Impacts of Elevated CO2 Levels on the Soil Bacterial Community in a Natural CO2-Enhanced Oil Recovery Area




Abstract
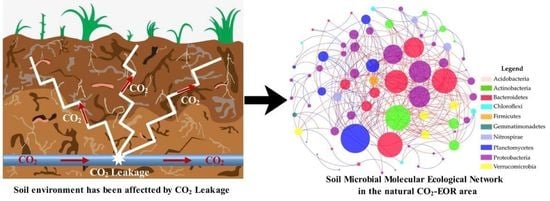
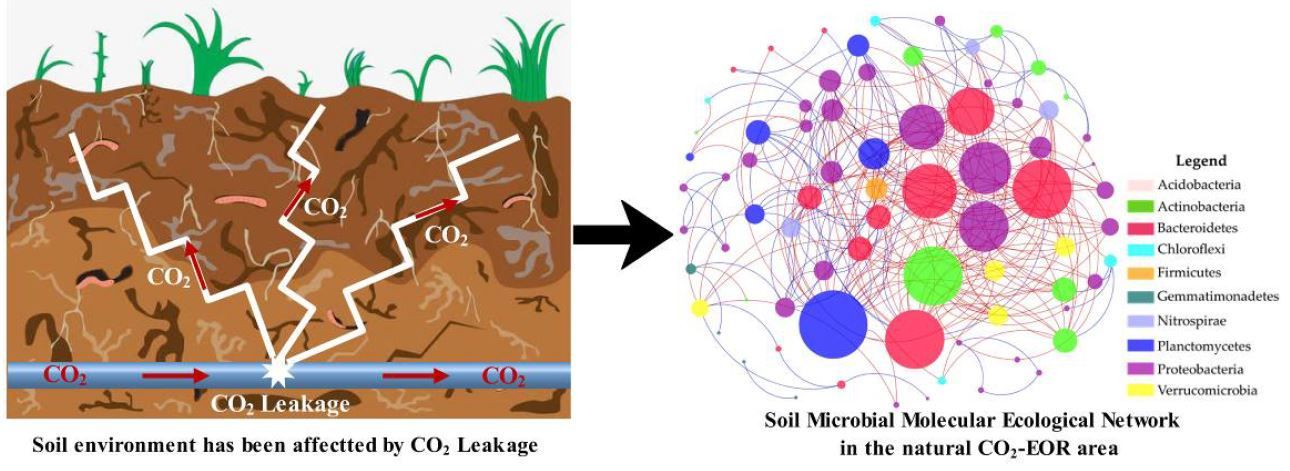
:
1. Introduction
2. Materials and Methods
2.1. Study Sites, Soil Sampling, and Measurment
