Can Unrestricted Density-Functional Theory Describe Open Shell Singlet Biradicals?

Abstract
:1 Introduction
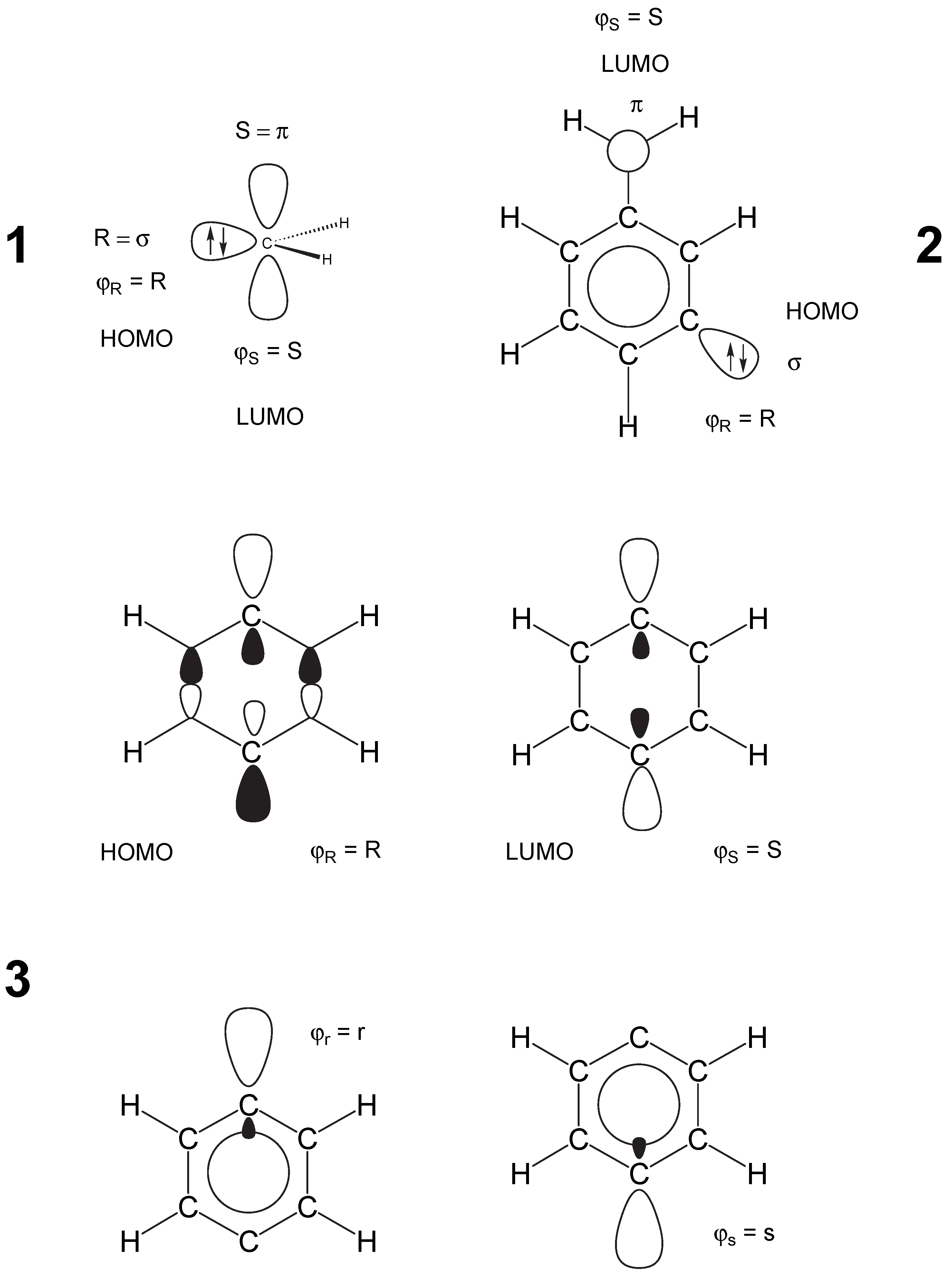
2 Ways of Describing Biradicals by Unrestricted Hartree-Fock Theory










3 Description of Open Shell Singlet Biradicals by DFT: Principal Shortcomings of UDFT


- The reference state is a single-determinant state. This assumption is essential for the calculation of EX.
- The correlation hole in the real system is described reasonably well by the model correlation hole from the homogeneous or weakly inhomogeneous electron gas. This assumption is essential for the calculation of EC.





