The B3LYP functional have already been shown to provide accurate geometries and vibrational frequencies for the phenolic systems [
15]. These aspects are, therefore, not discussed in the present article. However, it is important to check first whether the methodology used for calculating the bond dissociation energy can provide reliable BDE(O-H) values. Since our aim is to calculate the accurate absolute BDE(O-H) values for substituted phenols, the appropriate choice of the theoretical method is very important. To this end, we have calculated the BDE(O-H) values for three molecules, H
2O, CH
3OH and Ph-OH. The accurate BDE(O-H) values are known for the first two molecules, while reliable estimation is available for the BDE(O-H) value of PhO-H from a number of experimental studies.
Table 1 presents the calculated and experimental BDE(O-H) values for these three molecules. As we mentioned earlier, the experimental BDE(O-H) value of phenol varies within a wide range. However, Santos and Simoes suggested a value of 88.7±0.5 kcal/mol for the same analyzing the available experimental results [
6]. This value is close to the “best” experimental value of 87.3±1.5 kcal/mol for the BDE(O-H) of PhO-H, suggested from the experimental measurements by photoacoustic calorimetry in different solvents [
15,
23]. The B3LYP values, both for the 6-311G(d,p) and the 6-311++G(2df,2p), are found to be lower than the experimental values, whereas the (RO)B3LYP values with the larger basis set, 6-311++G(2df,2p), are in close agreement with the experimental values. For estimating BDE(O-H) values (RO)B3LYP method is superior to the usual B3LYP method. The (RO)B3LYP/6-311++G(2df,2p) has, therefore, subsequently been used for the BDE(O-H) calculations of all the substituted phenols (mentioned in
Table 2 and
Table 3).
A. BDE(O-H) of Substituted Phenols
Table 2 presents the calculated BDE(O-H) values for a series of para-substituted phenols along with the available experimental results. The results obtained from the (RO)B3LYP calculations with smaller 6-311G(d,p) basis set are nearly 1 kcal/mol lower than those obtained from the larger 6-311++G(2df,2p) basis set. Direct experimental gas-phase results are not available for these molecules. Bordwell and Cheng estimated the BDE(O-H) values from the oxidation potential measurements of phenoxide ions in DMSO and the pK
HA values in DMSO of their conjugate acids [
12]. In most of the cases these BDE(O-H) values are significantly higher than the corresponding calculated gas-phase BDE(O-H) values. Only for
para-hydroxy and
para-amino phenols, their estimated values are fairly close to our calculated results. Lind and co-workers [
11] calculated the BDE(O-H) of substituted phenols by calculating the free-energy for the reaction: X-C
6H
4OH(aq) → X-C
6H
4O(aq) + 1/2H
2(g) from the aqueous redox potentials and pK
a values and setting ΔG
osolv (the difference of solvation free energies between a phenol molecule and the corresponding phenoxide anion) to zero. They believed their calculated values can only be considered as an upper limit to the exact BDE(O-H) values [
11]. Our calculated values are found to be significantly lower than their estimated values. But as these estimations were made from the results of solution phase data (such as DMSO and water) under some assumptions, much emphasis should not be given to the correlation between our calculated BDE(O-H) values and these experimental values. However, our calculated BDE(O-H) values can be compared to the values predicted by Santos and Simoes [
6] from the extensive analysis of the available experimental results. The calculated and experimental relative BDE(O-H) values [ΔBDE(O-H)] are given in
Table 2 for comparison. The agreement between their ΔBDE(O-H) values and corresponding ours is found to be good. Only in the case of
para-cyanophenol the calculated relative BDE(O-H) value (1.9 kcal/mol) is lower than the value (4.3±1.9 kcal/mol) predicted by them [
6]. Our calculated absolute BDE(O-H) values are quite close to those reported by Wright and coworkers using (RO)B3LYP/6-311+G(2d,2p)//AM1 method [
19]. Since their method is computationally cheaper than the method used here, the former method may preferentially be applied to larger molecules. The B3LYP/6-31G(d,p) calculated ΔBDE(O-H) values for
para substituted phenols reported in the reference [
13] are not much different from those reported here and in the reference [
19]. The differences in our calculated ΔBDE(O-H) values and those reported in reference [
13] vary within a narrow range of 0.2 (
p-NO
2) to 1 kcal/mol (
p-NH
2). So, if one is interested only in ΔBDE(O-H) values, B3LYP/6-31G(d,p) procedure can be used for getting reasonable results.