2.2. DNA Extraction, PCR Amplification, and ILLUMINA MiSeq Sequencing
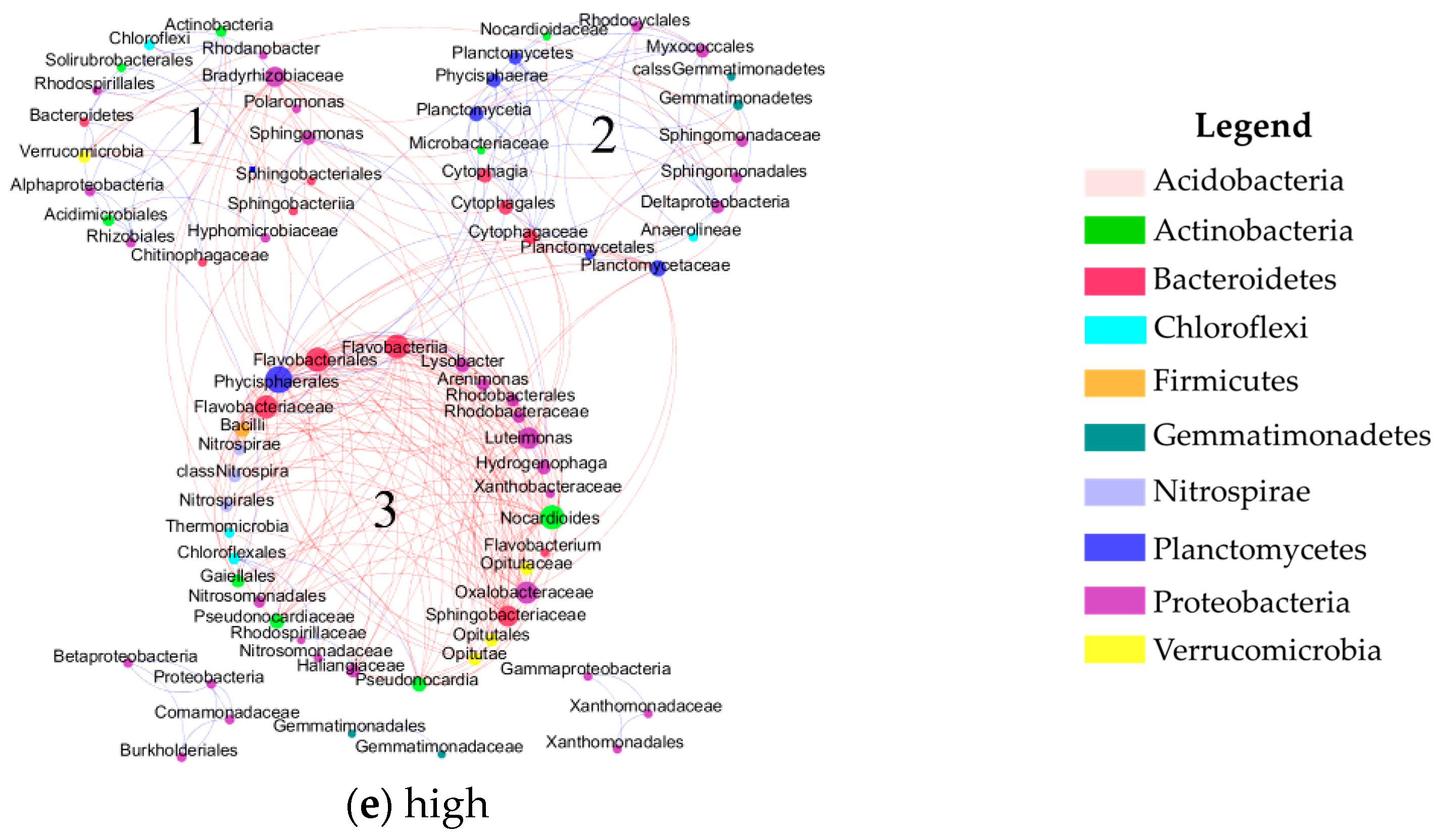
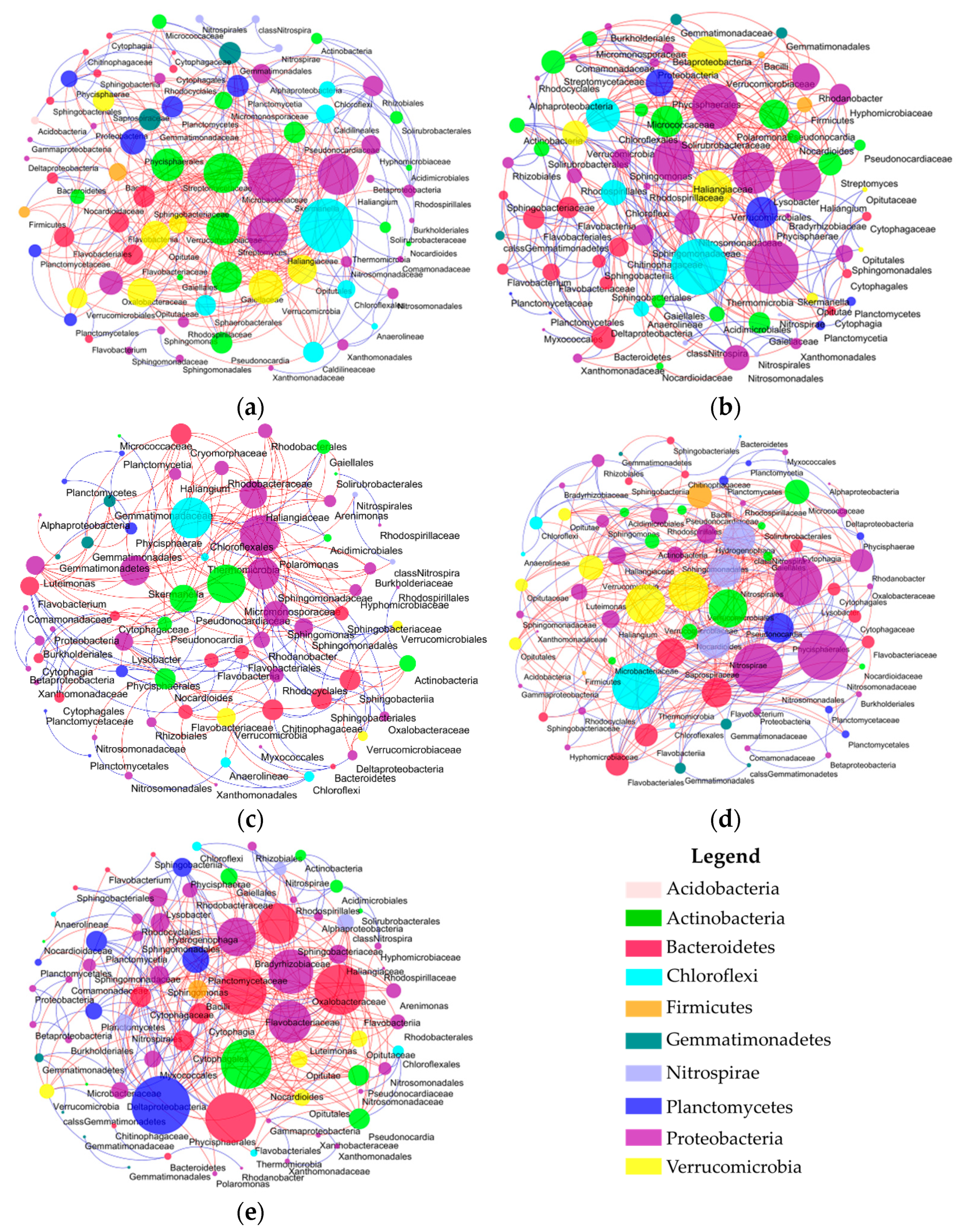
2.3. Network Construction and Analysis
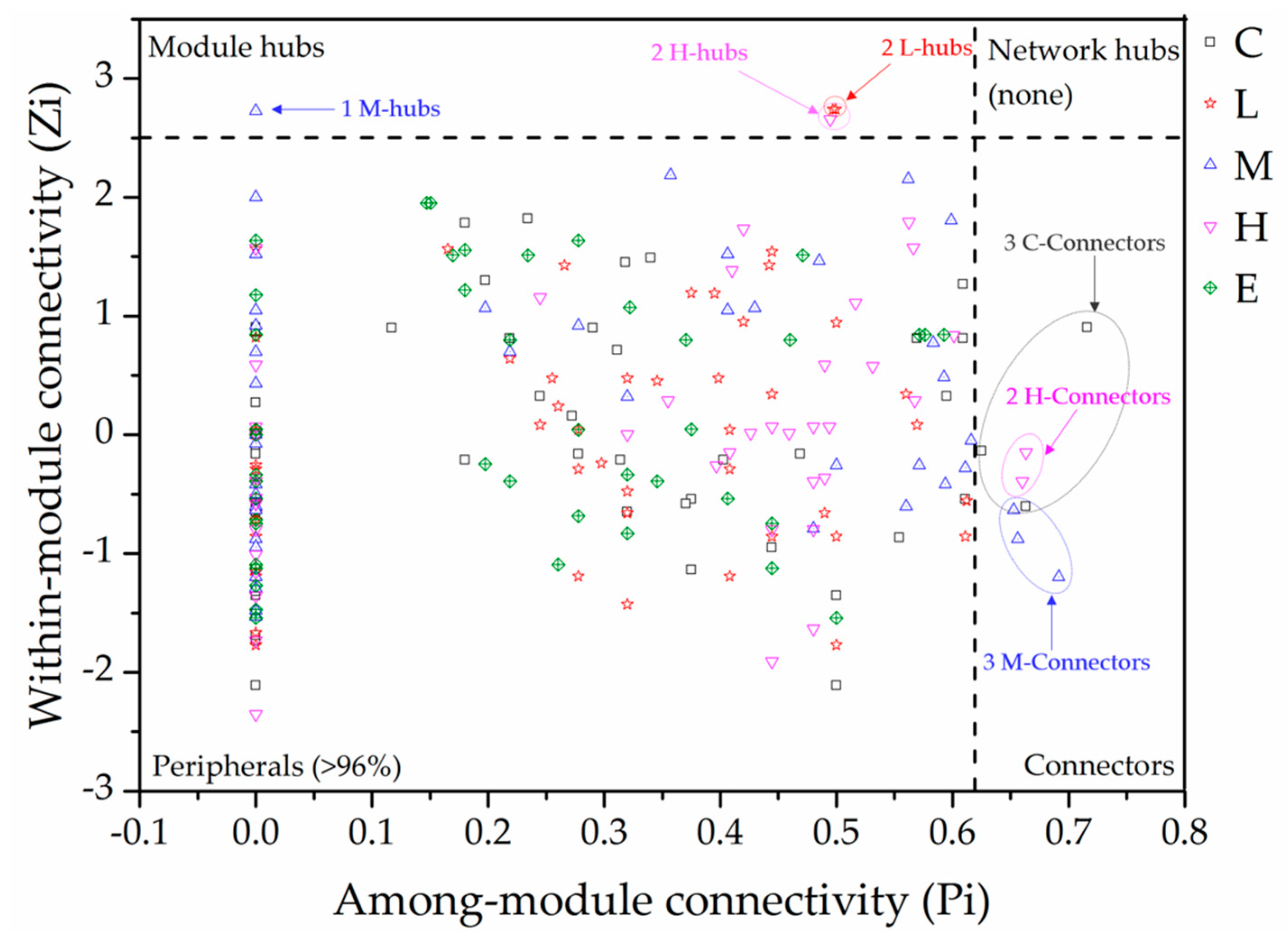
2.4. Characterization of the Molecular Ecological Networks and Statistical Analysis
3. Results