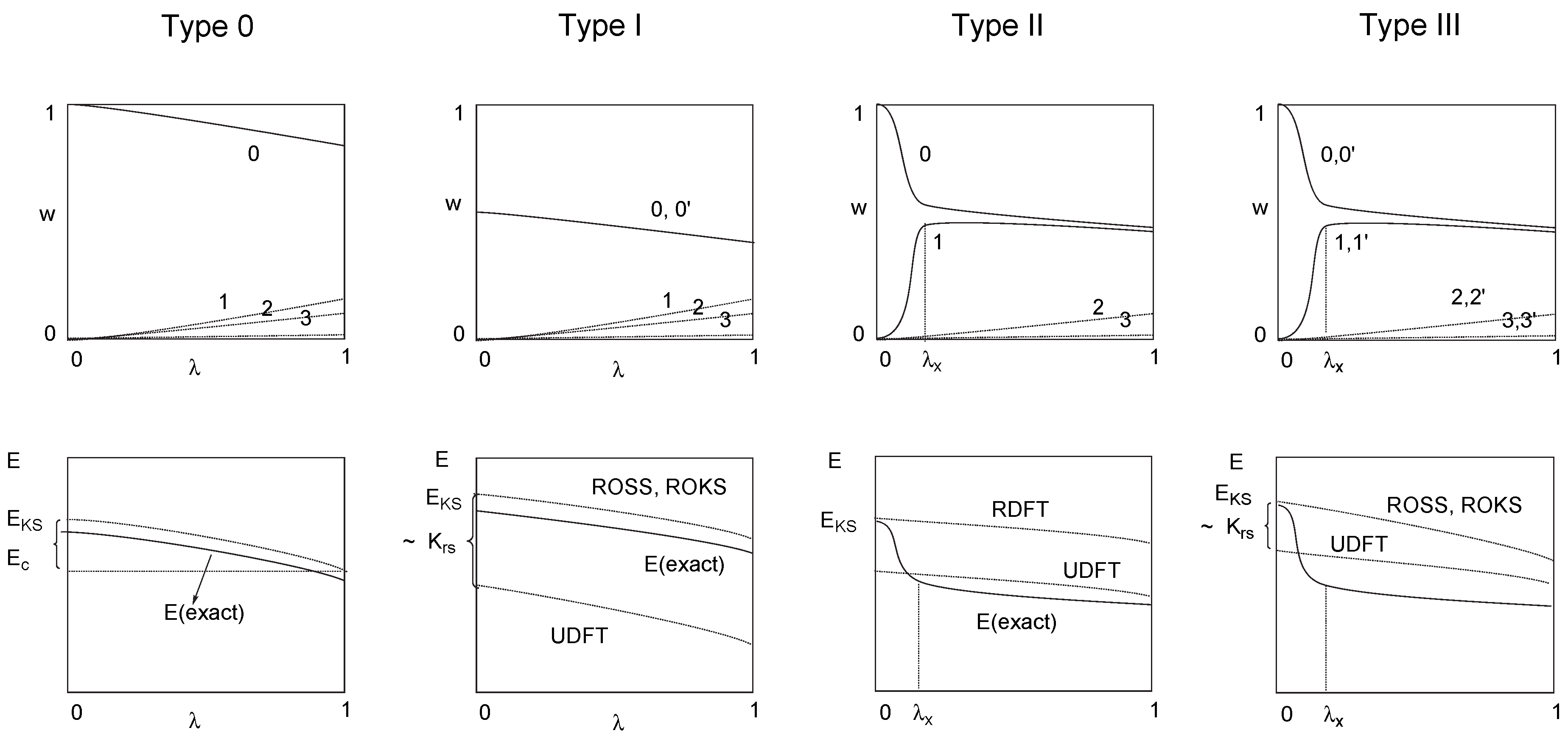
4 Why does UDFT Give Reasonable Results for Certain Open Shell Singlet Biradicals?
5 UDFT Description of Type II Biradicals





6 Use and Misuse of UDFT
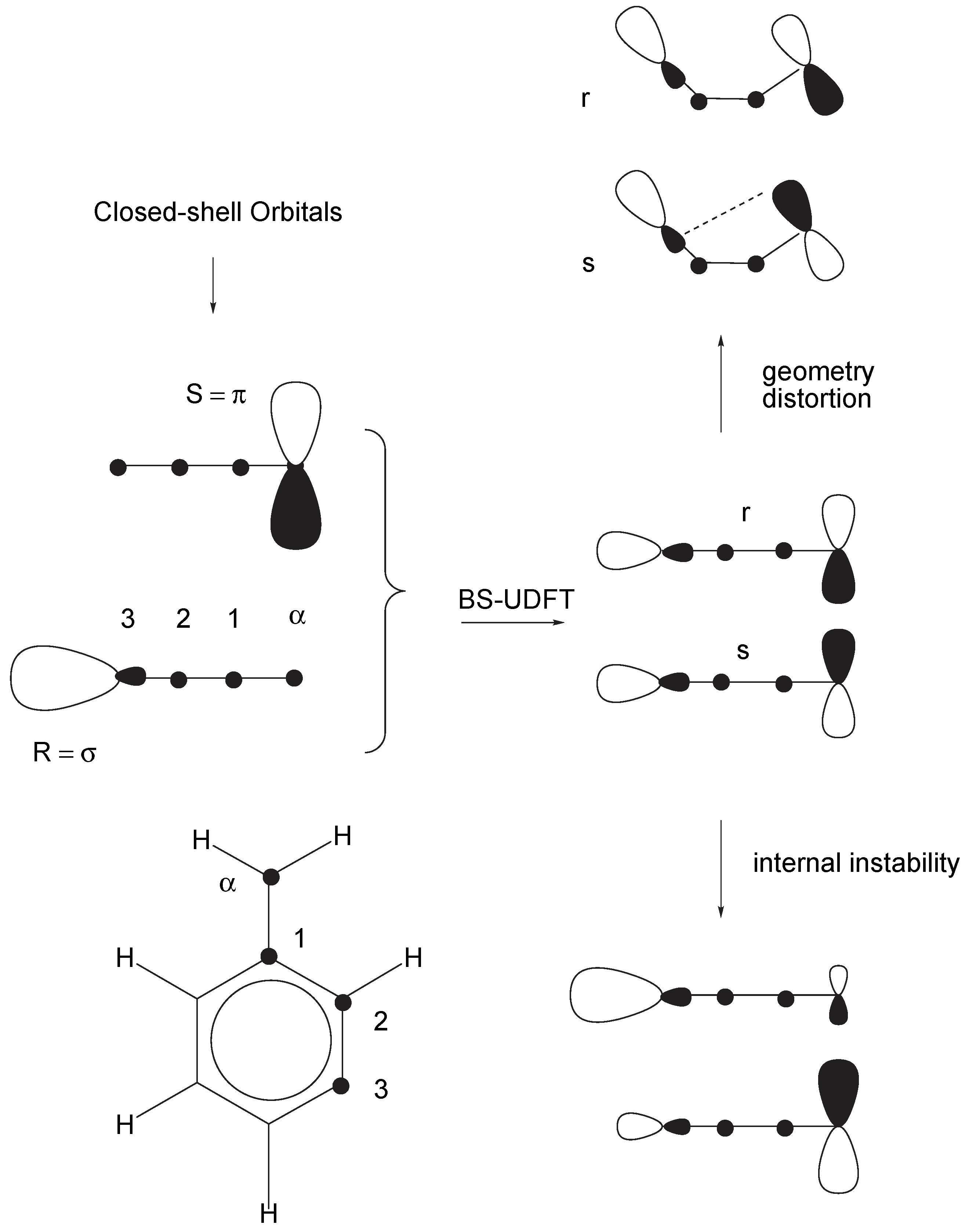
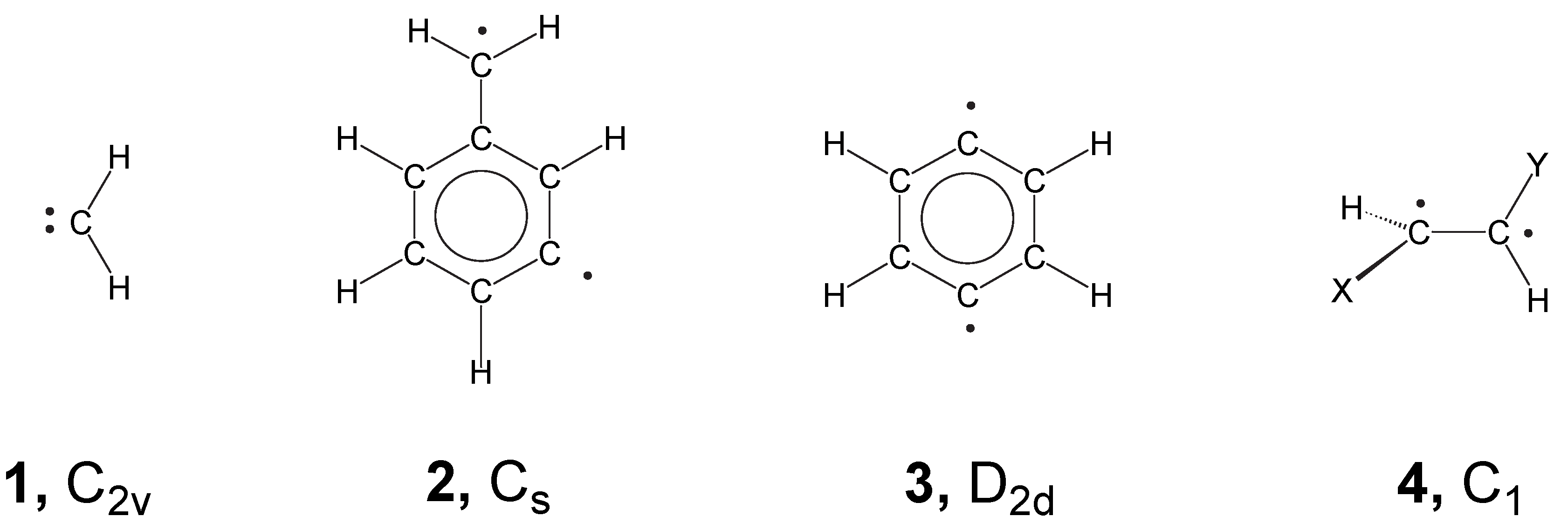
- For a type I system, UDFT fails for principal reasons and should not be used unless special precautions are taken. For example, PO-UDFT can be used in some cases if the overlap between the open-shell orbitals is small, i.e. the two unpaired electrons sit at different, preferably non-neighboring atoms. In this situation the exchange integral KRS will be negligible and the major deficiency of UDFT is disguised so that a reasonable description seems to be obtained. The PO-UDFT description of rotated alkene 4 represents such a case The use of BS-UDFT in these cases is always erroneous.