The effects of electron withdrawing and donating groups on the BDE(O-H) of the
para substituted phenol are opposite. Electron withdrawing groups (such as CF
3, CN, and NO
2) at the
para position increase the BDE(O-H) value from that in the parent phenol molecule, while
para-donor substituents (such as CH
3, OCH
3, and NH
2) tend to weaken the O-H bond of phenol. It is interesting to note that F
Table 2.
The homolytic bond dissociation enthalpies at 298K [BDE(O-H) in kcal/mol] of the O-H bonds of para substituted phenols (X-C6H4-OH) calculated by using the (RO)B3LYP procedures with two different basis sets, 6-311G(d,p) [sb] and 6-311++G(2df,2p) [lb].
Table 2.
The homolytic bond dissociation enthalpies at 298K [BDE(O-H) in kcal/mol] of the O-H bonds of para substituted phenols (X-C6H4-OH) calculated by using the (RO)B3LYP procedures with two different basis sets, 6-311G(d,p) [sb] and 6-311++G(2df,2p) [lb].
| Substituent (X) | BDE(O-H) | Calculated ΔBDEc | Expt.d ΔBDEc |
| sb | lb | Expt.a | Expt.b |
| H | 86.4 | 87.5 | 89.8 | 88.2 | 0.0 | 0.0 |
| F | 84.4 | 85.4 | | 87.4 | -2.1 | -1.0±1 |
| Cl | 85.5 | 86.1 | 90.3 | 87.6 | -1.4 | -0.2±1 |
| CH3 | 84.3 | 85.1 | 88.7 | 86.1 | -2.4 | -1.9±1 |
| OCH3 | 80.5 | 81.3 | 84.6 | 82.6 | -6.2 | -5.3±1 |
| OH | 80.6 | 81.7 | 81.5 | 80.2 | -5.8 | -6.5±2.4 |
| NH2 | 76.9 | 77.9 | 77.3 | 75.5 | -9.6 | -9.6±3.1 |
| CF3 | 89.2 | 90.4 | 95.3 | | 2.9 | 4.1±1 |
| CN | 88.6 | 89.4 | 94.2 | 92.9 | 1.9 | 4.3±1.9 |
| NO2 | 90.5 | 91.7 | 94.7 | 94.2 | 4.2 | 6.0±1.9 |
and Cl substitutions at the
para-position reduce the O-H bond strength of phenol, although they are generally considered as electron-withdrawing groups. Here, F and Cl behave like an electron donating substituent. It is generally believed that electron donating substituents at
para position decrease the BDEs of the O-H bonds of substituted phenols primarily by stabilizing the corresponding radicals and also to some extent by raising the ground-state energies [
5]. On the other hand, electron withdrawing substituents at the
para position interact with the O-H dipoles causing a lowering of ground-state energies and thereby increasing the BDE(O-H) values [
5]. The BDE(O-H) value of
para-aminophenol is 77.9 kcal/mol, which is 9.6 kcal/mol lower than that of the parent phenol molecule. Among the electron-withdrawing groups, NO
2 group affects the BDE(O-H) value most and the BDE(O-H) value increases by 4.2 kcal/mol.
