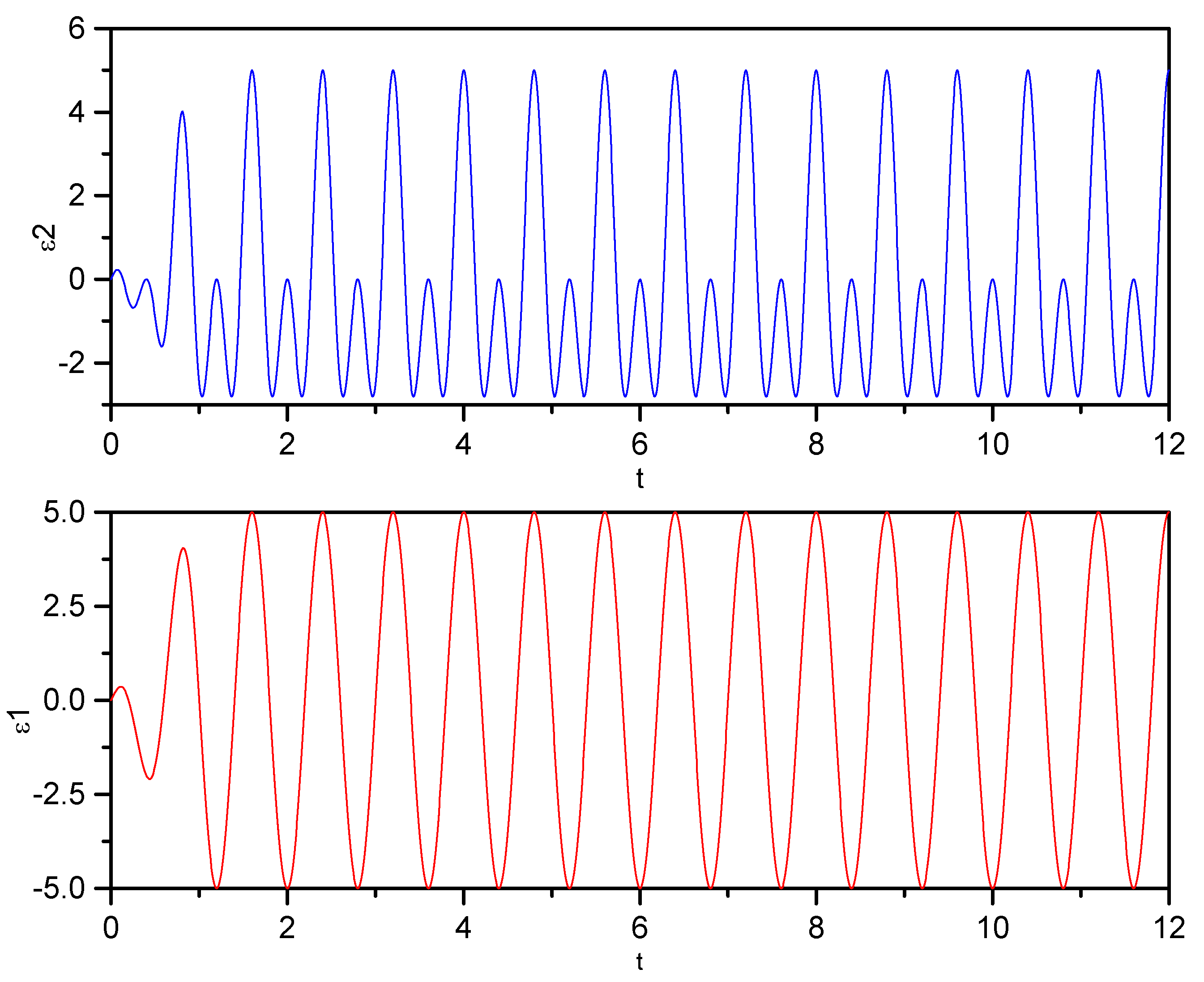
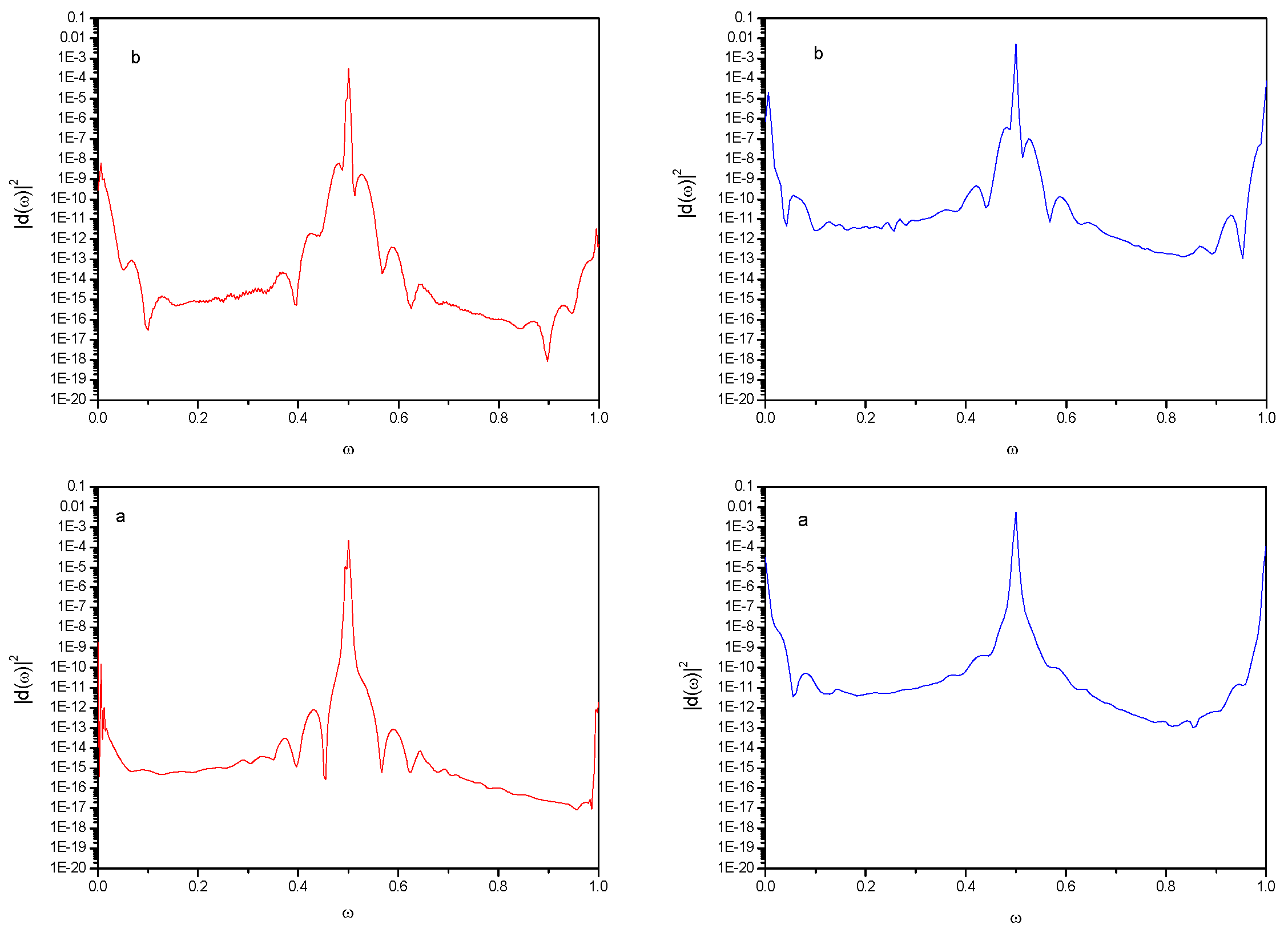
 ) monochromatic pulse, ε2 (
) monochromatic pulse, ε2 (  ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) monochromatic pulse, ε2 ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) monochromatic pulse, ε2 ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0. ) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0. ) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0. ) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0. ) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0. ) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0. ) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0. ) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
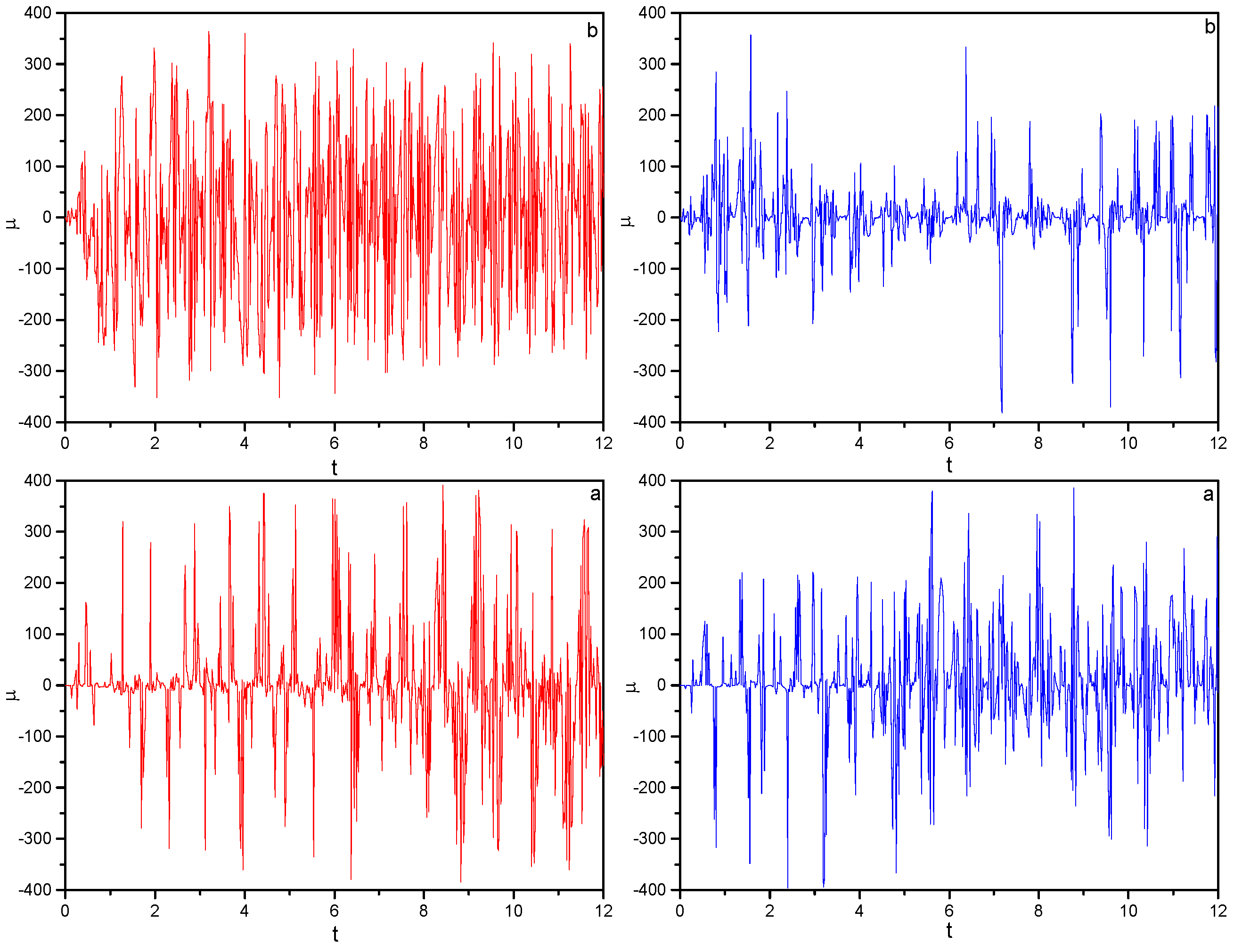
 ) Monochromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
 ) Bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0. ) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
