The Use of Isolated Human Lymphocytes in Mycotoxin Cytotoxicity Testing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Extraction of mycotoxins from barley samples
2.2. Cytotoxicity test
2.3. Analysis of the extracts
3. Results and Discussion
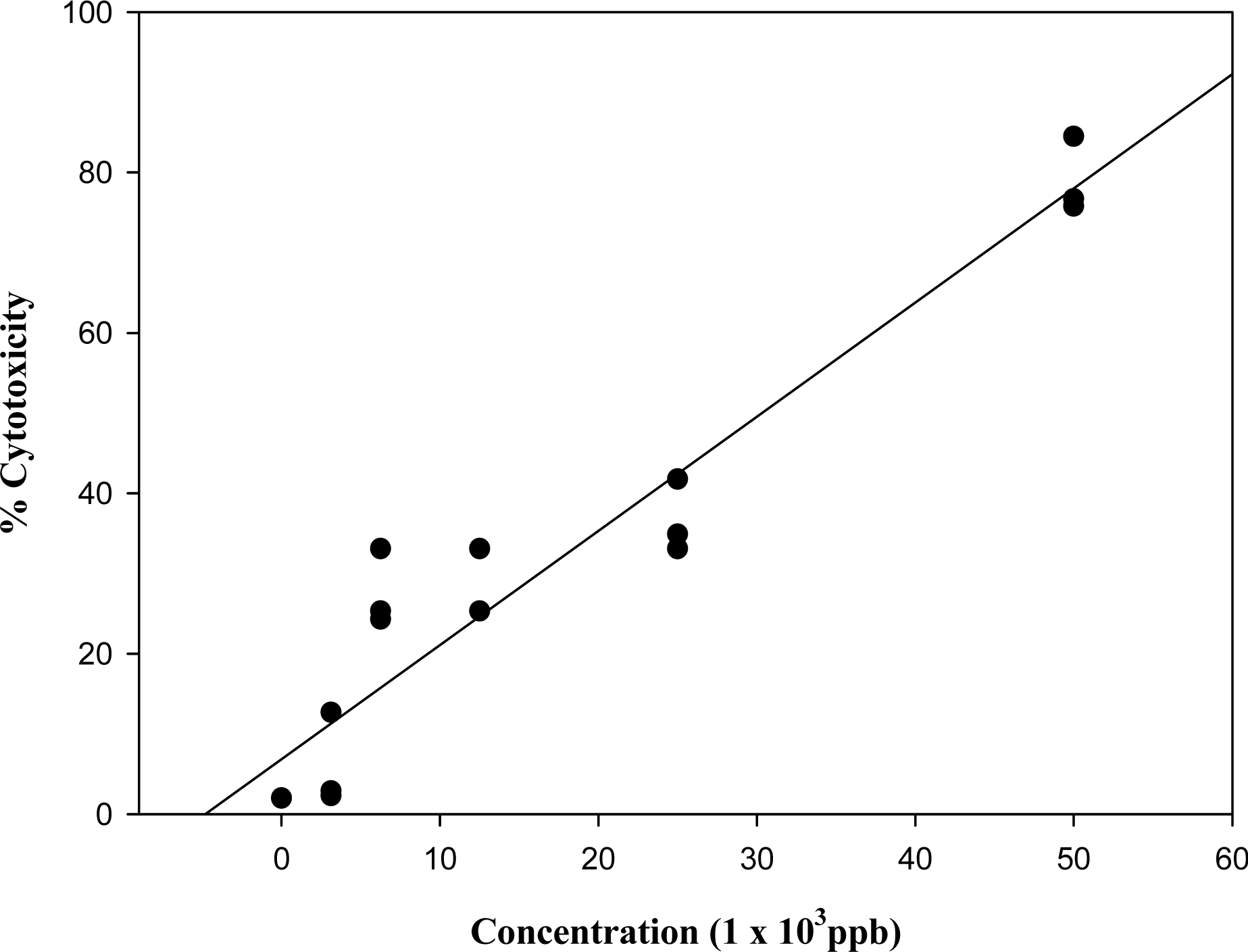
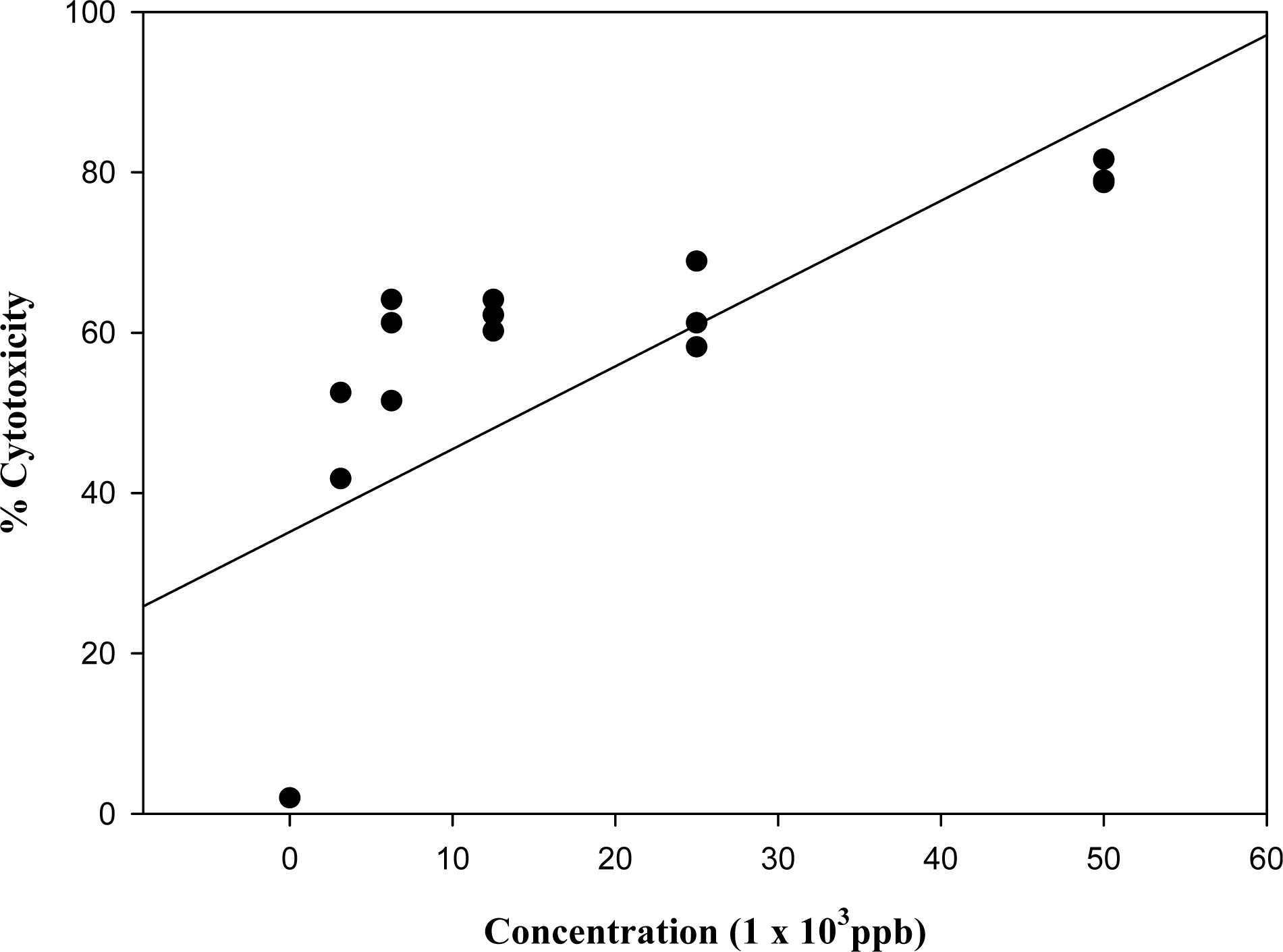
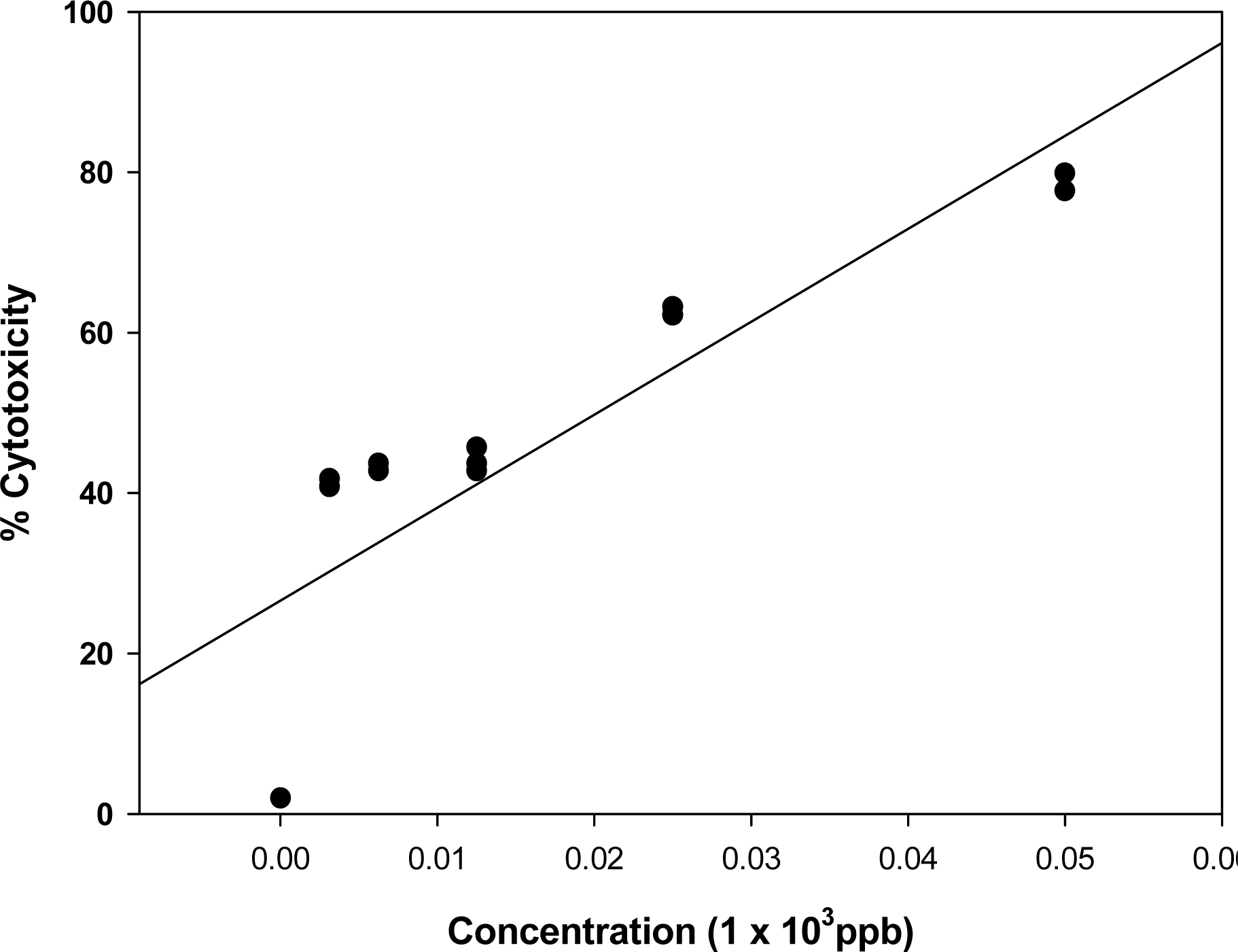
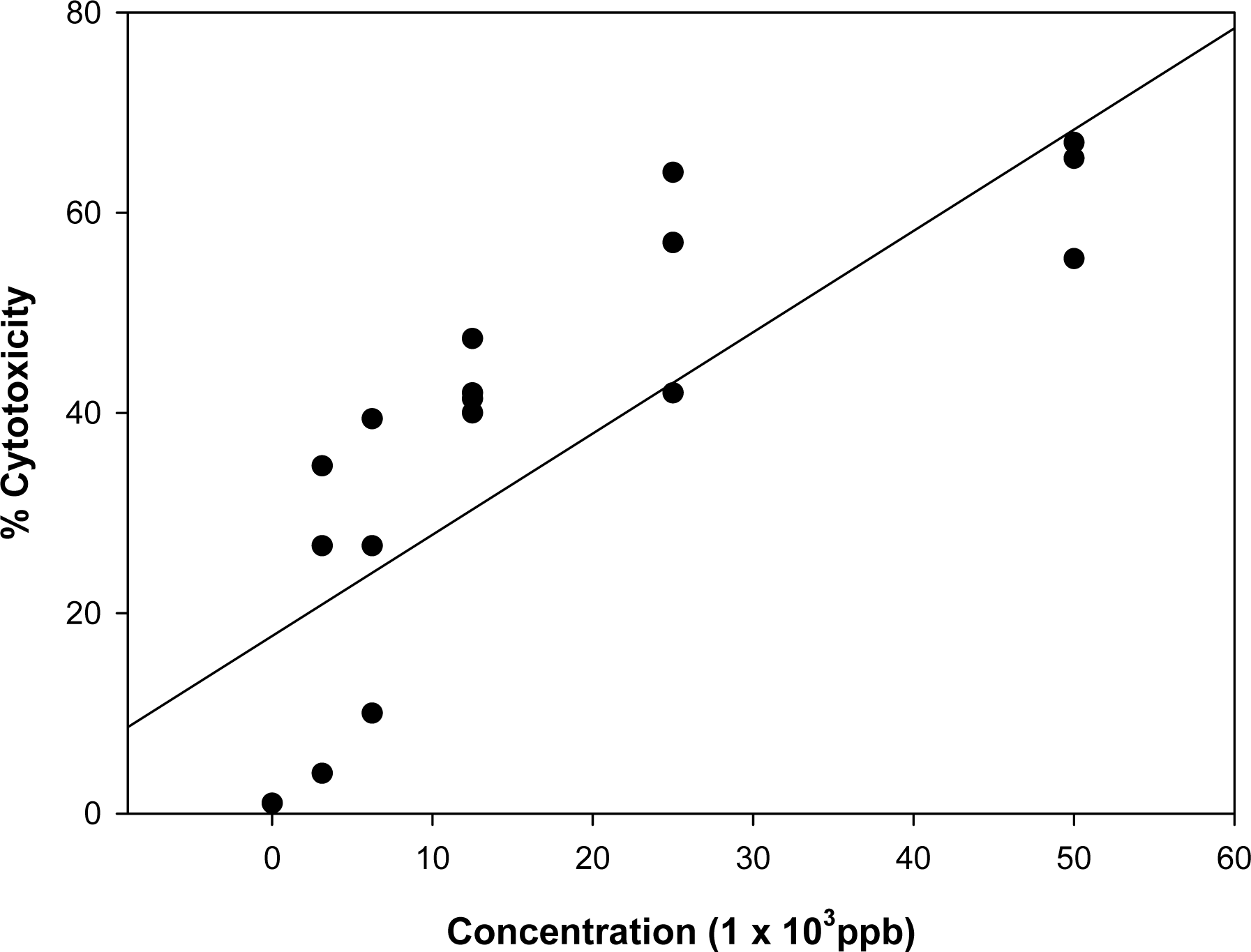
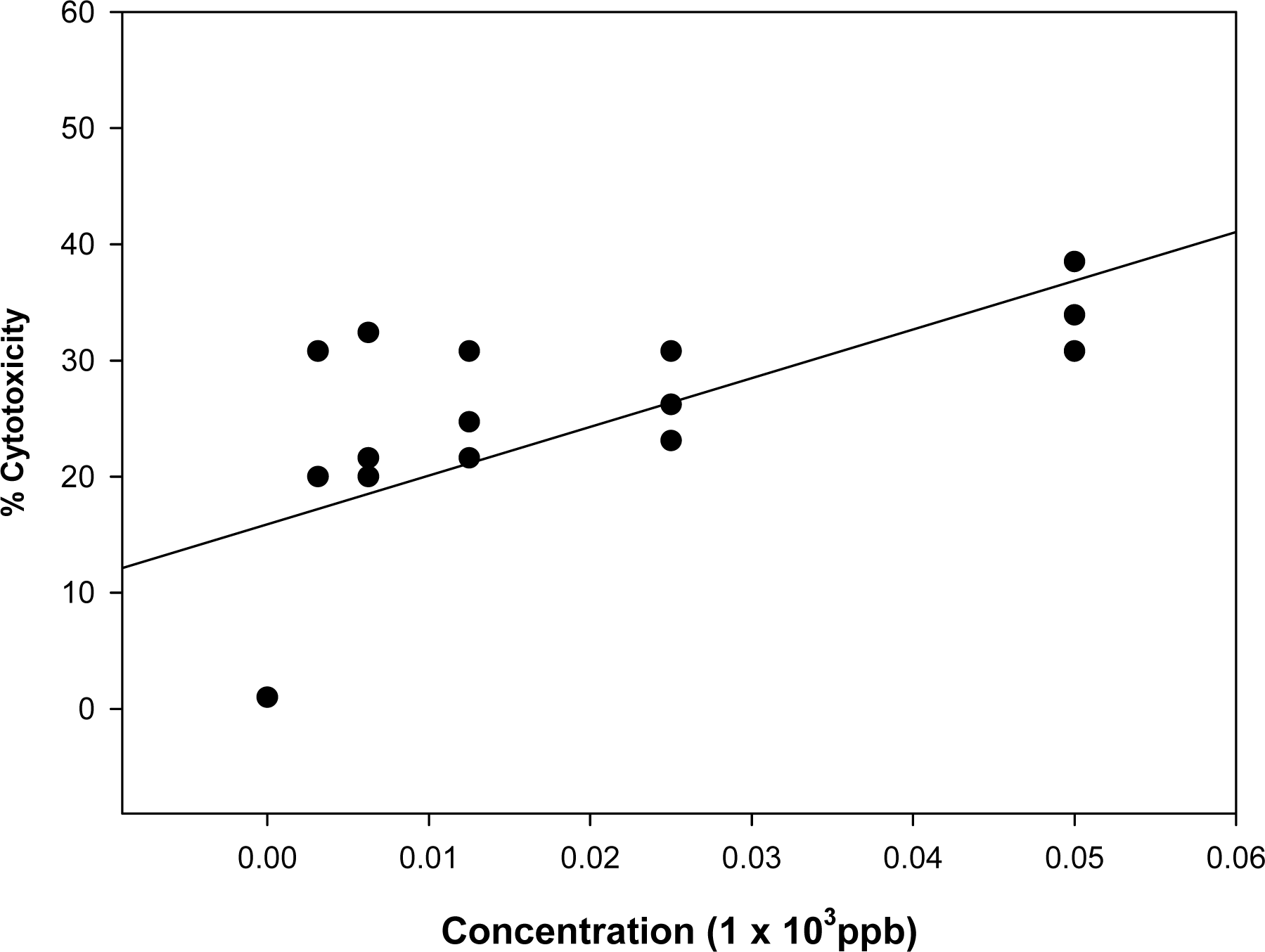
3.1. Effect of known mycotoxins and barley extracts on lymphocytes
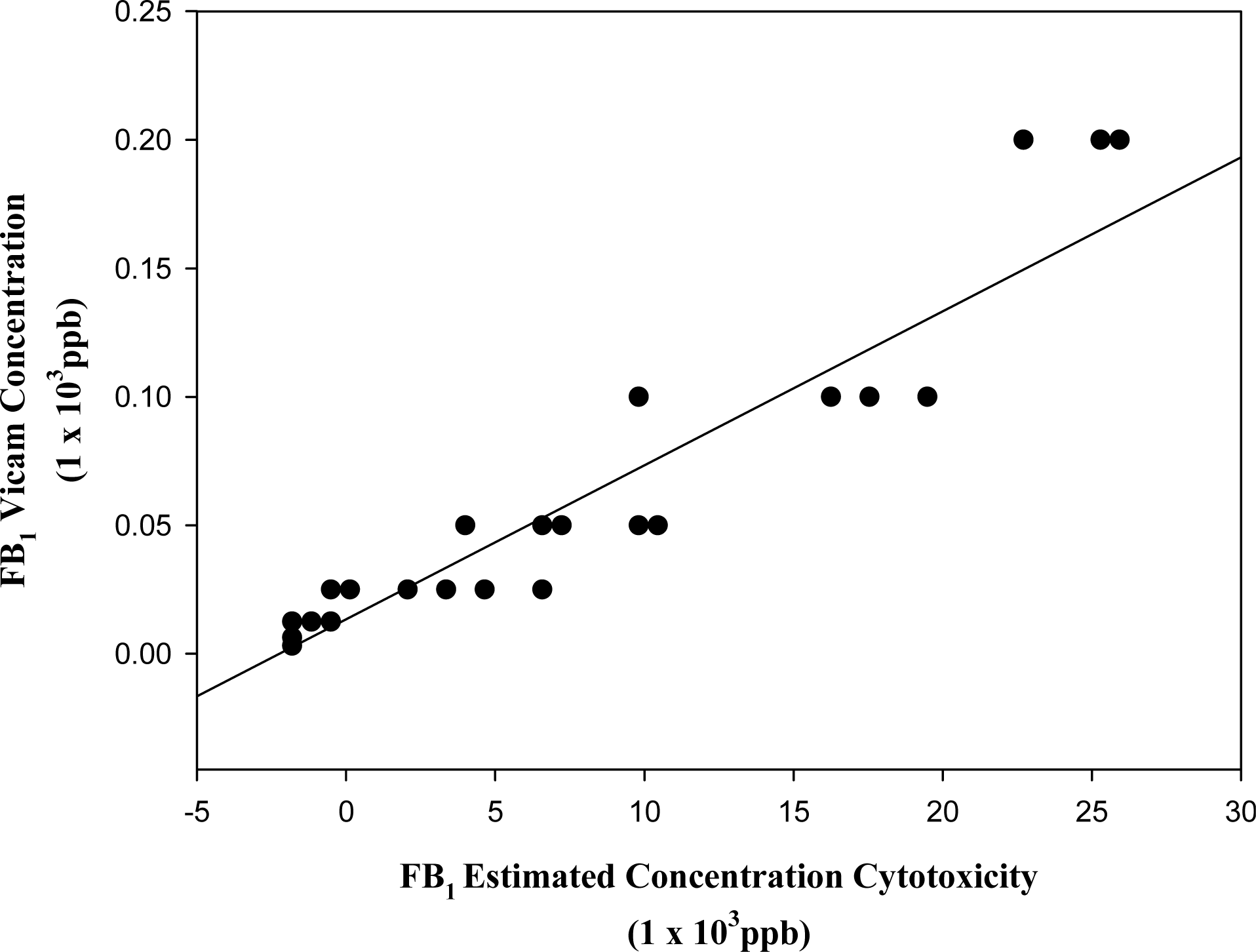
3.3. Estimation of toxin concentration in barley fractions using the cytotoxicity test
4. Conclusion
Acknowledgments
References
- Yu, JH; Keller, N. Regulation of secondary metabolism in filamentous fungi. Annu. Rev. Phytopathol 2005, 43, 437–458. [Google Scholar]