The calculated BDE(O-H) values for the
meta-substituted phenols are given in
Table 3. The predicted ΔBDE(O-H) values from the experimental results are also included in
Table 3. These values are found to be in good agreement with our calculated values. Our calculated values are also quite close to those calculated by Wright and co-workers [
19]. The effect of electron donor group at the
meta- and
para-position of phenol is strikingly different. Electron donor-group at the
meta position does not have any significant effect on the bond strength of the O-H bond in comparison to the parent unsubstituted phenol molecule, whereas the same at the
para position reduces the O-H bond strength significantly. Thus there is a substantial difference in the O-H bond strengths between the
para and
meta-substituted phenols for electron-donor substituents (see the values of ΔBDE
p-m in
Table 3). For
Table 3.
The homolytic bond dissociation enthalpies at 298 K [BDE(O-H) in kcal/mol] of the O-H bonds of meta substituted phenols (X-C6H4-OH) calculated by using the (RO)B3LYP procedures with two different basis sets, 6-311G(d,p) [sb] and 6-311++G(2df,2p) [lb].
Table 3.
The homolytic bond dissociation enthalpies at 298 K [BDE(O-H) in kcal/mol] of the O-H bonds of meta substituted phenols (X-C6H4-OH) calculated by using the (RO)B3LYP procedures with two different basis sets, 6-311G(d,p) [sb] and 6-311++G(2df,2p) [lb].
| Substituent (X) | BDE(O-H) | Calculated ΔBDEp-ma | Calculated ΔBDEb | Expt.c ΔBDEb |
| sb | lb |
| H | 86.4 | 87.5 | 0.0 | 0.0 | 0.0 |
| F | 87.5 | 88.4 | -3.0 | 0.9 | 1.4±1.9 |
| Cl | 87.5 | 88.4 | -2.3 | 0.9 | 1.2±1 |
| CH3 | 86.0 | 86.9 | -1.8 | -0.6 | -0.7±1 |
| OCH3 | 85.2 | 86.1 | -4.8 | -1.4 | 0.0±1 |
| OH | 86.1 | 87.0 | -5.3 | -0.5 | 0.2±1 |
| NH2 | 86.0 | 86.9 | -9.0 | -0.6 | -1.2±1 |
| CF3 | 88.3 | 89.5 | 0.9 | 2.0 | 3.1±1 |
| CN | 89.3 | 90.3 | -0.9 | 2.8 | 3.1±1.9 |
| NO2 | 89.7 | 90.7 | 1.0 | 3.2 | 4.5±1.9 |
example, the calculated BDE(O-H) value of