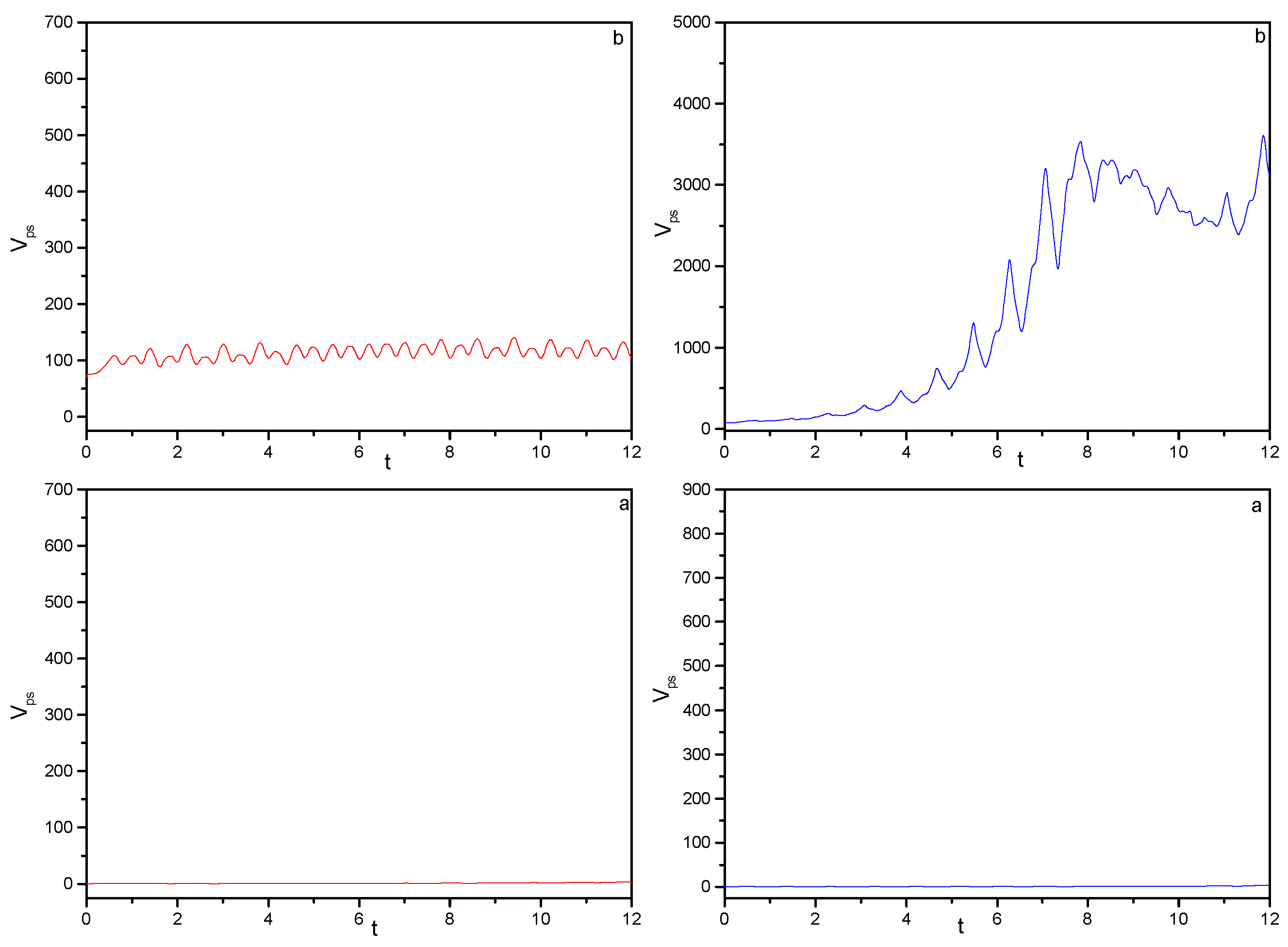
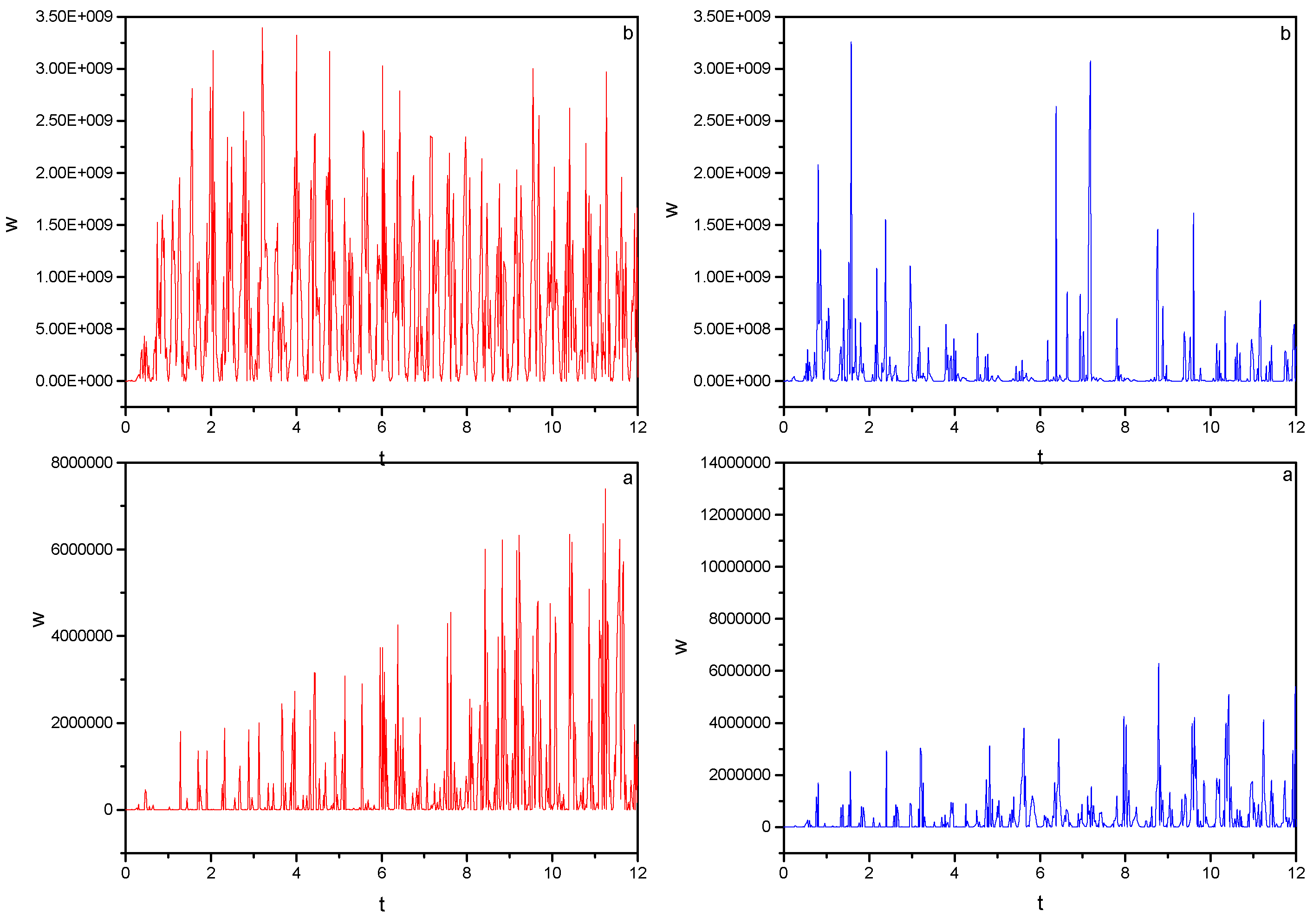
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0. ) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
) Monochromatic pulse, ( ) bichromatic pulse. Field parameters: ε0 = 5.0; ω0 = 2.5π, ω1 = 2ω0.
