Zeolite-Catalyzed Hydrocarbon Formation from Methanol: Density Functional Simulations

Abstract
:Introduction
Computational Details
Results
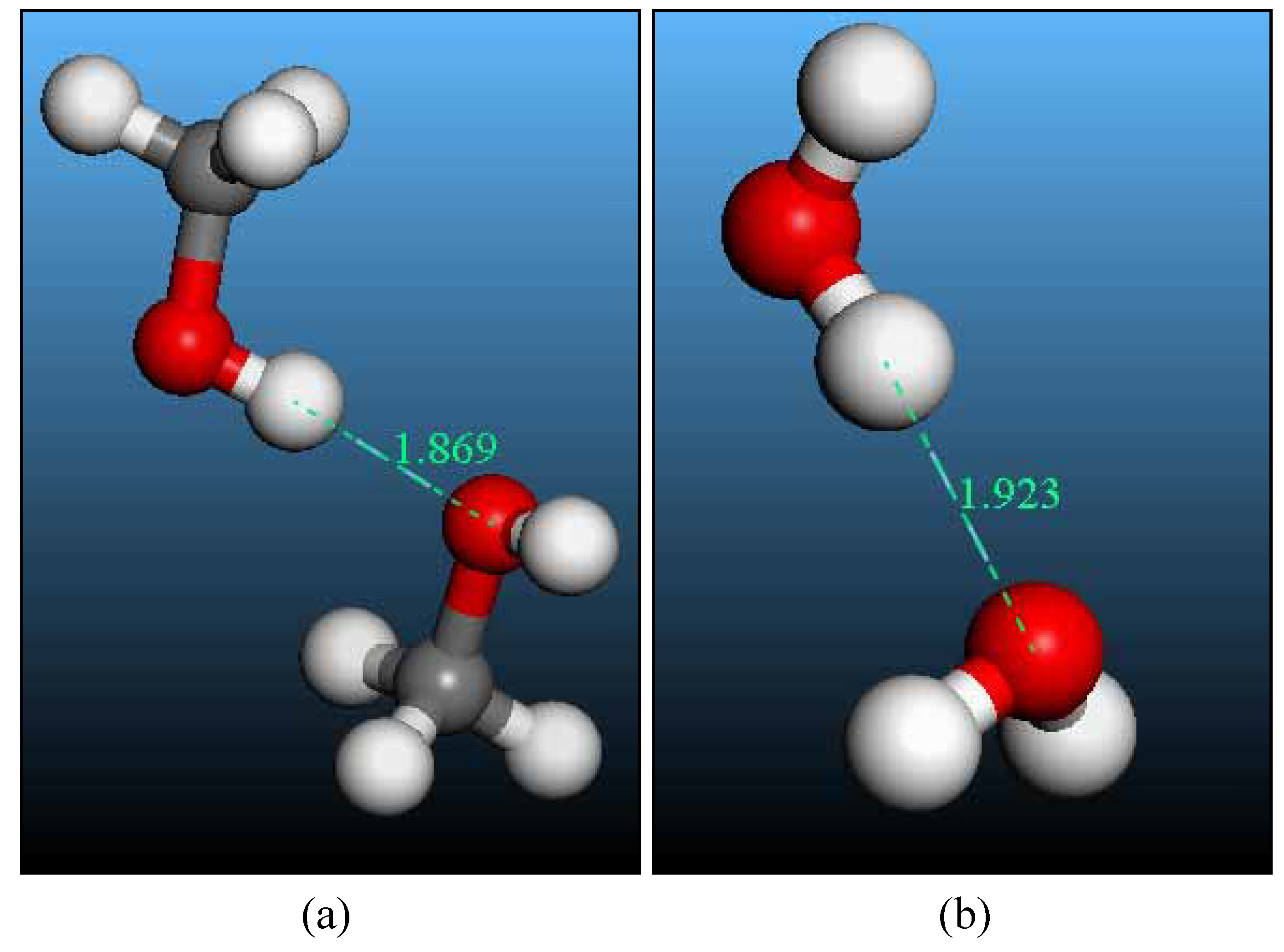
Hydrogen bonding

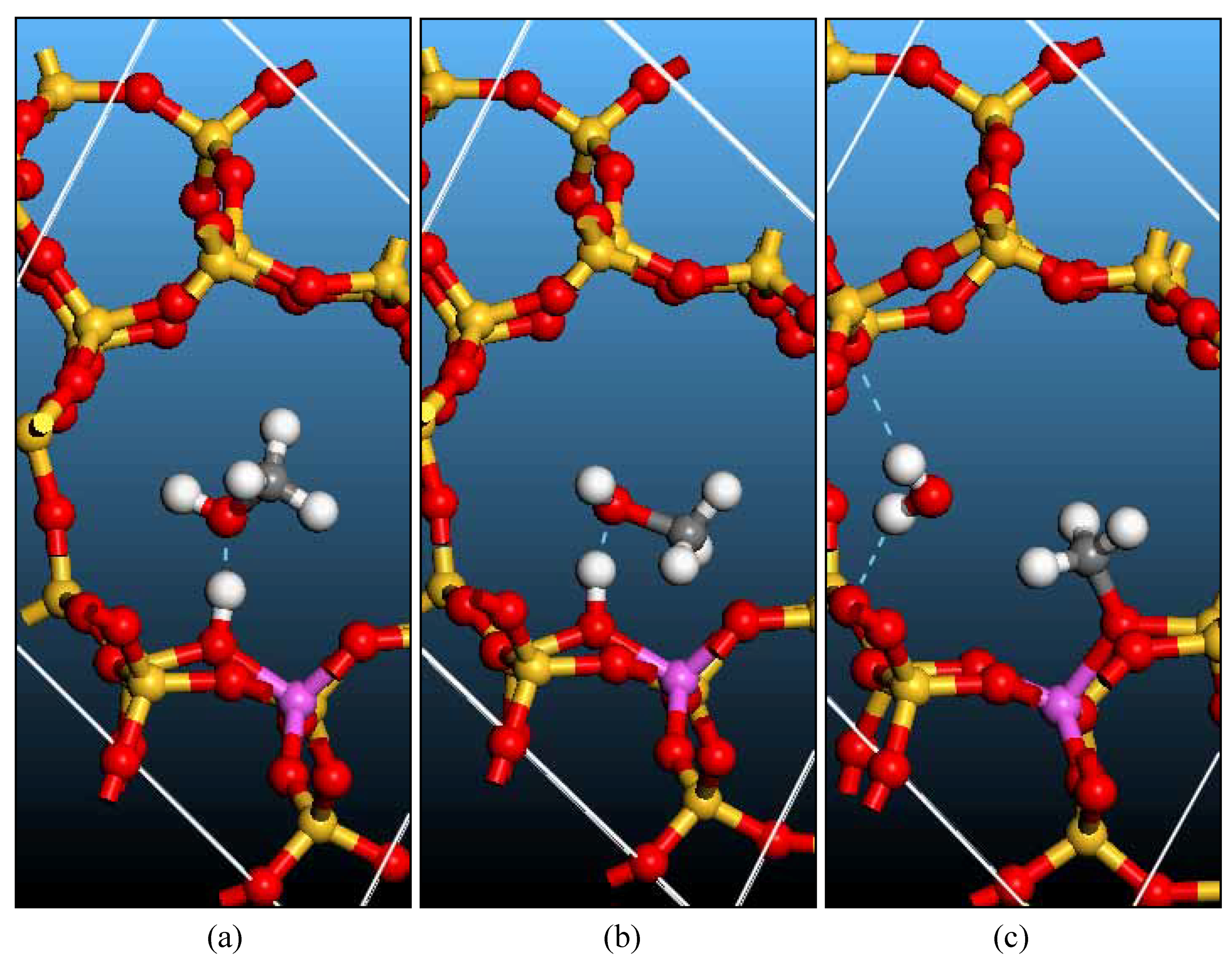
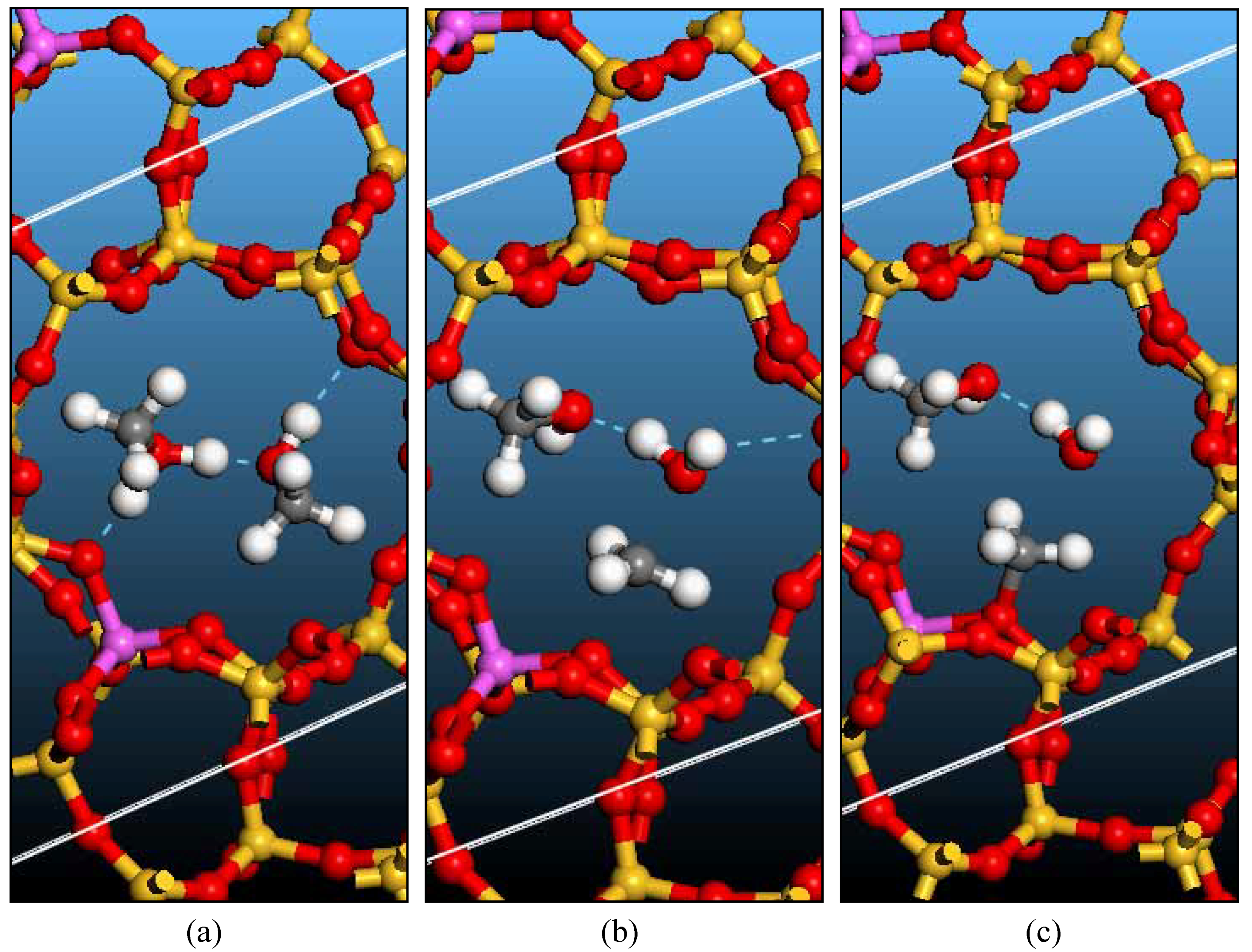
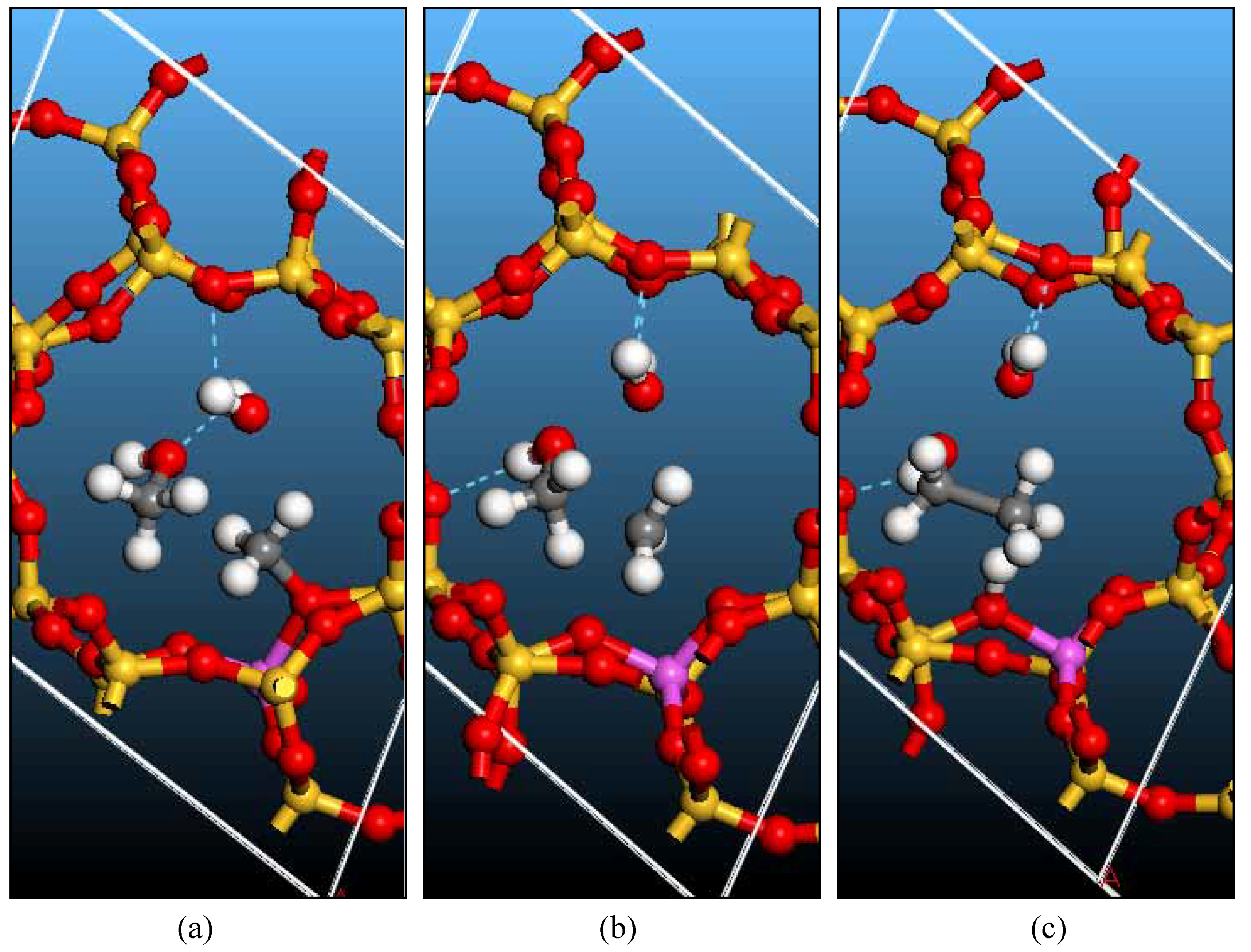