- For type II systems, BS-UDFT will give a reasonable description provided the corresponding singlet-triplet splitting is small (< 5 kcal/mol) while PO-UDFT is inappropriate because it describes an excited OSS state of the type II system.
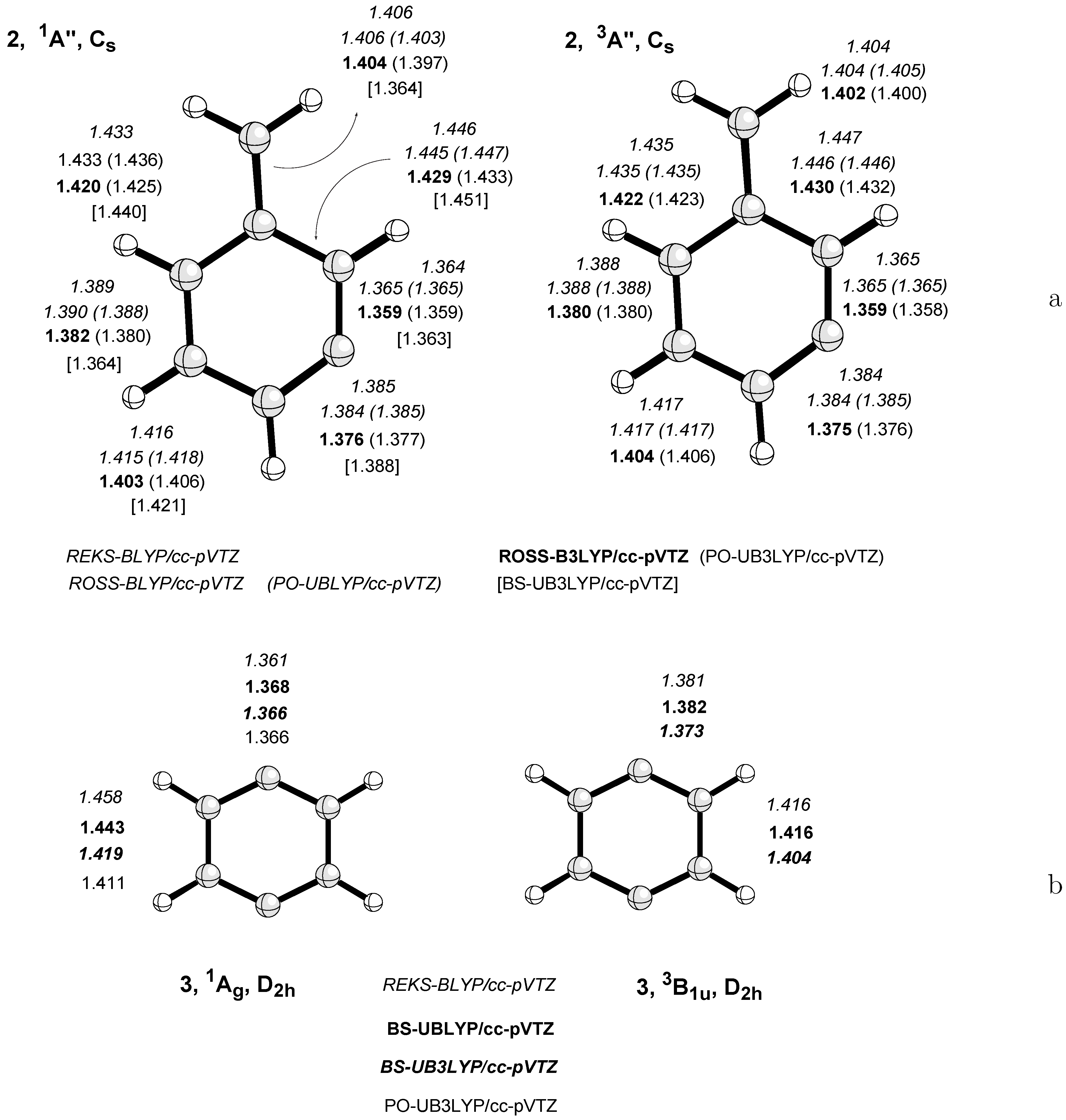
- Most difficult is always the description of type III systems because they possess a multiconfigurational-multideterminantal wave function. For a symmetric biradical such as 2, the PO-UDFT provides qualitatively correct energies and geometries because of a small value of KRS (related to a small singlet-triplet splitting) and the fact that UDFT describes spin polarization better than any restricted open-shell DFT method. BS-UDFT will always be erroneous as reflected by an unreasonably high OSS energy. Also, ROSS and REKS fail in these cases because they cannot describe spin-polarization correctly.
- Neither type I, type II or type III systems can be described by RDFT. This trivial fact was overlooked for some time (see, e.g. Ref. [20]). In the case that the biradical character of a given molecule is not known one can avoid an erroneous use of RDFT by making an appropriate stability test [11, 59] of the RDFT energy.
- The calculation of NOON values helps to determine the biradical character of a molecule and to find the appropriate method for description. This procedure is regularly used when analyzing CASSCF wave functions, however its usefulness in connection with BS-UHF or BS-UDFT descriptions is hardly exploited.
- Hybrid functionals such as B3LYP perform better than pure DFT functionals when describing OSS biradicals simply because they reduce the self-interaction error of commonly used DFT exchange functionals. Hence, the inclusion of static electron correlation via the wave function is more effective than for GGA functionals [59].
- Lowering the molecular symmetry for the initial guess (e.g., breaking the mirror symmetry) may remedy an incorrect choice of the UDFT method (i.e. PO-UDFT or BS-UDFT). However, this is neither necessary nor reasonable. Firstly, both the erroneous breaking of the initial symmetry and the transition from a BS-UDFT to a PO-UDFT wave function (or vice versa) require unnecessary computational expenses. Secondly, the final geometry and electronic structure in such an optimization will still be slightly asymmetric, which may lead to qualitatively incorrect results for symmetry-dependent quantities (e.g., infrared intensities).
- The differences between PO-UDFT and BS-UDFT vanish in the case of C1 symmetry of the biradical to be investigated. The question which of these approaches is more reasonable can only be answered by inspection of the resulting UDFT orbitals. However, it is always dangerous to consider the quality of calculated molecular properties as an appropriate criterion for the reliability of the UDFT approach chosen because this may disguise basic deficiencies of PO- or BS-UDFT.
- In any case it is advisable to use CAS-DFT as a more complete method to check the reliability of a given UDFT description. GVB-DFT is not reliable enough to fulfill this task as was demonstrated for type II and type III systems.