V. Concluding Remarks
Acknowledgment
References and Notes
- Jensen, R. V.; Susskind, S. M.; Sanders, M. M. Phys. Rep. 1991, 201, 1–56. [CrossRef]
- Koch, P. M.; van Leeuwen, K. A. H. Phys. Rep. 1995, 255, 289–403.
- Atoms in Intense Laser Fields; Gavrila, M. (Ed.) Academic Press: Boston, 1992.
- Sanders, M. M.; Jensen, R. V. Am. J. Phys. 1996, 64, 21–31, 1996, 64, 1013.
- Mariani, D. R. The Ionisation of Hydrogen and Helium Atoms by Static and Microwave ionization of highly excited hydrogen atoms: Experimental and theory, in the physics of phase space; Kim, Y. S., Zachary, W. W., Eds.; Springer: New York, 1987; pp. 106–113. [Google Scholar]
- Mariani, D. R.; van de Water, W.; Koch, P. M.; Bergeman, T. Phys. Rev. Lett. 1983, 50, 1261. [CrossRef]
- van de Water, W.; Yoakum, S.; van Leeuwen, K. A. H.; Sauer, B. E.; Moorman, L.; Galvez, E. J.; Mariani, D. R.; Koch, P. M. Phys. Rev. A. 1990, 42, 573.
- Bayfield, J. E.; Koch, P. M. Phys. Rev. Lett. 1974, 33, 258–261. [CrossRef]
- Koch, P. M.; van Leeuwen, K. A. H.; Rath, O.; Richards, D.; Jensen, R. V. Microwave ionization of highly excited hydrogen atoms: Experiment and theory, in the physics of phase space; Kim, Y. S., Zachary, W. W., Eds.; Springer: New York, 1987; pp. 106–113. [Google Scholar]
- van Leeuwen, K. A. H.; Oppen, G. V.; Renwick, S.; Bowlin, J. B.; Koch, P. M.; Jensen, R. V.; Rath, O.; Richards, D.; Leopold, J. G. Phys. Rev. Lett. 1985, 55, 2231–2234. [CrossRef] [PubMed]
- Galvez, E. J.; Sauer, B. B.; Moorman, L.; Koch, P. M.; Richards, D. Phys. Rev. Lett. 1988, 61, 2011–2014. [CrossRef]
- Leopold, J. G.; Richards, D. J. Phys. B At. Mol. Phys. 1989, 24, 1209–1240. [CrossRef]
- Sanders, M. M. Chaotic Ionisation of One and Two Electron Atom. Ph. D. thesis, Yale University, New Haven, CT, 1991. [Google Scholar]
- Bayfield, J. E. Am. Sci. 1983, 71, 375–383.
- Leopold, J. G.; Percival, I. C. J. Phys. B At. Mol. Phys. 1979, 12, 709–721. [CrossRef]
- Rath, O.; Richards, D. J. Phys. B. (submitted).
- Born, M. The Mechanics of the Atom; Frederick Ungar: New York, 1960. [Google Scholar]
- Leopold, J. G.; Richards, D. J. Phys. B. At. Mol. Phys. 1986, 19, 1125. [CrossRef]
- Jensen, R. V. Phys. Rev. Lett. 1982, 49, 1365–1368. [CrossRef]
- Jensen, R. V. Phys. Rev. A. 1982, 30, 386–397. [CrossRef]
- Schwieters, C. D.; Delos, J. B. Phys. Rev. A 1995, 51, 1030–1041. [CrossRef]
- Shepelyansky, D. L. Chaotic Behavior in Quantum System: Theory and Application. Plenum: New York, 1985; pp. 187–197. [Google Scholar]
- Lichtenberg, A. J.; Liebergman, M. A. Regular and Stochastic Motion; Springer: New York, 1983. [Google Scholar]
- Casati, G.; Chirikov, B. V.; Shepelyansky, D. L. Phys. Rev. Lett. 1984, 53, 2525–2528.
- Casati, G.; Guarneri, L.; Shepelyansky, D. L. IEEE J. Quantum Electron. 1988, QE – 24, 1420–1444.
- Jensen, R. V. Phys. Scr. 1987, 35, 668–673.
- Landau, L. D.; Lifshitz, E. M. Mechanics, Course of Theoretical PhysicsPergamon: New York, 1976; Vol. 1, 3rd ed. [Google Scholar]
- Goldstein, H. Classical MechanicsAddison – Wesley: Reading, MA, 1980, 2nd ed. [Google Scholar]
- Mcphersion, A.; Gibson, G.; Jara, H.; Johann, U.; Luk, T. S.; McIntyre, I. A.; Boyer, K.; Rhodes, C. K. J. Opt. Soc. Am. 1987, B4, 595. [CrossRef]
- Super – Intense Laser – Atom Physics; Piraux, B.; L’Huillier, A.; Rzazewski, K. (Eds.) NATO ASI Series B316; Plenum Press: New York, 1993.