meta-aminophenol is 9 kcal/mol higher than that of
para-aminophenol.
In the case of electron-withdrawing groups, the difference in the BDE(O-H) values for the meta and para substituent is less significant. An electron-withdrawing group at both the meta and para positions increases the BDE(O-H) values as compared with that for the unsubstituted phenol. Interestingly, however, F and Cl substituent at the meta position enhance the BDE(O-H) value from that in the parent phenol molecule. Therefore, these two atoms behave like weak electron withdrawing groups at the meta position, which is opposite to that observed at the para position.
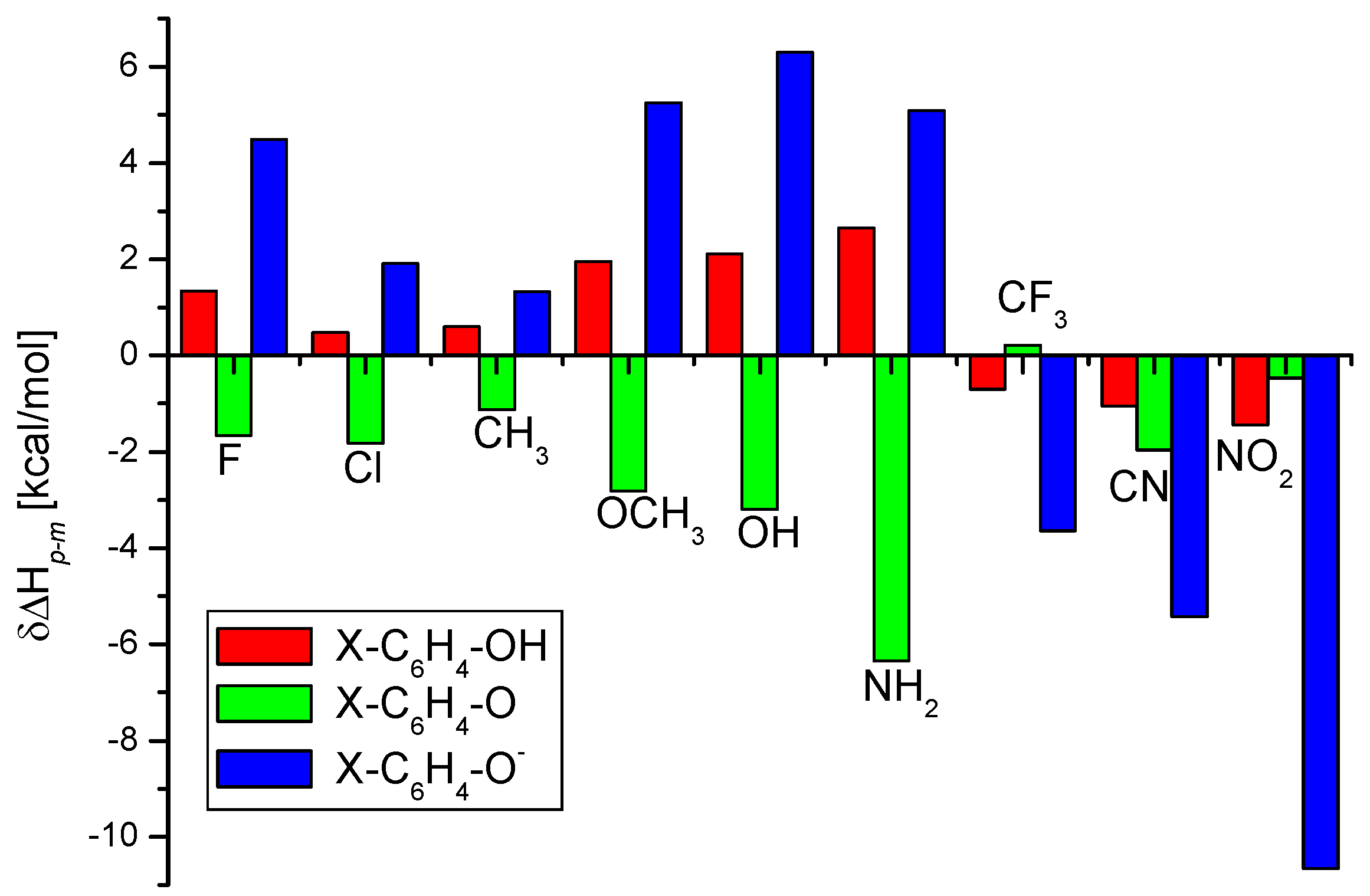
To analyze the origin of differences between the BDE(O-H) values of the
meta and
para substituted phenols, we have estimated the change in the enthalpy values (δΔH
p-m = ΔH
para − ΔH
meta) while going from
para to
meta substituted phenol, phenoxy radical, and phenoxide anion for each of the substituents. The δΔH
p-m values are shown in
Figure 1. The positive values of δΔH
p-m mean that the
meta substituted species is more stable than the corresponding
para substituted one.
In the case of electron-donating substituents, such as CH
3, OCH
3, OH, and NH
2, the substitutions at the
meta-position provide extra stability to the phenol molecule than the substitution at the
para position (see
Figure 1). The situation is opposite in the case of phenoxy radicals. Here the presence of electron donating group at the
para position provides additional stabilization to the system in comparison to that at the
meta position. These two opposing effects result in increase of O-H bond strength for an electron donating
meta substituent phenols compared to the corresponding
para
Figure 1.
The differences between the enthalpies (δΔHp-m) of para and meta substituted phenols (X-C6H4-OH), phenoxyl radicals (X-C6H4-O), and phenoxide ions (X-C6H4-O−) calculated at the (RO)B3LYP/6-311++G(2df,2p)//B3LYP/6-311G(d,p) level.