- Cole, RJ; Cox, RH. Handbook of Toxic Fungal Metabolites; Academic Press: New York, 1981. [Google Scholar]
- Frisvad, JC; Thrane, U; Samson, RA; Pitt, JI. Important mycotoxins and the fungi which produce them. Adv. Exp. Med. Biol 2006, 571, 3–31. [Google Scholar]
- Prelusky, DB; Rotter, BA; Rotter, RG. Toxicology of mycotoxins. In Mycotoxins in Grain. Compounds Other Than Aflatoxins; Miller, JD, Trenholm, HL, Eds.; Eagan Press: St. Paul, MN, USA, 1994; pp. 359–404. [Google Scholar]
- Veldman, V; Meijst, JAC; Borgreve, J; Heers-Van der Tol, JJ. Carry over of aflatoxin from cow’s food to milk. Anim. Prod 1992, 55, 163–168. [Google Scholar]
- Gelderblom, WCA; Jaskiewicz, K; Marasas, WFO; Thiel, PG; Horak, M; Vleggaar, R; Kriek, NPJ. Fumonisins- novel mycotoxins with cancer promoting activity produced by Fusarium moniliforme. Appl. Environ. Microbiol 4, 1806–1811.
- Robb, J; Norval, M. Comparison of cytotoxicity and thin-layer chromatography methods for detection of mycotoxins. Appl. Environ. Microbiol 1983, 46, 948–50. [Google Scholar]
- Cetin, Y; Bullerman, LB. Cytotoxicity of Fusarium mycotoxins to mammalian cell cultures as determined by the MTT assay. Food Chem. Toxicol 2005, 43, 755–764. [Google Scholar]
- Smith, JE; Anderson, JG; Lewis, CW; Murad, YM. Cytotoxic fungal spores in the indoor atmosphere of the damp domestic environment. FEMS Microbiol. Lett 1992, 100, 337–343. [Google Scholar]
- Widestrand, J; Lundh, T; Pettersson, H; Lindberg, JE. A rapid and sensitive cytotoxicity screening assay for trichothecenes in cereal samples. Food Chem. Toxicol 2003, 41, 1307–1313. [Google Scholar]