- Electronegativity: Struct. Bonding; Sen, K. D.; Jorgenson, C. K. (Eds.) Springer–Verlag: Berlin, 1987; Vol. 66.
- Chemical Hardness: Struct. Bonding; Sen, K. D; Mingos, D. M. P. (Eds.) Springer–Verlag: Berlin, 1987; Vol. 66.
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: Ithaca, NY, 1960. [Google Scholar]
- Pearson, R. G. Coord. Chem. Rev. 1990, 100, 403.Hard and Soft Acids and Bases; Dowden, Hutchinson and Ross: Stroudsberg, PA, 1973.
- Hohenberg, P.; Kohn, W. Phys. Rev. B 1964, 136, 864.Kohn, W.; Sham, L. J. Phys. Rev. A 1965, 140, 1133.Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, 1989. [Google Scholar]Annu. Rev. Phys. Chem. 1995, 46, 701.Chattaraj, P. K. J. Indian. Chem. Soc. 1992, 69, 173.Kohn, W.; Becke, A. D.; Parr, R. G. J. Phys. Chem. 1996, 100, 12974.
- Pearson, R. G. J. Chem. Educ. 1987, 64, 561. Acc. Chem. Res. 1993, 26, 250. J. Chem. Educ. 1999, 76, 267.
- Parr, R. G.; Chattaraj, P. K. J. Am. Chem. Soc. 1991, 113, 1854.Chattaraj, P. K.; Liu, G. H.; Parr, R. G. Chem. Phys. Lett. 1995, 237, 171.Pearson, R. G. Chemtracts Inorg. Chem. 1991, 3, 317.Liu, S.; Parr, R. G. J. Chem. Phys. 1997, 106, 5578. Chattaraj, P. K. Proc. Indian Natl. Sci. Acad. Part A 1996, 62, 513. Pearson, R. G. Chemical Hardness: Application from Molecules to Solid; Wiley–VCH Verlag GMBH: Weinheim, 1997. [Google Scholar] Ayers, P. W.; Parr, R. G. J. Am. Chem. Soc. 2000, 122, 2010.
- Parr, R. G.; Donnelly, D. A.; Levy, M.; Palke, W. E. J. Chem. Phys. 1978, 68, 3801.
- Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105, 7512. [CrossRef]
- Berkowitz, M.; Ghosh, S. K.; Parr, R. G. J. Am. Chem. Soc. 1985, 107, 6811. Ghosh, S. K.; Berkowitz, M. J. Chem. Phys. 1985, 83, 2976.
- Parr, R. G.; Yang, W. J. Am. Chem. Soc. 1984, 106, 4049. [CrossRef]
- Sanderson, R. T. Science 1951, 114, 670. Science 1955, 121, 207. J. Chem. Educ. 1954, 31, 238. Chattaraj, P. K.; Maiti, B. J. Chem. Educ. 2001, 78, 811–813.
- Politzer, P.; Weinstein, H. J. Chem. Phys. 1979, 70, 3680. Parr, R. G.; Bartolotti, L. J. J. Am. Chem. Soc. 1982, 104, 3081. Nalewajski, R. F. J. Phys. Chem. 1985, 89, 2831. Mortier, W. J.; Ghosh, S. K.; Shankar, S. J. Am. Chem. Soc. 1986, 108, 4315.
- Chattaraj, P. K.; Sengupta, S. J. Phys. Chem. 1996, 100, 16126. [CrossRef]
- Ghanty, T. K.; Ghosh, S. K. J. Phys. Chem. 1996, 100, 12296.
- Chattaraj, P. K.; Sengupta, S. J. Phys. Chem. A 1997, 101, 7893. [CrossRef]
- Chattaraj, P. K.; Poddar, A. J. Phys. Chem. A 1998, 102, 9944, 1999, 103, 1274. Chattaraj, P. K.; Fuentealba, P; Gomez, B.; Contreas, R. J. Am. Chem. Soc. 2000, 122, 348. Fuentealba, P.; Simon – Manso, Y.; Chattaraj, P. K. J. Phys. Chem. A 2000, 104, 3185. Chattaraj, P. K.; Fuentealba, P.; Jaque, P.; Toro – Labbe, A. J. Phys. Chem. A 1999, 103, 9307. Chattaraj, P. K.; Maiti, B. J. Phys. Chem. A 2001, 105, 169–183.
- Jaynes, E. T. Statistical Physics; Ford, K. W., Ed.; Brandeis Lectures, Vol – 3; Benjamin: New York, 1963. [Google Scholar] Levine, R. D.; Bernstein, R. B. Dynamics of Molecular Collisions; Miller, W. H., Ed.; Plenum Press: New York, 1976. [Google Scholar] Gadre, S. R.; Bendale, R. D. Curr. Sci. 1985, 54, 970.