Acknowledgment
References
- Marquardt, R.; Balster, A.; Sander, W.; Kraka, E.; Cremer, D.; Radziszewiski, J. G. p-benzyne. Angew. Chem. Int. Ed. 1998, 37, 955–958. [Google Scholar] [CrossRef]
- (a) Myers, A. G. Proposed structure of the neocarzinostatin chromophore-Methyl thioglycolate adduct - a mechanism for the nucleophilic activation of neocarzinostatin. Tetrahedron Lett. 1987, 28, 4493–4496. [Google Scholar] (b) Myers, A. G.; Proteau, P. J.; Handel, T. M. Stereochemical assignment of neocarzinostatin chromophore - structures of neocarzinostatin chromophore methyl thioglycolate adducts. J. Am. Chem. Soc. 1988, 110, 7212–7214. [Google Scholar] (c) Myers, A. G.; Dragovich, P. S.; Kuo, E. Y. Studies on the thermal generation and reactivity of a class of (σ, π)-1,4-biradicals. J. Am. Chem. Soc. 1992, 114, 9369–9386. [Google Scholar]
- Sander, W.; Wandel, H.; Bucher, G.; Gräfenstein, J.; Kraka, E.; Cremer, D. α, 3-didehydro-5-methyl-6-hydroxytoluene: Matrix isolation of a diradical related to the neocarzinostatin chromophore. J. Am. Chem. Soc. 1998, 120, 8480–8485. [Google Scholar] [CrossRef]
- (a) Jones, R. R.; Bergman, R. G. p-Benzyne. Generation as an intermediate in a thermal isomerization reaction and trapping evidence for the 1,4-benzenediyl structure. J. Am. Chem. Soc. 1972, 94, 660–661. [Google Scholar] (b) Bergman, R. G. Reactive 1,4-dehydroaromatics. Acc. Chem. Res. 1973, 6, 25–31. [Google Scholar] (c) Lockhart, T. P.; Comita, P. B.; Bergman, R. G. Kinetic evidence for the formation of discrete 1,4-dehydrobenzene intermediates. Trapping by inter- and intramolecular hydrogen atom transfer and observation of high-temperature CIDNP. J. Am. Chem. Soc. 1981, 103, 4082–4090. [Google Scholar] (d) Lockhart, T. P.; Bergman, R. G. Evidence for the reactive spin state of 1,4-dehydrobenzenes. J. Am. Chem. Soc. 1981, 103, 4091–4096. [Google Scholar] (e) Gleiter, R.; Kratz, D. Conjugated enediynes - An old topic in a different light. Angew. Chem. Int. Ed. 1993, 32, 842–845. [Google Scholar] (f) Turro, N. J.; Evenzahav, A.; Nicolaou, K. C. Photochemical analog of the bergman cycloaromatization reaction. Tetrahedron Lett. 1994, 35, 8089–8092. [Google Scholar] (g) Kuwatani, Y.; Ueda, I. Synthesis of new Sworski-type dehydroannulenes - 3,4-benz-1,2,5,6,8,9,12,13-octadehydro[14]annulenes. Angew. Chem. Int. Ed. 1995, 34, 1892–1894. [Google Scholar]
- For reviews see: (a) Enediyne Antibiotics as Antitumor Agents; Borders, D. B.; Doyle, T. W. (Eds.) Marcel Dekker: New York, 1995. (b) Neocarzinostatin: The Past, present, and Future of an Anticancer Drug; Maeda, H.; Edo, K.; Ishida, N. (Eds.) Springer: New York, 1997. (c) Nicolaou, K. C.; Smith, A. L. Molecular design, chemical synthesis, and biological action of enediynes. Acc. Chem. Res. 1992, 25, 497–503. [Google Scholar] (d) Nicolaou, K. C.; Dai, W.-M. Chemistry and biology of the enediyne anticancer antibiotics. Angew. Chem. Int. Ed. 1991, 30, 1387–1416. [Google Scholar] (e) Pogozelski, W. K.; Tullius, T. D. Oxidative strand scission of nucleic acids: Routes initiated by hydrogen abstraction from the sugar moiety. Chem. Rev. 1998, 98, 1089–1107. [Google Scholar] (f) Maier, M. E.; Boße, F.; Niestroj, A. J. Design and synthesis of dynemicin analogs. Eur. J. Org. Chem. 1999, 1, 1–13. [Google Scholar] (g) Thorson, J. S.; Shen, B.; Whitwam, R. E.; Liu, W.; Li, Y.; Ahlert, J. Enediyne biosynthesis and self-resistance: A progress report. Bioorganic Chemistry 1999, 27, 172–188. [Google Scholar] (h) Wisniewski Grimssom, J.; Gunawardena, G. U.; Klingberg, D.; Huang, D. The chemistry of enediynes, enyne allenes and related compounds. Tetrahedron 1996, 19, 6453–6518. [Google Scholar] (i) Fallis, A. G. 1998 Alfred Bader Award Lecture - Tangents and targets: The synthetic highway from natural products to medicine. Can. J. Chem. 1999, 77, 159–177. [Google Scholar] (j) Caddick, S.; Delisser, V. M.; Doyle, V. E.; Khan, S.; Avent, A. G.; Vile, S. Studies toward the synthesis of natural and unnatural dienediynes 1. Approaches to a functionalised bicyclic ring system. Tetrahedron 1999, 55, 2737–2754. [Google Scholar]
- Wenthold, P. G.; Squires, R. R.; Lineberger, W. C. Ultraviolet photoelectron spectroscopy of the o-, p-, and p-benzyne negative ions. Electron affinities and singlet-triplet splittings for o-, m-, and p-benzyne. J. Am. Chem. Soc. 1998, 120, 5279–5290. [Google Scholar] [CrossRef]
- (a) Wenthold, P. G.; Wierschke, S. G.; Nash, J. J.; Squires, R. R. α, 3-dehydrotoluene - Experimental and theoretical evidence for a singlet ground-state. J. Am. Chem. Soc. 1993, 115, 12611–12612. [Google Scholar] (b) Logan, C. F.; Ma, J. C.; Chen, P. The photoelectron-spectrum of the α, 3-dehydrotoluene biradical. J. Am. Chem. Soc. 1994, 116, 2137–2138. [Google Scholar]
- (a) Hoffner, J. H.; Schottelius, M. J.; Feichtinger, D.; Chen, P. Chemistry of the 2,5-didehydropyridine biradical: Computational, kinetic, and trapping studies toward drug design. J. Am. Chem. Soc. 1998, 120, 376–385. [Google Scholar] (b) Logan, C. F.; Chen, P. Ab initio calculation of hydrogen abstraction reactions of phenyl radical and p-benzyne. J. Am. Chem. Soc. 1996, 118, 2113–2114. [Google Scholar] (c) Schottelius, M. J.; Chen, P. 9,10-dehydroanthracene: p-benzyne-type biradicals abstract hydrogen unusually slowly. J. Am. Chem. Soc. 1996, 118, 4896–4903. [Google Scholar] (d) Chen, P. Design of diradical-based hydrogen abstraction agents. Angew. Chem. Int. Ed. 1996, 35, 1478–1480. [Google Scholar]
- Roth, W. R.; Hopf, H.; Horn, C. The energy well of diradicals. 5. 1,3,5-Cyclohexatriene-1,4-diyl and 2,4-cyclohexadiene-1,4-diyl. Chem. Ber. 1994, 127, 1765–1779. [Google Scholar] [CrossRef]