- Hanelt, M; Garies, M; Kollarczik, B. Cytotoxicity of mycotoxins evaluated by the MTT-cell culture assay. Mycopathology 1994, 128, 167–174. [Google Scholar]
- Patterson, DSP; Roberts, BA. Mycotoxins in animal feedstuffs: Sensitive thin layer chromatographic detection of aflatoxin, ochratoxin A, sterigmatocystin, zearalenone and T2 toxin. J. Assoc. Off.. Anal. Chem 1979, 62, 1265–1267. [Google Scholar]
- Boyum, A. Isolation of mononuclear cells and granulocytes from human blood. J. Soc. Clin. Labs 1968, 21, 77–78. [Google Scholar]
- Sydenham, EW; Shephard, GS; Thiel, PG. Liquid chromatographic determination of fumonisins B1 B2 and B3 in foods and feeds. J. Assoc. Off. Anal. Chem. Internat 1992, 75, 313–318. [Google Scholar]
- Eke, Z; Torkos, KN. N-dimethyl-trimethylsilyl-carbamate as derivatizing agent in gas chromatography. Microchem. J 2003, 44, 43–46. [Google Scholar]
- Myburg, RB; Dutton, MF; Chuturgoon, AA. Cytotoxicity of fumonisin B1, diethylnitrosamine and catechol on the SNO oesophageal cancer cell line. Environ. Health Perspec 2002, 110, 813–815. [Google Scholar]
- Wang, E; Norred, WP; Bacon, CW; Riley, RT; Merrill, AH. Inhibition of sphingolipid biosynthesis by fumonisins: Implications for diseases associated with Fusarium moniliforme. J. Biol. Chem 1991, 266, 14486–14490. [Google Scholar]
- Riley, RT; Wang, E; Schroeder, JJ; Smith, ER; Plattner, RD; Abbas, H; Yoo, HS; Merrill, AH. Evidence for disruption of sphingolipid metabolism as a contributing factor in the toxicity and carcinogenicity of fumonisins. Nat. Tox 1996, 4, 3–15. [Google Scholar]
- Rheeder, IP; Marasas, WFO; Thiel, PG; Sydenham, EW; Shephard, GS; Van Schalkwyk, DJ. Fusarium moniliforme and fumonisins in corn in relation to human esophageal cancer in Transkei. Pathophysiology 1992, 82, 353–357. [Google Scholar]