- Parr, R. G.; Szentpaly, L. v.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922. [CrossRef]
- Chattaraj, P. K.; Sengupta, S. J. Phys. Chem. A 1999, 103, 6122. [CrossRef]
- Chattaraj, P. K.; Nath, S. Int. J. Quantum Chem. 1994, 49, 705. [CrossRef]
- Runge, E.; Gross, E. K. U. Phys. Rev. Lett.; 1984; Volume 52, p. 997. [Google Scholar] Dhara, A. K.; Ghosh, S. K. Phys. Rev. A; 1987; Volume 35, p. 442. [Google Scholar] Erhard, S.; Gross, E. K. U. Multiphoton Processes 1996; Lambropoulos, P., Walther, H., Eds.; IOP Publishing: London, 1997; pp. 37–45, and reference therein. [Google Scholar]
- Chattaraj, P. K.; Sengupta, S.; Poddar, A. Int. J. Quantum Chem. 1998, 69, 279. Nonlinear Dynamics and Computational Physics; Sheorey, V. B. (Ed.) Narosa: New Delhi, 1999; pp. 45–53.
- Lakshmanan, M.; Ganeshan, K. Curr. Sci. 1995, 68, 38.
- Casati, G.; Chirikov, B. V.; Guarneri, I; Shepelyansky, D. L. Phys. Rep. 1987, 154, 77. Hasegawa, H.; Robnik, M.; Wunner, G. Prog. Theo. Phys. Suppl. 1989, 98, 198. Ganeshan, K.; Lakshmanan, M. Phys. Rev. A 1990, 42, 3940. Howard, J. E.; Farelly, D. Phys. Lett. A 1993, 178, 62. Delande, D.; Gay, J. C. Phys. Rev. Lett. 1987, 59, 1809. Delande, D. Chaos and Quantum Physics; Elsevier: Amsterdam, 1991. [Google Scholar] Friedrich, H.; Wintgen, D. Phys. Rep. 1989, 183, 37. Holle, A.; Weibusch, G.; Main, J.; Hager, B.; Rottke, H.; Welge, K. H. Phys. Rev. Lett. 1986, 56, 2594. Holle, A; Marini Rottke, H.; Welge, K. H. Phys. Rev. Lett. 1986, 56, 2594. Holle, A.; Marini, J.; Weibusch, G.; Rottke, H.; Welge, K. H. Phys. Rev. Lett. 1988, 61, 161.
- Madelung, E. Z. Phys. 1926, 40, 322. Ghosh, S. K.; Deb, B. M. Phys. Rep. 1982, 92, 1–44.
- Chattaraj, P. K.; Sengupta, S. Phys. Lett. A 1993, 181, 225. Ind. J. Pure Appl. Phys. 1996, 34, 518. Chattaraj, P. K. Ind. J. Pure Appl. Phys. 1994, 32, 101.
- Holland, P. R. The Quantum Theory of Motion; Cambridge University Press: Cambridge, U. K., 1993. [Google Scholar]
- Sengupta, S.; Chattaraj, P. K. Phys. Lett. A 1996, 215, 119. Chattaraj, P. K.; Sengupta, S. Curr. Sci. 1996, 71, 134. Chattaraj, P. K.; Sengupta, S. J. Phys. Chem. A 1999, 103, 6122–6126.
- Belinfante, F. J. A Survey of Hidden Variable Theories; Pergamon Press: New York, 1973. [Google Scholar]
- Deb, B. M.; Chattaraj, P. K. Phys. Rev. A 1989, 39, 1696–1713.
- Deb, B. M.; Chattaraj, P. K. Chem. Phys. Lett. 1988, 148, 550–556.
- Deb, B. M.; Chattaraj, P. K.; Mishra, S. Phys. Rev. A 1991, 43, 1248–1257.
- Chattaraj, P. K. Int. J. Quant. Chem. 1992, 41, 845–859.
- Deb, B. M.; Dey, B. K. Int. J. Quant. Chem. 1995, 56.
- Nath, S.; Chattaraj, P. K. Pramana 1995, 45, 65–73.
- Chattaraj, P. K.; Nath, S. Chem. Phys. Lett. 1994, 217, 342–348.
- Chattaraj, P. K.; Nath, S. Proc. Indian. Acad. Sci. (Ch. Sci.) 1994, 106, 229–249.
- de Broglie, L. Nonlinear Wave Mechanics: A Causal Interpretation; Elsevier: Amsterdam, 1993. [Google Scholar]
- Bohm, D. Phys. Rev. 1952, 85, 166 – 179, 180 – 193.
- Gutzwiller, M. C. Chaos in Classical and Quantum Mechanics. Springer: Berlin, 1990. [Google Scholar]
- Eckhardt, B. Phys. Rep. 1988, 103, 205–297.
- Jensen, R. V. Nature 1992, 355, 311–317, 1995, 373, 16 and references therein.