- (a) Kraka, E.; Cremer, D. CCSD(T) investigation of the bergman cyclization of enediyne - Relative stability of o-didehydrobenzene, m-didehydrobenzene, and p-didehydrobenzene. J. Am. Chem. Soc. 1994, 116, 4929–4936. [Google Scholar] (b) Kraka, E.; Cremer, D. Ortho-benzyne, meta-benzyne, and para-benzyne - A comparative CCSD(T) investigation. Chem. Phys. Lett. 1993, 216, 333–340. [Google Scholar]
- Gräfenstein, J.; Hjerpe, A. M.; Kraka, E.; Cremer, D. An accurate description of the Bergman reaction using restricted and unrestricted DFT: Stability test, spin density, and on-top pair density. J. Phys. Chem. 2000, 104, 1748–1761. [Google Scholar] [CrossRef]
- Kraka, E.; Cremer, D. Computer design of anticancer drugs. A new enediyne warhead. J. Am. Chem. Soc. 2000, 122, 8245–8264. [Google Scholar] [CrossRef]
- (a) Kraka, E.; Cremer, D. Structure and stability of enediynes containing heteroatoms - A quantum chemical investigation. Theochem 2000, 506, 191–211. [Google Scholar] (b) Kraka, E.; Cremer, D. The para-didehydropyridine, para-didehydropyridinium, and related biradicals - A contribution to the chemistry of enediyne antitumor drugs. J. Comp. Chem. 2001, 22, 216–229. [Google Scholar]
- Koga, N.; Morokuma, K. Comparison of biradical formation between enediyne and enyne allene - abinitio CASSCF and MRSDCI study. J. Am. Chem. Soc. 1991, 113, 1907–1911. [Google Scholar] [CrossRef]
- (a) Wenthold, P. G.; Paulino, J. A.; Squires, R. R. The absolute heats of formation of ortho-benzyne, meta-benzyne, and para-benzyne. J. Am. Chem. Soc. 1991, 113, 7414–7415. [Google Scholar] (b) Wenthold, P. G.; Squires, R. R. Biradical thermochemistry from collision-induced dissociation threshold energy measurements - Absolute heats of formation of ortho-benzyne, meta-benzyne, and para-benzyne. J. Am. Chem. Soc. 1994, 116, 6401–6412. [Google Scholar] (c) Wierschke, S. G.; Nash, J. J; Squires, R. R. A multiconfigurational SCF and correlation-consistent CI study of the structures, stabilities, and singlet-triplet splittings of o-benzyne, m-benzyne, and p-benzyne. J. Am. Chem. Soc. 1993, 115, 11958–11967. [Google Scholar]
- (a) Lindh, R.; Persson, B. J. Ab-initio study of the Bergman reaction - the autoaromatization of hex-3-ene-1,5-diyne. J. Am. Chem. Soc. 1994, 116, 4963–4969. [Google Scholar] (b) Lindh, R.; Lee, T. J.; Bernhardsson, A.; Persson, B. J.; Karlström, G. Extended ab-initio and theoretical thermodynamics studies of the Bergman reaction and the energy splitting of the singlet o-benzynes, m-benzynes, and p-benzynes. J. Am. Chem. Soc. 1995, 117, 7186–7194. [Google Scholar] (c) Lindh, R.; Ryde, U.; Schütz, M. On the significance of the trigger reaction in the action of the calicheamicin anti-cancer drug. Theor. Chem. Acta 1997, 97, 203–210. [Google Scholar]
- (a) Cramer, C. J.; Nash, J. J.; Squires, R. R. A reinvestigation of singlet benzyne thermochemistry predicted by CASPT2, coupled-cluster and density functional calculations. Chem. Phys. Lett. 1997, 277, 311–320. [Google Scholar] (b) Cramer, C. J.; Debbert, S. Heteroatomic substitution in aromatic sigma biradicals: the six pyridynes. Chem. Phys. Lett. 1998, 287, 320–326. [Google Scholar]
- Cramer, C. J. Bergman, aza-Bergman, and protonated aza-Bergman cyclizations and intermediate 2,5-arynes: Chemistry and challenge to computation. J. Am. Chem. Soc. 1998, 120, 6261–6269. [Google Scholar] [CrossRef]
- (a) Schreiner, P. R. Monocyclic enediynes: Relationships between ring sizes, alkyne carbon distances, cyclization barriers, and hydrogen abstraction reactions. Singlet-triplet separations of methyl-substituted p-benzynes. J. Am. Chem. Soc. 1998, 120, 4184–4190. [Google Scholar] (b) Schreiner, P. R. Cyclic enediynes: relationship between ring size, alkyne carbon distance, and cyclization barrier. Chem. Commun. 1998, 4, 483–484. [Google Scholar]
- Chen, W.-C.; Chang, N.-Y.; Yu, C.-H. Density functional study of Bergman cyclization of enediynes. J. Phys. Chem. A 1998, 102, 2584–2593. [Google Scholar] [CrossRef]
- (a) Engels, B.; Lennartz, C.; Hanrath, M.; Schmittel, M.; Strittmatter, M. Regioselectivity of biradical cyclizations of enyne-allenes: Influence of substituents on the switch from the Myers-Saito to the novel C2–C6 cyclization. Angew. Chem. Int. Ed. 1998, 37, 1960–1963. [Google Scholar] (b) Engels, B.; Hanrath, M. A theoretical comparison of two competing diradical cyclizations in enyne-allenes: The Myers-Saito and the novel C2–C6 cyclization. J. Am. Chem. Soc. 1998, 120, 6356–6361. [Google Scholar]
- Cramer, C. J.; Squires, R. R. Quantum chemical characterization of the cyclization of the neocarzinostatin chromophore to the 1,5-didehydroindene biradical. Organic Lett. 1999, 1, 215–218. [Google Scholar] [CrossRef]
- Schreiner, P. R.; Prall, M. Myers-Saito versus C2–C6 (“Schmittel”) cyclizations of parent and monocyclic enyne-allenes: Challenges to chemistry and computation. J. Am. Chem. Soc. 1999, 121, 8615–8627. [Google Scholar] See also (b) Prall, M.; Wittkopp, A.; Schreiner, P. R. Can fulvenes form from enediynes? A systematic high-level computational study on parent and benzannelated enediyne and enyne-allene cyclizations. J. Phys. Chem. A 2001, 105, 9265–9274. [Google Scholar] (c) Prall, M.; Wittkopp, A.; Fokin, A. A.; Schreiner, P. R. Substituent effects on the Bergman cyclization of (Z)-1,5-hexadiyne-3-enes: A systematic computational study. J. Comp. Chem. 2001, 22, 1605–1614. [Google Scholar]
- Raghavachari, K.; Trucks, G. W.; Pople, J. A.; Head-Gordon, M. A 5th-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Handy, N. C.; Pople, J. A.; Head-Gordon, M.; Raghavachari, K.; Trucks, G. W. Sizeconsistent Brueckner theory limited to double substitutions. Chem. Phys. Lett. 1989, 164, 185–192. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A 1133–1138. [Google Scholar] [CrossRef]