Bridging hydroxyl groups and zeolite models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy (kJ/mol) | |||||
|---|---|---|---|---|---|
| Site | rAlH (Å) | αSiO(H)Al (deg) | Fixed Cell | Ref. [26] | Optimized Cell |
| O1H | 2.54 (2.48±04) | 130.1 (135.7) | 0.0 | 0.0 | 0.0 |
| O2H | 2.43 | 141.1 (144.6) | 7.8 | 9.8 | 8.0 |
| O3H | 2.46 (2.40±04) | 136.8 (139.8) | 4.1 | 4.9 | 4.3 |
| O4H | 2.44 | 135.9 (141.9) | 9.4 | 7.9 | 9.1 |
Surface methoxyl: Formation and Reactions






Conclusions
Acknowledgement
References
- Andzelm, J.; Govind, N.; Fitzgerald, G.; Maiti, A. Int. J. Quantum Chem. submitted. 2001.
- Chang, C.D.; Silvestri, A.J. J. Catal. 1977, 47, 249. [CrossRef]
- Sauer, J. Chem. Rev. 1989, 89, 199.
- Hutchings, G.J.; Hunter, R. Catal. Today 1990, 6, 279. [CrossRef]
- Haase, F.; Sauer, J. Micro. Meso. Mat. 2000, 35, 379. [CrossRef]
- Sinclair, P.E.; Catlow, C.R.A. J. Chem. Soc. Faraday Trans. 1996, 92, 2099. [CrossRef]
- Shah, R.; Gale, J.D.; Payne, M.C. J. Phys. Chem. 1996, 100, 11688. [CrossRef]
- Sandre, E.; Payne, M.C.; Gale, J.D. Chem. Commun. 1998, 22, 2445.