- Pestka, JJ; Bondy, GS. Alteration of immune function following dietary mycotoxin exposure. Can. J. Physiol. Pharmacol 1990, 66, 1009–1016. [Google Scholar]
- Rotter, BA; Prelusky, DB; Pestka, JJ. Toxicology of deoxynivalenol (vomitoxin). J. Toxicol. Environ. Health 1996, 48, 1–34. [Google Scholar]
- Visconti, A; Minervini, F; Lucivero, G; Gambatesa, V. Cytotoxic and immune-effects of Fusarium mycotoxins using rapid colorimetric bioassay. Mycopathol 1991, 113, 181–186. [Google Scholar]
- Bungar, J; Westphal, G; Monnich, A; Hhinnendahl, B; Hallier, E; Muller, M. 2004 Cytotoxicity of occupationally and environmentally relevant mycotoxins. Toxicol 2004, 202, 199–211. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Cytotoxicity* ppb | FB1 ppb | DON ppb | OTA ppb |
|---|---|---|---|---|
| B4 N fraction | 25,000 | n.d. | 4,800 | − |
| B4 A fraction | >50 | − | − | 4 |
| B7 A fraction | >50 | − | − | 1 |
| B9 A fraction | 50 | − | − | 2 |
| B10 N fraction | 6,250 | n.d. | n.d. | − |
| B10 A fraction | >50 | − | − | 6 |
| B11 N fraction | ||||
| FB1 fraction | 50,000 | 40 | 900 | − |
| B11 A fraction | >50 | − | − | 4 |
| B14 N & A fractions | n.d. | n.d. | n.d. | n.d. |
| B21 N fraction | 20,000 | n.d. | 7,500 | − |
| FB1 fraction | ||||
| B30 | 18,000 | n.d. | 7,500 | − |
| B31 N&A fractions | n.d. | n.d. | n.d. | n.d. |
| MB5 N&A fractions | >50,000 | n.d. | n.d. | − |
| MB24 | ||||
| N fraction | >50,000 | 71 | 6,200 | − |
| FB1 fraction | ||||
| PB4 A fraction | 20 | − | − | 2 |
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maenetje, P.W.; De Villiers, N.; Dutton, M.F. The Use of Isolated Human Lymphocytes in Mycotoxin Cytotoxicity Testing. Int. J. Mol. Sci. 2008, 9, 1515-1526. https://doi.org/10.3390/ijms9081515
Maenetje PW, De Villiers N, Dutton MF. The Use of Isolated Human Lymphocytes in Mycotoxin Cytotoxicity Testing. International Journal of Molecular Sciences. 2008; 9(8):1515-1526. https://doi.org/10.3390/ijms9081515
Chicago/Turabian StyleMaenetje, Pholo W., Neil De Villiers, and Mike F. Dutton. 2008. "The Use of Isolated Human Lymphocytes in Mycotoxin Cytotoxicity Testing" International Journal of Molecular Sciences 9, no. 8: 1515-1526. https://doi.org/10.3390/ijms9081515
APA StyleMaenetje, P. W., De Villiers, N., & Dutton, M. F. (2008). The Use of Isolated Human Lymphocytes in Mycotoxin Cytotoxicity Testing. International Journal of Molecular Sciences, 9(8), 1515-1526. https://doi.org/10.3390/ijms9081515