- For reviews on DFT methods, see for example (a) Parr, R. G.; Yang, W. International Series of Monographs on Chemistry 16: Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [Google Scholar] (b) Density Functional Methods in Chemistry; Labanowski, J. K.; Andzelm, J. W. (Eds.) Springer: Heidelberg, 1990. (c) Theoretical and Computational Chemistry, Vol. 2, Modern Density Functional Theory - A Tool For Chemistry; Seminario, J. M.; Politzer, P. (Eds.) Elsevier: Amsterdam, 1995. (d) Chemical Applications of Density Functional Theory; ACS Symposium Series 629; Laird, B. B.; Ross, R. B.; Ziegler, T. (Eds.) American Chemical Society: Washington, DC, 1996. (e) Lecture Notes in Physics, Density Functionals: Theory and Applications; Joubert, D. (Ed.) Springer: Hei-delberg, 1997. (f) Recent Advances in Computational Chemistry, Vol. 1, Recent Advances in Density Functional Methods, Part II; Chong, D. P. (Ed.) World Scientific: Singapore, 1997. (g) Electronic Density Functional Theory, Recent Progress and New Directions; Dobson, J. F.; Vignale, G.; Das, M. P. (Eds.) Plenum Press: New York, 1998. (h) Gill, P. Encyclopedia of Computational Chemistry; Schleyer, P. v. R., Allinger, N. L., Clark, T., Gasteiger, J., Kollman, P. A., Schaefer, H. F., III, Schreiner, P. R., Eds.; Wiley: Chichester, UK, 1998; Vol. 1, p. 678. [Google Scholar]
- Szalay, P. G. Modern Ideas in Coupled-Cluster Methods; Bartlett, R. J., Ed.; World Scientific: Singapore, 1997; p. 81. [Google Scholar]
- Kraka, E.; Cremer, D.; Bucher, G.; Wandel, H.; Sander, W. A CCSD(T) and DFT investigation of m-benzyne and 4-hydroxy-m-benzyne. Chem. Phys. Lett. 1997, 268, 313–320. [Google Scholar] [CrossRef]
- For a recent review, see Bally, T.; Borden, W. T. Reviews Comp. Chem.; Lipkowitz, K. B., Boyd, D. B., Eds.; Wiley: New York, 1999; Vol. 13, p. 1. [Google Scholar]
- For related work, see (a) Ziegler, T.; Rauk, A.; Baerends, E. J. On the calculation of multiplet energies by the Hartree-Fock-Slater method. Theor. Chim. Acta 1977, 43, 261–271. [Google Scholar] (b) Noodleman, L.; Case, D. A. Density-functional theory of spin polarization and spin coupling in iron-sulfur clusters. Adv. Inorg. Chem. 1992, 38, 423–470. [Google Scholar] (c) Lovell, T.; McGrady, J. E.; Stranger, R.; MacGregor, S. Optimized structures of bimetallic systems: A comparison of full- and broken-symmetry density functional calculations. Inorg. Chem. 1996, 35, 3079–3080. [Google Scholar] (d) Noodleman, L.; Post, D.; Baerends, E. J. Symmetry breaking and ionization from symmetry equivalent inner shells and lone pairs in Xα theory. Chem. Phys. 1982, 64, 159–166. [Google Scholar] (e) Cramer, C. J.; Dulles, F. J.; Giesen, D. J.; Almlöf, J. Density-functional theory - Excited-states and spin annihilation. Chem. Phys. Lett. 1995, 245, 165–170. [Google Scholar]
- Gräfenstein, J.; Cremer, D. Can density functional theory describe multi-reference systems? Investigation of carbenes and organic biradicals. Phys. Chem. Chem. Phys. 2000, 2, 2091–2103. [Google Scholar] [CrossRef]
- (a) Langreth, D. C.; Perdew, J. P. The exchange correlation energy of a metallic surface. Solid State Commun. 1975, 17, 1425–1429. [Google Scholar] (b) Gunnarsson, O.; Lundqvist, B. I. Exchange and correlation in atoms, molecules, and solids by the spin-density-functional formalism. Phys. Rev. B 1976, 13, 4274–98. [Google Scholar]
- (a) Fukutome, H. Unrestricted Hartree-Fock theory and its applications to molecules and chemical reactions. Int. J. Quantum Chem. 1981, 20, 955–1065. [Google Scholar] (b) Yamaguchi, K. Symmetry and broken symmetry in molecular orbital (MO) descriptions of unstable molecules. Generalized MO theoretical studies on 1,3-dipolar species. J. Mol. Struct. (THEOCHEM) 1983, 12, 101–120. [Google Scholar]
- Gräfenstein, J.; Cremer, D.; Kraka, E. Density functional theory for open-shell singlet biradicals. Chem. Phys. Lett. 1998, 288, 593–602. [Google Scholar] [CrossRef]
- Frank, I.; Hutter, J.; Marx, D.; Parrinello, M. Molecular dynamics in low-spin excited states. J. Chem. Phys. 1998, 108, 4060–4069. [Google Scholar] [CrossRef]
- Filatov, M.; Shaik, S. Spin-restricted density functional approach to the open-shell problem. Chem. Phys. Lett. 1998, 288, 689–697. [Google Scholar] Application of spin-restricted open-shell Kohn-Sham method to atomic and molecular multiplet states. J. Chem. Phys. 1999, 110, 116–125.
- Lim, M. H.; Worthington, S. E.; Dulles, F. J.; Cramer, C .J. Chemical Applications of Density Functional Theory; ACS Symposium Series 629; Laird, B. B., Ross, R. B., Ziegler, T., Eds.; American Chemical Society: Washington, DC, 1996; p. 402. [Google Scholar]
- Dunning, T. H., Jr. Gaussian-basis sets for use in correlated molecular calculations. 1. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- (a) Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] (b) Lee, C.; Yang, W.; Parr, R. P. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- (a) Becke, A. D. Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] See also (b) Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. Ab-initio calculation of vibrational absorption and circular-dichroism spectra using density-functional force-fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- Kraka, E.; Gräfenstein, J.; Gauss, J.; Reichel, F.; Olsson, L.; Konkoli, Z; He, Z; Cremer, D. COLOGNE 99; Göteborg University: Göteborg, 1999. [Google Scholar]
- Amos, R. D. CADPAC 5: The Cambridge Analytic Derivatives Package, issue 5, University of Cambridge, 1992. with contributions from Alberts, I. L.; Andrews, J. S.; Colwell, S. M.; Handy, N. C.; Jayatilaka, D.; Knowles, P. J.; Kobayashi, R.; Koga, N.; Laidig, K. E.; Maslen, P. E.; Murray, C. W.; Rice, J. E.; Sanz, J.; Simandiras, E. D.; Stone, A. J.; Su, M. D.
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A., Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98, Revision A.5, Gaussian, Inc.: Pittsburgh PA, 1998.