- Hutchings, G.J.; Watson, G.W.; Willock, D.J. Micro. Meso. Mat. 1999, 29, 67. [CrossRef]
- Blaszkowski, S.R.; van Santen, R.A. J. Phys. Chem. 1995, 99, 11728. [CrossRef]
- Zicovich-Wilson, C.M.; Viruela, P.; Corma, A. J. Phys. Chem. 1995, 99, 13224. [CrossRef]
- Sinclair, P.E.; Catlow, C.R.A. J. Chem. Soc. Faraday Trans. 1997, 93, 333. [CrossRef]
- Blaszkowski, S.R.; van Santen, R.A. J. Am. Chem. Soc. 1997, 119, 5020. [CrossRef] [Green Version]
- Delley, B. J.Chem.Phys. 1990, 92, 508. J. Phys. Chem. 1996, 100, 6107. J. Chem. Phys. 2000, 113, 7756.
- DMol3, Materials Studio 2.0. Accelrys Inc.: San Diego, CA, USA.
- Milman, V.; Winkler, B.; White, J.A.; Pickard, C.J.; Payne, M.C.; Akhmatskaya, E.V.; Nobes, R.H. Int. J. Quant. Chem. 2000, 77, 895. [CrossRef]
- CASTEP. Materials Studio 2.0, Accelrys Inc.: San Diego, CA, USA.
- Andzelm, J.; King-Smith, D.; Fitzgerald, G. Chem. Phys. Lett. 2001, 335, 321, and to be published.
- Govind, N.; Fitzgerald, G.; King-Smith, D. to be published.
- Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865. [CrossRef]
- Monkhorst, H. J.; Pack, J. D. Phys. Rev. B 1976, 13, 5188. [CrossRef]
- Szalewicz, K.; Jeziorski, B. Molecular Interactions; Scheiner, S., Ed.; John Wiley & Sons: New York, 1997; p. 3. [Google Scholar]
- Klamt, A.; Jonas, V.; Burger, T.; Lohrenz, J. J. Phys. Chem. 1998, 102, 5074. [CrossRef]
- Kristian, S.; Pulay, P. Chem. Phys. Lett. 1994, 175, 229.
- Czjzek, M.; Jobic, H.; Fitch, A.; Vogt, T. J. Phys. Chem. 1992, 96, 1535. [CrossRef]
- Hill, J-R.; Freeman, C.; Delley, B. J. Phys. Chem. A 1999, 103, 3772.
- Haase, F.; Sauer, J. J. Am. Chem. Soc. 1995, 117, 3780. [CrossRef]
- Sinclair, P.E.; Catlow, C.R.A. J. Phys. Chem. 1997, 101, 295. [CrossRef]
© 2002 by MDPI (http://www.mdpi.org), Basel, Switzerland.
Share and Cite
Govind, N.; Andzelm, J.; Reindel, K.; Fitzgerald, G. Zeolite-Catalyzed Hydrocarbon Formation from Methanol: Density Functional Simulations. Int. J. Mol. Sci. 2002, 3, 423-434. https://doi.org/10.3390/i3040423
Govind N, Andzelm J, Reindel K, Fitzgerald G. Zeolite-Catalyzed Hydrocarbon Formation from Methanol: Density Functional Simulations. International Journal of Molecular Sciences. 2002; 3(4):423-434. https://doi.org/10.3390/i3040423
Chicago/Turabian StyleGovind, Niranjan, Jan Andzelm, Kurt Reindel, and George Fitzgerald. 2002. "Zeolite-Catalyzed Hydrocarbon Formation from Methanol: Density Functional Simulations" International Journal of Molecular Sciences 3, no. 4: 423-434. https://doi.org/10.3390/i3040423
APA StyleGovind, N., Andzelm, J., Reindel, K., & Fitzgerald, G. (2002). Zeolite-Catalyzed Hydrocarbon Formation from Methanol: Density Functional Simulations. International Journal of Molecular Sciences, 3(4), 423-434. https://doi.org/10.3390/i3040423