Figure 1.
The differences between the enthalpies (δΔHp-m) of para and meta substituted phenols (X-C6H4-OH), phenoxyl radicals (X-C6H4-O), and phenoxide ions (X-C6H4-O−) calculated at the (RO)B3LYP/6-311++G(2df,2p)//B3LYP/6-311G(d,p) level.
substituted counterpart. In the case of electron-withdrawing groups, such as CN, CF3, and NO2, para-substituted phenols are more stable than the corresponding meta substituted phenols. The situation is the same for the substituted phenoxy radicals, except for the CF3 group. As a result, the difference in the BDE(O-H) values between the para and meta substituted cyanophenol (nitrophenol as well) is found to be small. However, in the case of CF3 group, meta substituted phenoxy radical is slightly more stable than the para substituted one.
B. Proton Affinities of Substituted Phenoxide Ions
The gas-phase acidities of substituted phenols are estimated from the proton affinities of the corresponding phenoxide anions (X-ph-O
−). The lower the PA value of the phenoxide anion is the greater should be the acidity of the corresponding parent phenol molecule [
20]. The PAs are calculated from the enthalpies of the substituted phenols and phenoxide anions by using equation (2).
Table 4 presents the PA values for both the
para and
meta substituted phenols along with the available experimental values. The experimental PA values are obtained by adding the ionization potential of hydrogen IP(H) (313.6 kcal/mol) to the difference of BDE(O-H) and the electron affinity of phenoxyl radical [
8]. The PA values calculated at the B3LYP/6-311G(d,p) level are almost 5-6 kcal/mol larger than those obtained from the B3LYP/6-311++G(2df,2p) results and also from the experimental values. This is primarily due to the poor representation of the electronic structure of the phenoxide anions when 6-311G(d,p) basis set is used. Because it is well known that diffuse functions in the basis set is
Table 4.
Proton affinities [PA in kcal/mol] of the para and meta substituted phenoxide ions (X-C6H4-O−) calculated by using the B3LYP/6-311++G(2df,2p) method. The ΔPAp-m and q(O-) represent the difference of the calculated PA values between the para and meta-substituted phenoxide ions and the net Mulliken charge on the oxygen atom at the B3LYP/6-311G(d,p) level.
Table 4.
Proton affinities [PA in kcal/mol] of the para and meta substituted phenoxide ions (X-C6H4-O−) calculated by using the B3LYP/6-311++G(2df,2p) method. The ΔPAp-m and q(O-) represent the difference of the calculated PA values between the para and meta-substituted phenoxide ions and the net Mulliken charge on the oxygen atom at the B3LYP/6-311G(d,p) level.
| Substituent (X) | PA | ΔPAp-m | q(O-) |
| para | Expt.a | ΔPApb | meta | Expt.a | ΔPAmb | | para | meta |
| H | 347.8 | 346.9 | 0.0 | 347.8 | 346.9 | 0.0 | 0.0 | -0.5188 | |
| F | 345.2 | 344.3 | -2.6 | 342.0 | 341.1 | -5.8 | 3.2 | -0.5209 | -0.5098 |
| Cl | 341.3 | 340.3 | -6.5 | 339.8 | 339.0 | -8.0 | 1.5 | -0.5053 | -0.4982 |
| CH3 | 349.0 | 348.2 | 1.2 | 348.3 | 347.3 | 0.5 | 0.7 | -0.5185 | -0.5165 |
| OCH3 | 350.2 | 347.6 | 2.4 | 346.9 | 345.4 | -0.9 | 3.3 | -0.5262 | -0.5105 |
| OH | 350.1 | | 2.3 | 345.9 | 341.8 | -1.9 | 4.2 | -0.5300 | -0.5163 |
| NH2 | 352.3 | 351.1 | 4.5 | 349.8 | 347.8 | 2.0 | 2.5 | -0.5193 | -0.5185 |
| CF3 | 334.6 | | -13.2 | 337.5 | | -10.3 | -2.9 | -0.4856 | -0.5000 |
| CN | 329.4 | 329.2 | -18.4 | 333.7 | 332.6 | -14.1 | -4.3 | -0.4733 | -0.4951 |
| NO2 | 323.6 | | -24.2 | 332.8 | 331.2 | -15.0 | -9.2 | -0.4528 | -0.4927 |
necessary for the proper calculation of electronic structure of an anionic system. Indeed, the PA values obtained from the latter method, which includes diffuse functions in the basis set, agree quite well with the experimental results. Only in the case of
para-methoxy phenol and
meta-hydroxy phenol, our calculated PA values are significantly larger than those obtained from the experiment.