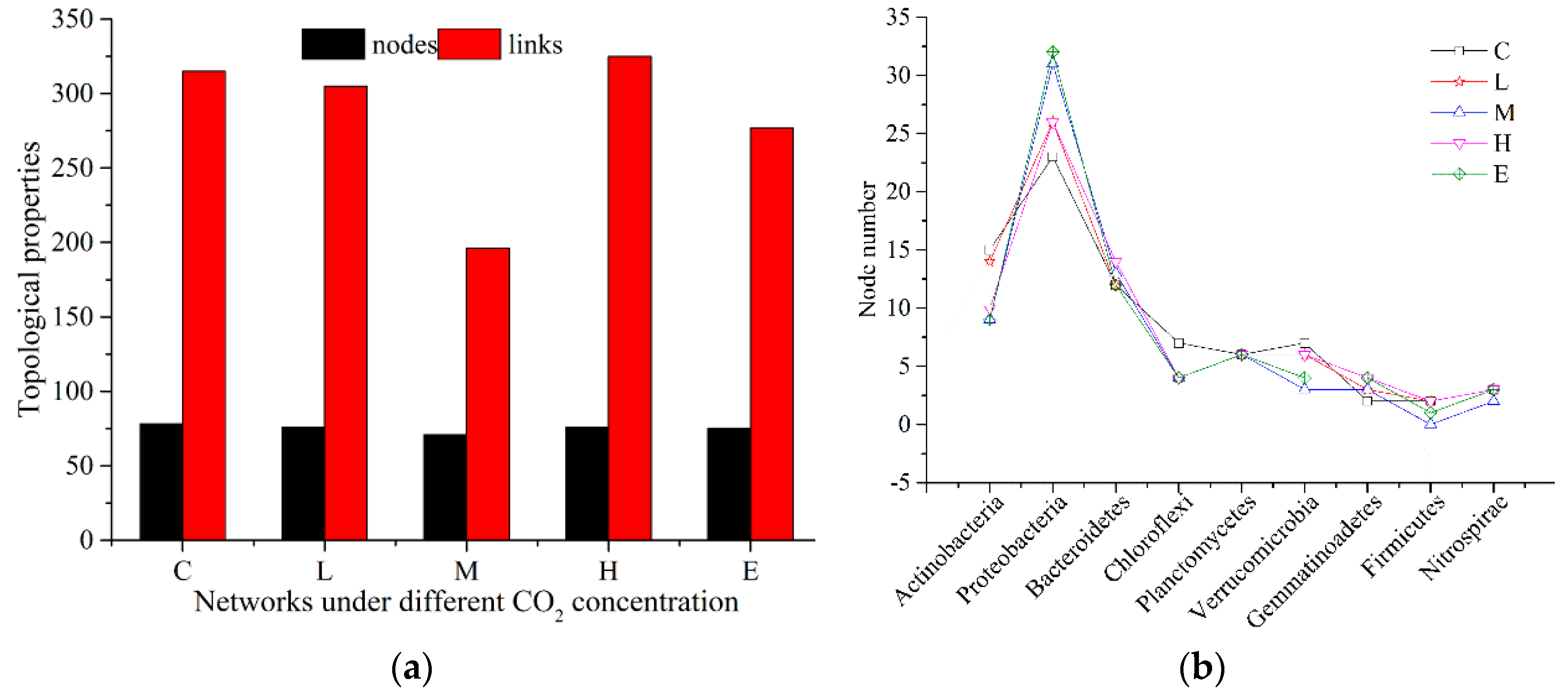
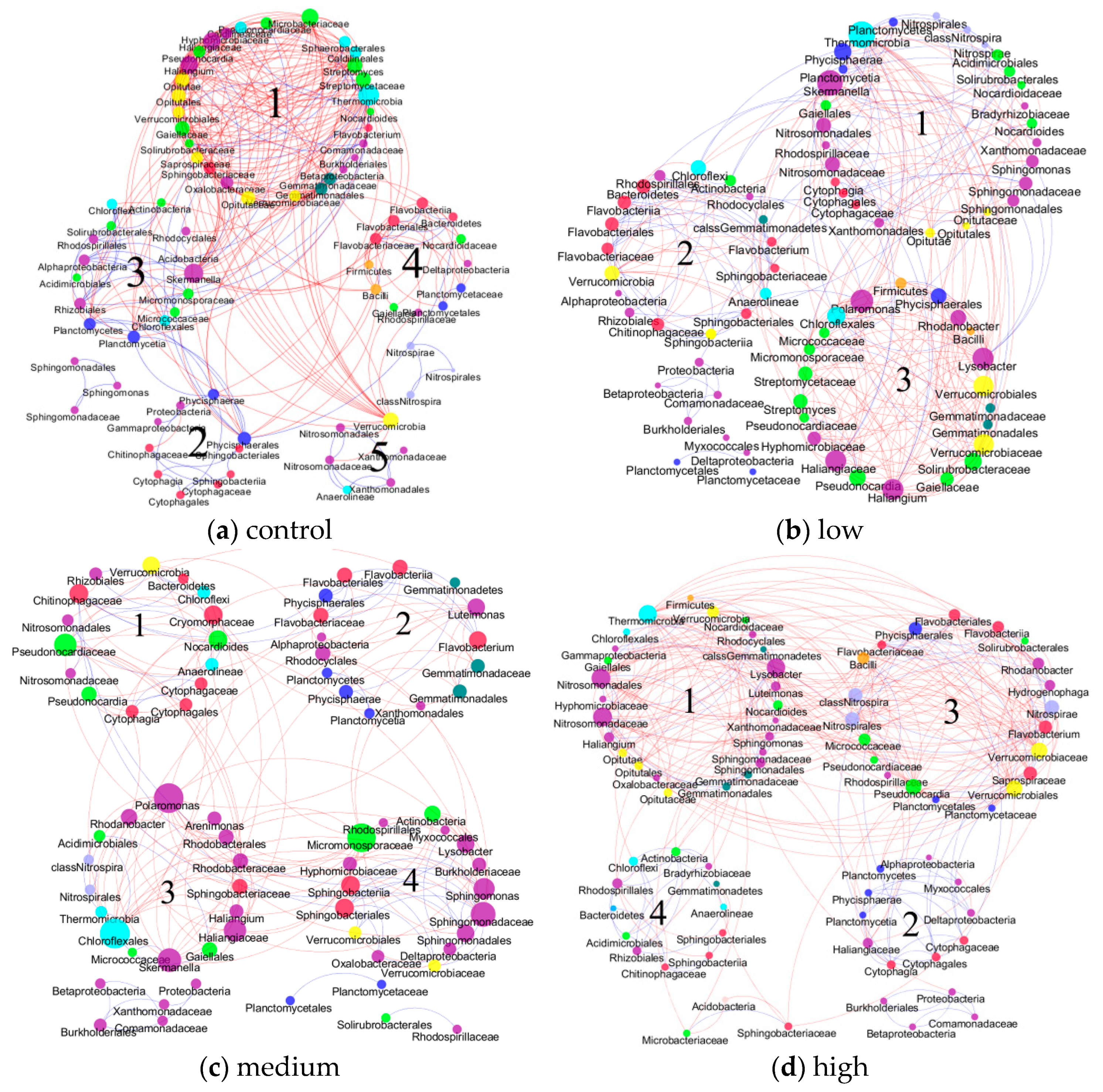
3.1. Topological Properties of Molecular Ecological Networks (MENs) under Different CO2 Leakage Concentrations
3.2. Dominant Microbial Taxa under Different CO2 Leakage Gradient
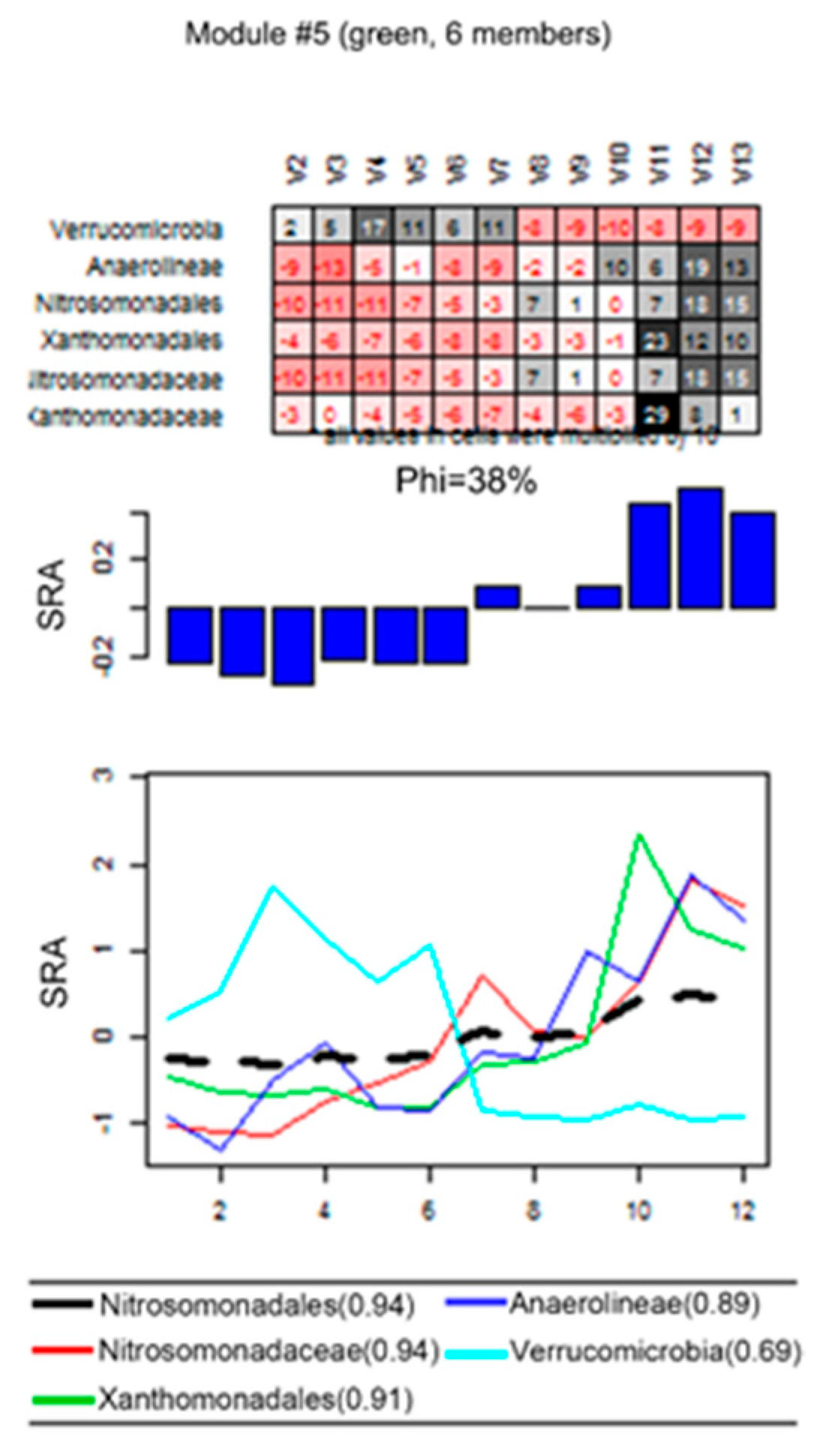