- Dewdney, C.; Hiley, B. J. Found. Phys. 1982, 12, 27–48.
- Takabayasi, T. Prog. Theor. Phys. 1952, 8, 143–182, 1953, 9, 187 – 222; 1955, 14, 283 – 302. [Google Scholar]
- Kan, K. K.; Griffin, J. Phys. Rev. C 1977, 15, 1126–1157.
- Weiner, J. H.; Partom, Y. Phys. Rev. 1969, 187, 1134–1147. Weiner, J. H.; Askar, A. J. Chem. Phys. 1972, 54, 3534–3541.
- McCullough, E. A.; Wyatt, R. E. J. Chem. Phys. 1971, 54, 3534–3541.
- Hirschfelder, J. O.; Christoph, A. C.; Palke, W. E. J. Chem. Phys. 1974, 61, 5435–5455. Hirschfelder, J. O.; Tang, K. T. J. Chem. Phys. 1976, 64, 760–785, 1976, 65, 470 – 486.
- Skodie, R. T.; Rohrs, H. W.; VanBuskirk, J. Phys. Rev. A 1989, 40, 2894–2916.
- Parmenter, R. H.; Valentine, R. W. Phys. Lett. A 1995, 201, 1–8.
- Schwengelbeck, U.; Faisal, F. H. M. Phys. Lett. A 1995, 199, 281–286.
- Faisal, F. H. M.; Schwengelbeck, U. Phys. Lett. A, 1995; 207, 31–36.
- Misner, C. W.; Thorne, K. S.; Wheeler, J. A. Gravitation; W. H. Freeman and Company: San Francisco, 1973. [Google Scholar]
- McDonald, S. W.; Kaufman, A. N. Phys. Rev. Lett. 1979, 42, 1189–1191.
- Berry, M. V. Proc. R. Soc. A, London 1987, 413, 183–198.
- Chattaraj, P. K. Symmetries and Singularity Structures: Intregability and Chaos in Nonlinear Dynamical Systems; Lakshmanan, M., Daniel, M., Eds.; Springer–Verlag: Berlin, 1990; pp. 172–182. [Google Scholar]
- Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, U. K., 1989. [Google Scholar]
- Fuentealba, P. J. Chem. Phys. 1995, 103, 6571.
- Ghosh, S. K.; Deb, B. M. J. Phys. B 1994, 27, 381. [CrossRef]
- Parr, R. G. J. Phys. Chem. 1988, 92, 3060. [CrossRef]
- Feit, M. D.; Fleck, J. A., Jr. J. Chem. Phys. 1984, 80, 2578. Choudhury, S.; Gangopadhayay, G.; Ray, D. S. Ind. J. Phys. B 1995, 69, 507. Graham, R.; Hohnerbach, M. Phys. Rev. A 1991, 43, 3966. Idem. Phys. Rev. Lett. 1990, 64, 637.
- Pearson, R. G. Chemical Hardness: Applications from Molecules to Solids; Wiley – VCH Verlag GMBH: Weinheim, 1997; pp. 116–119. [Google Scholar]
- L’Huillier, A.; Lompre, L. A.; Mainfray, G.; Manus, C. Atoms in Intense Laser Fields; Gavrila, M., Ed.; Academic Press: Boston, 1992; p. 139. [Google Scholar]
- Ames, W. F. Numerical Methods for Partial Differential Equations. Academic: New York, 1977; p. 252. [Google Scholar]
- Chattaraj, P. K.; Rao, K. S.; Deb, B. M. J. Comput. Phys. 1987, 72, 504. [CrossRef]
© 2002 by MDPI (http://www.mdpi.org), Basel, Switzerland.
Share and Cite
Chattaraj, P.K.; Maiti, B. Chemical Reactivity Dynamics and Quantum Chaos in Highly Excited Hydrogen Atoms in an External Field: A Quantum Potential Approach. Int. J. Mol. Sci. 2002, 3, 338-359. https://doi.org/10.3390/i3040338
Chattaraj PK, Maiti B. Chemical Reactivity Dynamics and Quantum Chaos in Highly Excited Hydrogen Atoms in an External Field: A Quantum Potential Approach. International Journal of Molecular Sciences. 2002; 3(4):338-359. https://doi.org/10.3390/i3040338
Chicago/Turabian StyleChattaraj, P. K., and B. Maiti. 2002. "Chemical Reactivity Dynamics and Quantum Chaos in Highly Excited Hydrogen Atoms in an External Field: A Quantum Potential Approach" International Journal of Molecular Sciences 3, no. 4: 338-359. https://doi.org/10.3390/i3040338
APA StyleChattaraj, P. K., & Maiti, B. (2002). Chemical Reactivity Dynamics and Quantum Chaos in Highly Excited Hydrogen Atoms in an External Field: A Quantum Potential Approach. International Journal of Molecular Sciences, 3(4), 338-359. https://doi.org/10.3390/i3040338