The electron donating group (such as CH
3, OCH
3, etc.) at the
para-position of phenoxide anion increases the PA value slightly from that of the parent unsubstituted phenoxide anion (see
Table 4). On the other hand, the effect of the presence of electron donating group at the
meta-position of phenoxide anion depends upon the nature of the substituent, while CH
3 and NH
2 groups tend to increase the PA value from the parent unsubstituted phenoxide anion, OCH
3 and OH groups work in the opposite direction. On the other hand, the electron withdrawing groups at the
meta or
para-position have a strong lowering effect (from 10 to 24 kcal/mol) on the PA values of the phenoxide anion. Electron withdrawing group at the
para-position reduces the PA value much more than that caused by the same group at the
meta-position. The difference of PA values between the
para and
meta substituted phenoxide anions are given in
Table 4.
It is clear that in the case of electron donating groups, meta substituted phenol is more acidic than the corresponding para substituted phenols, whereas the opposite is true for the strong electron withdrawing groups. In the cases of F and Cl, meta substitution increases the acidity much more than para substitution does. But in both the positions, F and Cl behave similarly as the electron withdrawing groups, like CN and NO2.
To understand clearly the reason behind the variation of PA with the position of substituent, we have calculated the enthalpy differences between
meta and
para substituted phenols and phenoxide anions.
Figure 1 displays the graphical representation of the same. As can be seen from the figure, the change in the stability order with the change in the position (
meta/para) of the substituent remains the same for both phenol molecules and phenoxide anions. Like the substituted phenol molecules, electron donating
meta substituent stabilizes the phenoxide anion much more than that at the
para position, whereas the opposite is true for the electron withdrawing substituents. The F and Cl act as an electron donating substituent for this case. Of course, the difference in the enthalpy values of
meta and
para substituted phenoxide anions is always much more than that for the corresponding phenol molecules. As a result, there is a substantial difference in the PA values of the
meta and
para substituted phenoxide anions.
In search for a molecular parameter which can be correlated to the PA values of phenoxide anions, the first obvious choice is the point charge on the oxygen atom. The PA value is expected to increase (conversely, the acidity to decrease) with the increase in electronic population on the oxygen atom.
Table 4 presents the net electronic charge (Mulliken population) on the oxygen atoms of the substituted phenoxide anions. As expected, generally the electron donating groups increase the net electronic charge on the oxygen atom, while the electron withdrawing groups decrease the net charge on the oxygen atom.