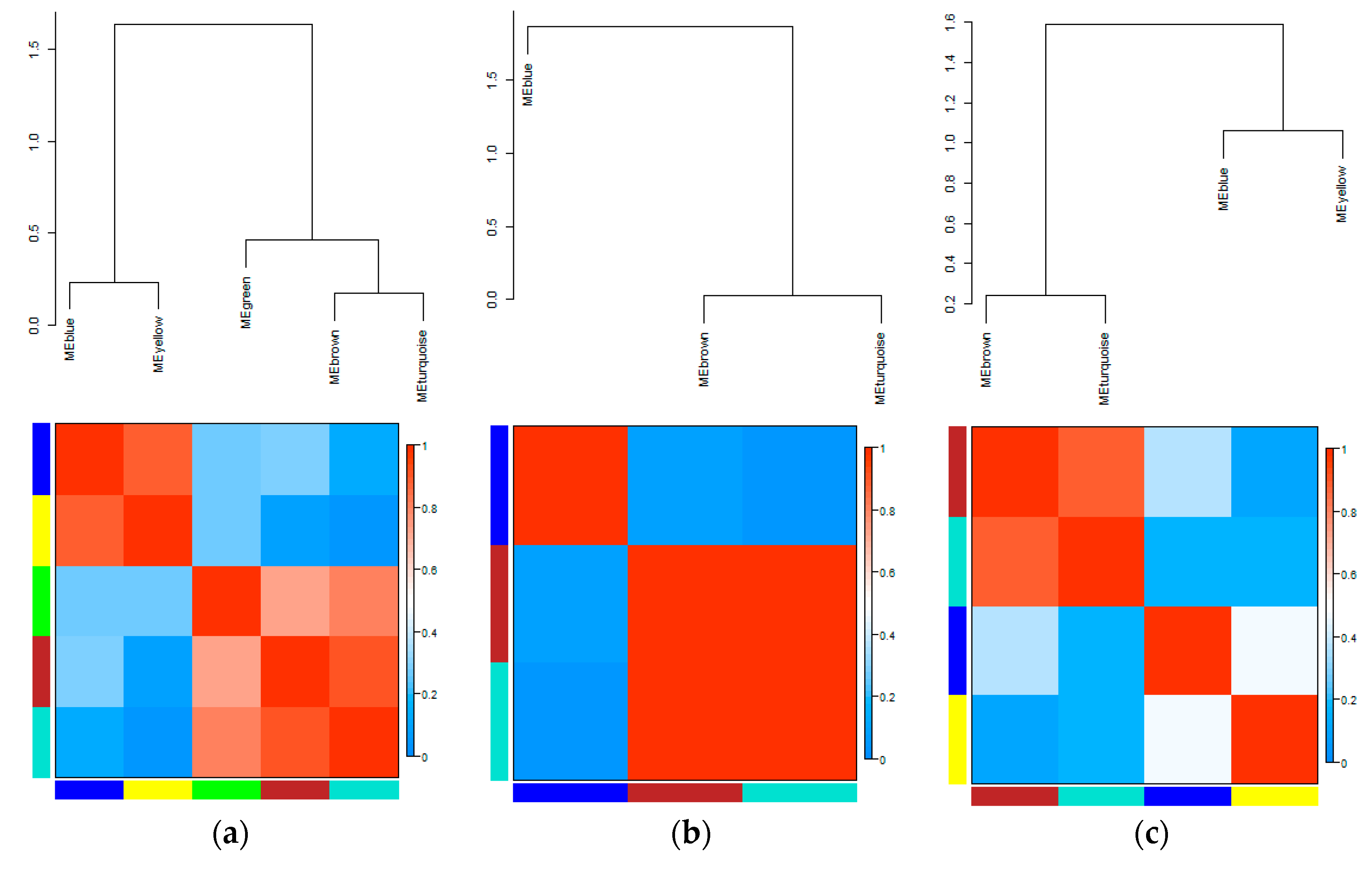
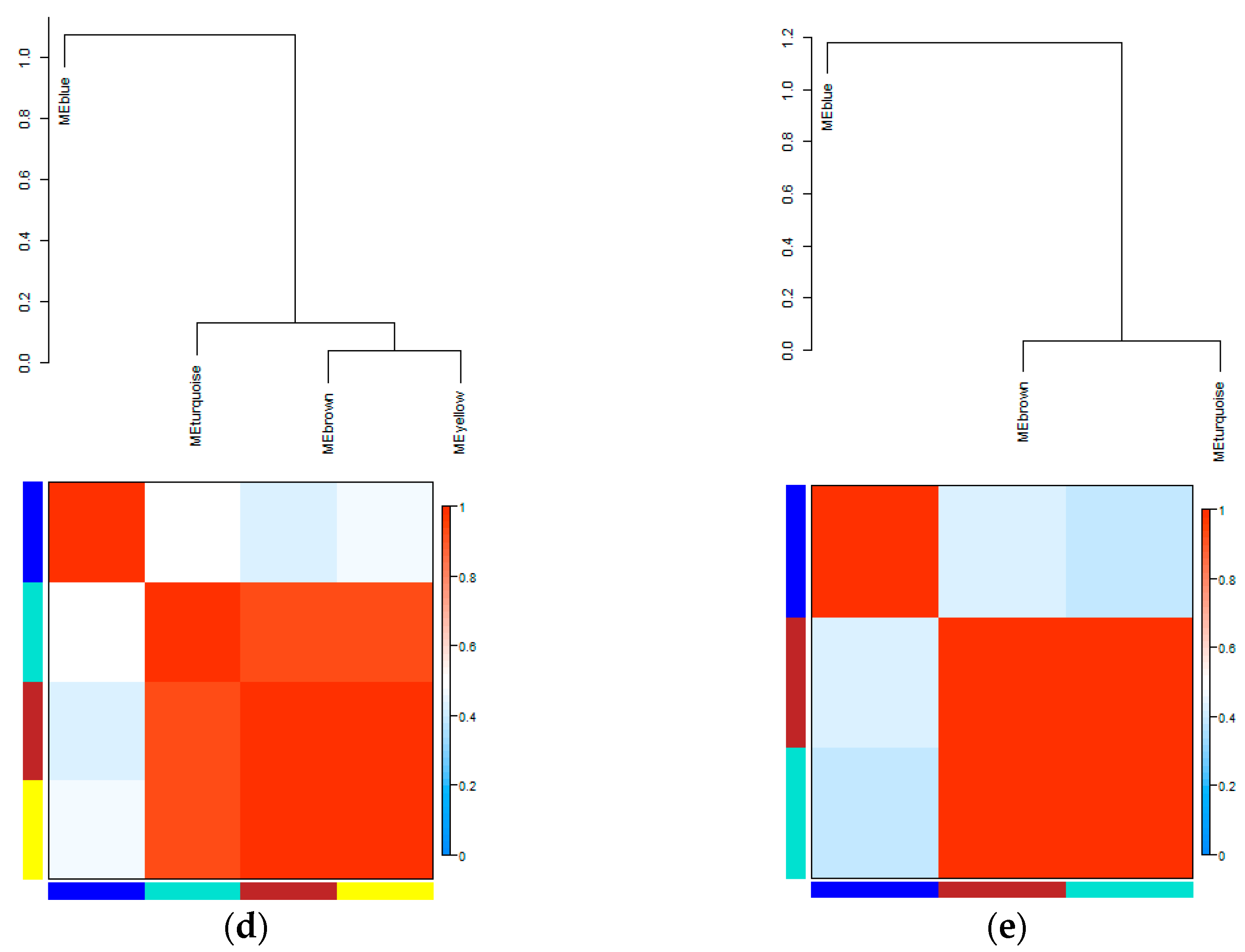
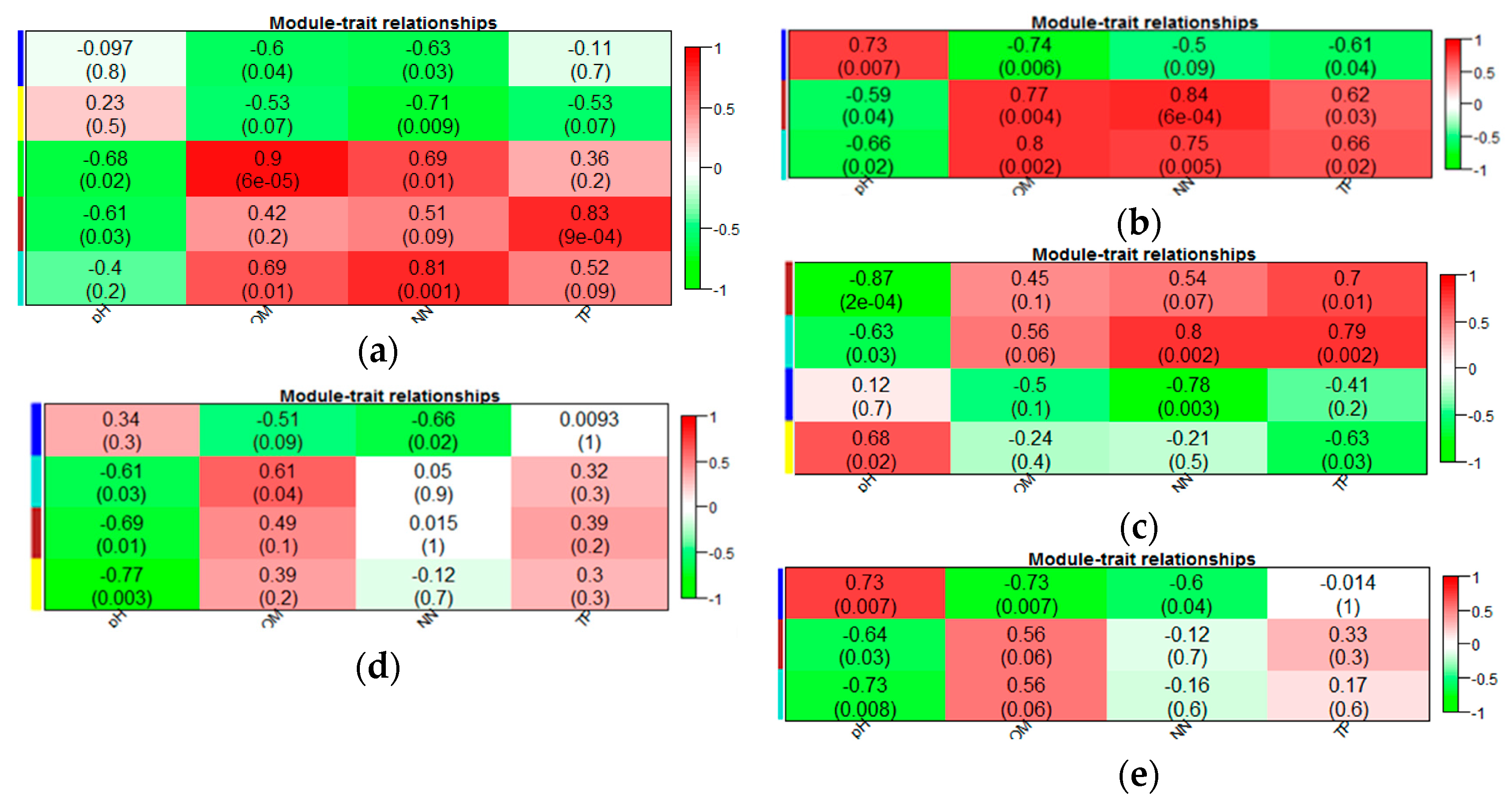
3.3. Eigengene Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- IPCC. Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2014; pp. 18–28. [Google Scholar]