- Gräfenstein, J; Cremer, D. The combination of density functional theory with multiconfiguration methods - CAS-DFT. Chem. Phys. Lett. 2000, 316, 569–577. [Google Scholar]
- (a) Bauschlicher, C. W. Accurate ab initio calculations for the 1B1–1A1 separation in methylene. Chem. Phys. Lett. 1980, 74, 273–276. [Google Scholar] (b) Bauschlicher, C. W.; Langhoff, S. R. On the 1A1–3B1 separation in CH2 and SiH2. J. Chem. Phys. 1987, 87, 387–391. [Google Scholar]
- McKellar, A. R. W.; Bunker, P. R.; Sears, T. J.; Evenson, K. M.; Saykally, R. J.; Langhoff, S. R. Far infrared laser magnetic resonance of singlet methylene: singlet-triplet perturbations, singlet-triplet transitions, and the singlet-triplet splitting. J. Chem. Phys. 1983, 79, 5251–64. [Google Scholar] [CrossRef]
- Bettinger, H. F.; Schleyer, P. v. R.; Schreiner, P. R.; Schaefer, H. F., III. The Encyclopedia of Computational Chemistry; Schleyer, P. v. R., Allinger, N., Clark, T., Gasteiger, J., Kollman, P. A., Schaefer, H. F., III, Schreiner, P. R., Eds.; Wiley-Interscience: Chichester, 1998; p. 183. [Google Scholar]
- Petek, H.; Nesbitt, D. J.; Darwin, D. C.; Ogilby, P. R.; Moore, C. B.; Ramsay, D. A. Analysis of CH2 A1A1 (1,0,0) and (0,0,1) Coriolis-coupled states, A1A1–X 3B1 spin orbit coupling, and the equilibrium structure of CH2 X 1A1 state. J. Chem. Phys. 1989, 91, 6566–6578. [Google Scholar] [CrossRef]
- Jensen, P.; Bunker, P. R. The potential surface and stretching frequencies of X3B1 methylene (CH2) determined from experiment using the Morse oscillator-rigid bender internal dynamics Hamiltonian. J. Chem. Phys. 1988, 89, 1327–1332. [Google Scholar] [CrossRef]
- Andersson, K.; Roos, B. Multiconfigurational 2nd-order perturbation-theory - A test of geometries and binding-energies. Int. J. Quant. Chem. 1993, 45, 591–607. [Google Scholar] [CrossRef]
- Bobrowicz, F. W.; Goddard, W. A., III. Methods of Electronic Structure Theory, Modern Theoretical Chemistry; Vol. 3, Schaefer, III, Ed.; Plenum Press: New York, 1977; p. 79. [Google Scholar]
- (a) Kraka, E. Homolytic dissociation-energies from GVB-LSDC calculations. Chem. Phys. 1992, 161, 149–153. [Google Scholar] (b) Kraka, E.; Cremer, D.; Nordholm, S. Molecules in Natural Science and Biomedicine; Maksic, Z., Eckert-Maksic, M., Eds.; Ellis Horwood: Chichester, 1971; p. 351. [Google Scholar] (c) In the case of the OSS state of 1 or 2, the GVB-DFT description simplifies to a ROHF-DFT description. See, Davidson, E. R. Spin-restricted open-shell self-consistent-field theory. Chem. Phys. Lett. 1973, 21, 565–567. [Google Scholar]
- Cabrero, J.; Ben-Amor, N.; Caballol, R. Singlet-triplet gap in α-n-dehydrotoluene and related biradicals: An ab initio configuration interaction study. J. Phys. Chem. A 1999, 103, 6220–6224. [Google Scholar] [CrossRef]
- Wenthold, P. G.; Wierschke, S. G.; Nash, J. J.; Squires, R. R. Biradical thermochemistry from collision-induced dissociation threshold energy measurements 2. Experimental and theoretical studies of the mechanism and thermochemistry of formation of α, n-dehydrotoluene biradicals from gas-phase halide elimination reactions. J. Am. Chem. Soc. 1994, 116, 7378–7392. [Google Scholar]
- Wu, W.; Shaik, S. VB-DFT: A nonempirical hybrid method combining valence bond theory and density functional energies. Chem. Phys. Lett. 1999, 301, 37–42. [Google Scholar] [CrossRef]
- Borowski, B.; Jordan, K. D.; Nichols, J.; Nachtigall, P. Investigation of a hybrid TCSCF-DFT procedure. Theor. Chem. Acc. 1998, 99, 135–140. [Google Scholar] [CrossRef]
- Filatov, M.; Shaik, S. A spin-restricted ensemble-referenced Kohn-Sham method and its application to diradicaloid situations. Chem. Phys. Lett. 1999, 304, 429–437. [Google Scholar] [CrossRef]
- Cremer, D. Mol. Phys. in press.
- (a) Bauernschmitt, R.; Ahlrichs, R. Stability analysis for solutions of the closed shell Kohn-Sham equation. J. Chem. Phys. 1996, 104, 9047–9052. [Google Scholar] (b) Seeger, R.; Pople, J. A. Self-consistent molecular orbital methods. XVII. Constraints and stability in Hartree-Fock theory. J. Chem. Phys. 1977, 66, 3045–3050. [Google Scholar]
- Wenthold, P. G.; Lipton, M. A. A density functional molecular orbital study of the C2–C7 and C2–C6 cyclization pathways of 1,2,4-heptatrien-6-ynes. The role of benzannulation. J. Am. Chem. Soc. 2000, 122, 9265–9270. [Google Scholar] [CrossRef]