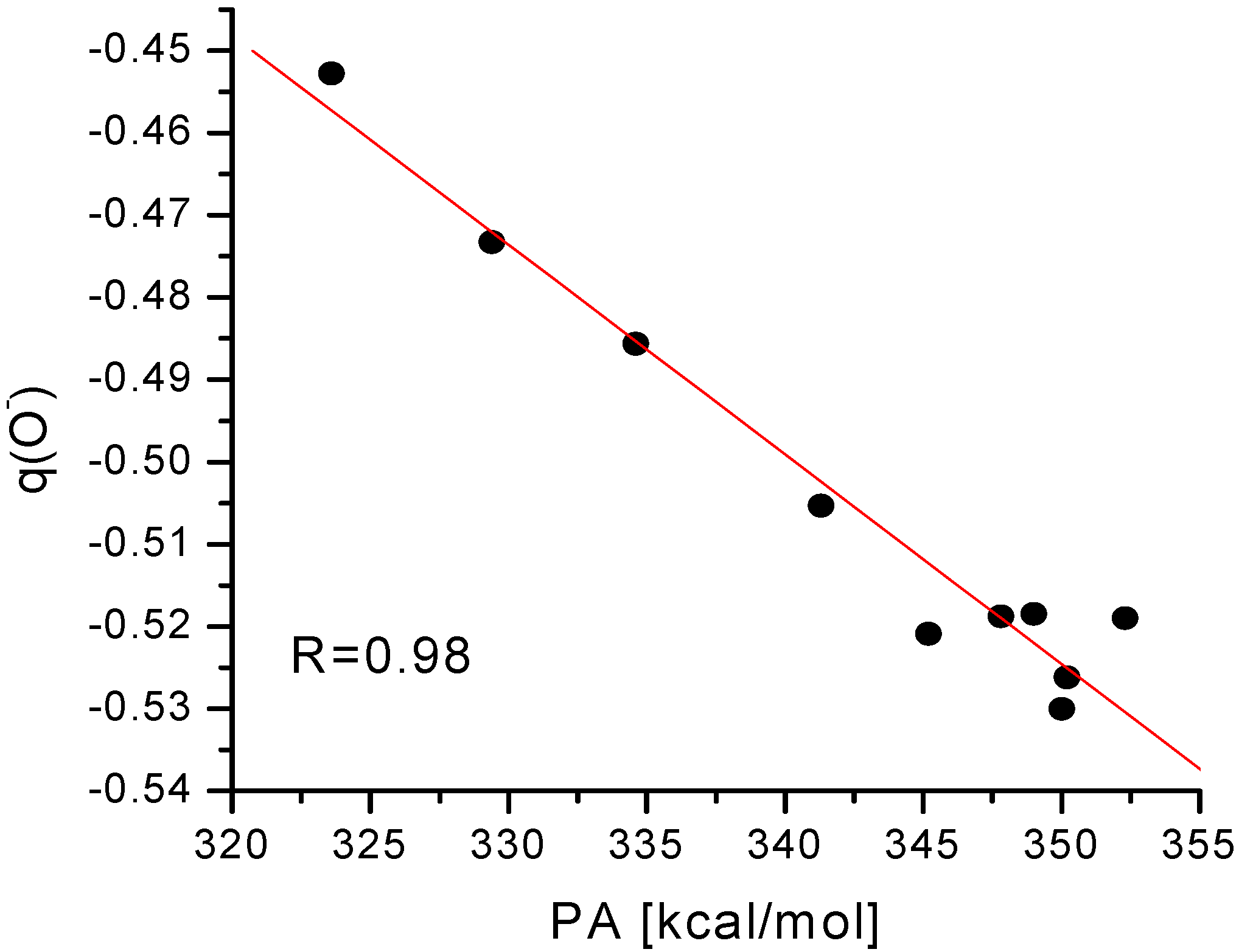
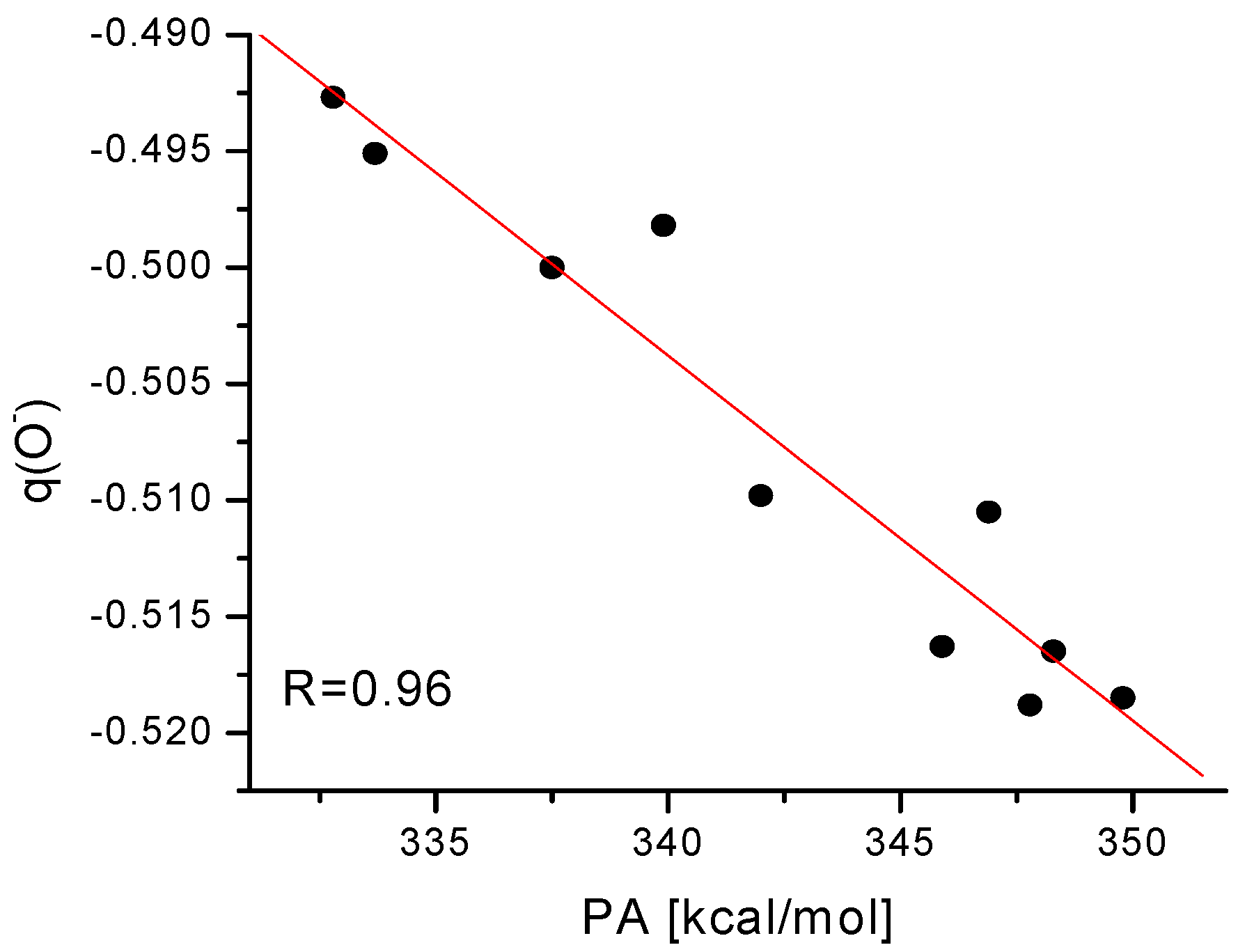
Figure 2 and
Figure 3 display the correlation between the net Mulliken charge on the oxygen atom and the PA of the
para and
meta substituted phenoxide anions, respectively.
Figure 2.
Plot of proton affinities (PA) of
para substituted phenoxide ions (mentioned in
Table 4) against the net electronic population on the oxygen atom of the phenoxide ions.
Figure 2.
Plot of proton affinities (PA) of
para substituted phenoxide ions (mentioned in
Table 4) against the net electronic population on the oxygen atom of the phenoxide ions.
Figure 3.
Plot of proton affinities (PA) of
meta substituted phenoxide ions (mentioned in
Table 4) against the net electronic population on the oxygen atom of the phenoxide ions.
Figure 3.
Plot of proton affinities (PA) of
meta substituted phenoxide ions (mentioned in
Table 4) against the net electronic population on the oxygen atom of the phenoxide ions.
Although there are some deviations, the overall correlation is reasonably well, especially in the case of para substituted phenoxide anions. Such correlation can be used for the approximate estimation of PA value (hence acidity as well) from the point charge on the oxygen atom of substituted phenoxide anion.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