- Pople, J.; Gill, P.M.W.; Handy, N.C. Spin-unrestricted character of Kohn-Sham orbitals for open-shell systems. Int. J. Quant. Chem. 1995, 56, 303–305. [Google Scholar] [CrossRef]
- Gräfenstein, J.; Cremer, D. On the diagnostic value of in Kohn-Sham density functional theory. Mol. Phys. 2001, 99, 981–989. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
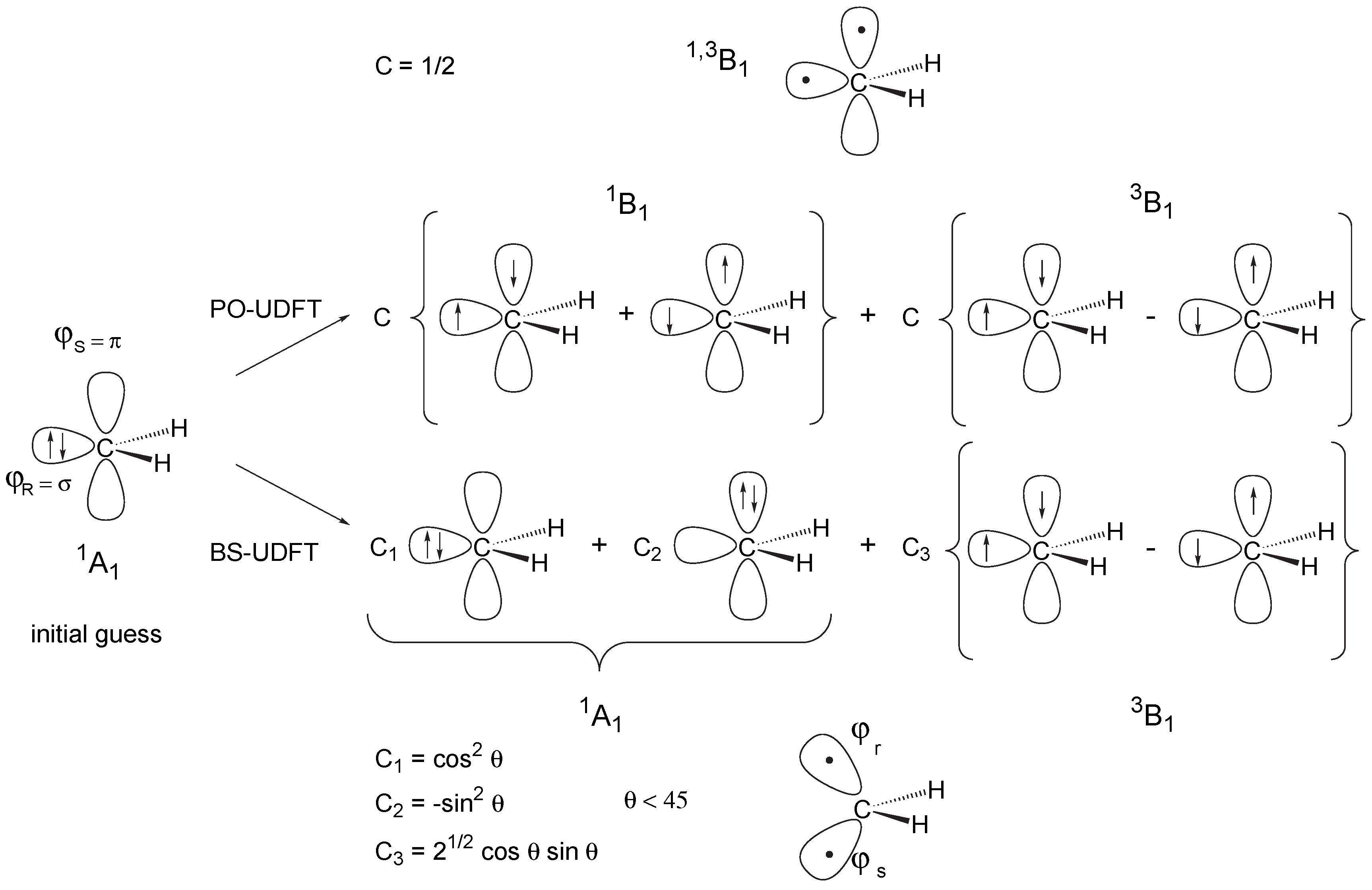{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Functional | E or ∆ E | rCH | ∠HCH | NOON(a1) | NOON(b1) |
| 3B1 State | ||||||
| RODFT | B3LYP | −39.16668 | 1.077 | 133.9 | 1 | 1 |
| BLYP | −39.14139 | 1.084 | 134.4 | 1 | 1 | |
| ROKS | BLYP | −39.14214 | 1.083 | 134.6 | 1 | 1 |
| PO-UDFT | B3LYP | −39.16874 | 1.077 | 135.0 | 1.000 | 1.000 |
| BLYP | −39.14396 | 1.083 | 135.5 | 1.000 | 1.000 | |
| ROHF-DFT b | LYP | −39.14608 | 1.07 | 133.9 | 1 | 1 |
| CAS(6,6)-DFT c | CS | −39.12067 | 1.102 | 131.0 | 1.000 | 1.000 |
| Expt. d | 1.070 | 134.0 | 1 | 1 | ||
| 1A1 State | ||||||
| RDFT | B3LYP e | 10.5 (11.8) | 1.110 | 101.6 | 2 | 0 |
| BLYP e | 9.4 (10.4) | 1.120 | 100.9 | 2 | 0 | |
| REKS | BLYP | 8.8 | 1.119 | 101.1 | 1.980 | 0.020 |
| GVB-DFT b | LYP | 6.3 | 1.110 | 101.6 | 1.986 | 0.014 |
| CAS(6,6)-DFT c | CS | 6.9 | 1.138 | 100.0 | 1.909 | 0.088 |
| Expt. d | 9.1 | 1.111 | 101.9 | N/A | N/A | |
| 1B1 State | ||||||
| ROSS-DFT | B3LYP | 30.4 | 1.072 | 147.5 | 1 | 1 |
| BLYP | 29.8 | 1.078 | 148.6 | 1 | 1 | |
| ROKS | BLYP | 30.8 | 1.077 | 148.6 | 1 | 1 |
| PO-UDFT | B3LYP | 11.4 | 1.077 | 138.1 | 1.000 | 1.000 |
| BLYP | 10.1 | 1.083 | 138.3 | 1.000 | 1.000 | |
| BS-UDFT | B3LYP | 5.3 | 1.101 | 114.0 | 1.528 | 0.472 |
| BLYP | 4.4 | 1.083 | 114.3 | 1.525 | 0.475 | |
| ROHF-DFT b | LYP | 25.8 | 1.072 | 147.5 | 1 | 1 |
| CAS(6,6)-DFT c | CS | 35.2 | 1.131 | 134.1 | 1.000 | 1.000 |
| CI f | 35.4 | 1.077 | 141.5 | N/A | N/A | |
| NOON values | |||||||
| Method | Functional | E(3A″) | ∆E(1A′) | 3A″, a’ | 3A″, a” | 1A′, a’ | 1A′, a” |
| ROSS | B3LYP | -270.31971 | 0.8 | 1 | 1 | 1 | 1 |
| BLYP | −270.19931 | 0.9 | 1 | 1 | 1 | 1 | |
| REKS | BLYP | −270.20613 | 0.9 | 1 | 1 | 1 | 1 |
| PO-UDFT | B3LYP | −270.32322 | −1.0 | 1.000 | 1.000 | 1.000 | 1.000 |
| BLYP | −270.20152 | −0.5 | 1.000 | 1.000 | 1.000 | 1.000 | |
| BS-UDFT | B3LYP | −270.32322 | 22.5 | 1.000 | 1.000 | 1.423 | 0.577 |
| GVB-DFT b | LYP | −270.11665 | −7.4 | 1 | 1 | 1 | 1 |
| CAS(8,8)-DFT c | CS | −270.03909 | −2.6 | 1 | 1 | 1 | 1 |
| DDCI2 d | −2.0 | ||||||
| Expt. e | −5... 0 | 1 | 1 | 1 | 1 | ||
| NOON values | |||||||
| Method | Functional | E(3Blu) | ∆E(1Ag) | 3Blu, blu | 3Blu, ag | 1Ag, blu | 1Ag, ag |
| RDFT | B3LYP | −230.95254 | 13.1 | 1 | 1 | 2 | 0 |
| BLYP | −230.85665 | −0.1 | 1 | 1 | 2 | 0 | |
| REKS | BLYP | −230.86028 | −5.7 | 1 | 1 | 1.723 | 0.277 |
| BS-UDFT | B3LYP | −230.95254 | −2.6 | 1.000 | 1.000 | 1.281 | 0.719 |
| BLYP | −230.85606 | −4.5 | 1.000 | 1.000 | 1.492 | 0.508 | |
| BS-UDFT + SFb | B3LYP | −230.95254 | −4.9 | ||||
| BLYP | −230.85606 | −7.3 | |||||
| PO-UDFT | B3LYP | −230.95254 | 38.9 | 1.000 | 1.000 | 1.000 | 1.000 |
| GVB-DFT | LYP | −230.77758 | −10.4 | 1 | 1 | 1.179 | 0.821 |
| CAS(8,8)-DFTc | CS | −230.70904 | −2.5 | 1.000 | 1.000 | 1.242 | 0.758 |
| Expt. d | −3.5 ± 0.5 | 1 | 1 | not known | |||
© 2002 by MDPI, Basel, Switzerland. Reproduction for noncommercial purposes permitted.
Share and Cite
Gräfenstein, J.; Kraka, E.; Filatov, M.; Cremer, D. Can Unrestricted Density-Functional Theory Describe Open Shell Singlet Biradicals? Int. J. Mol. Sci. 2002, 3, 360-394. https://doi.org/10.3390/i3040360
Gräfenstein J, Kraka E, Filatov M, Cremer D. Can Unrestricted Density-Functional Theory Describe Open Shell Singlet Biradicals? International Journal of Molecular Sciences. 2002; 3(4):360-394. https://doi.org/10.3390/i3040360
Chicago/Turabian StyleGräfenstein, Jürgen, Elfi Kraka, Michael Filatov, and Dieter Cremer. 2002. "Can Unrestricted Density-Functional Theory Describe Open Shell Singlet Biradicals?" International Journal of Molecular Sciences 3, no. 4: 360-394. https://doi.org/10.3390/i3040360
APA StyleGräfenstein, J., Kraka, E., Filatov, M., & Cremer, D. (2002). Can Unrestricted Density-Functional Theory Describe Open Shell Singlet Biradicals? International Journal of Molecular Sciences, 3(4), 360-394. https://doi.org/10.3390/i3040360