- Chen, F.; Zhang, W.; Ma, J.; Yang, Y.; Zhang, S.; Chen, R. Experimental study on the effects of underground CO2 leakage on soil microbial consortia. Int. J. Greenh. Gas Control 2017, 63, 241–248. [Google Scholar] [CrossRef]
- Zhou, X.; Apple, M.E.; Dobeck, L.M.; Cunningham, A.B.; Spangler, L.H. Observed response of soil O2 concentration to leaked CO2 from an engineered CO2 leakage experiment. Int. J. Greenh. Gas Control 2013, 16, 116–128. [Google Scholar] [CrossRef]
- Chen, F.; Yang, Y.; Ma, Y.; Hou, H.; Zhang, S.; Ma, J. Effects of CO2 leakage on soil bacterial communities from simulated CO2-EOR areas. Environ. Sci. Process. Impacts 2016, 18, 547–554. [Google Scholar] [CrossRef]
- Herbert, E.R.; Boon, P.; Burgin, A.J.; Neubauer, S.C.; Franklin, R.B.; Hopfensperger, K.N.; Lamers, L.P.M.; Gell, P.; Ardón, M. A global perspective on wetland salinization: Ecological consequences of a growing threat to freshwater wetlands. Ecosphere 2016, 6, 1–43. [Google Scholar] [CrossRef]
- Li, Q.; Song, R.; Liu, X.; Liu, G.; Sun, Y. Monitoring of Carbon Dioxide Geological Utilization and Storage in China: A Review. Acid Gas Extr. Dispos. Relat. Top. 2016, 33, 1–358. [Google Scholar] [CrossRef]
- Sáenz de Miera, L.E.; Arroyo, P.; de Luis Calabuig, E.; Falagán, J.; Ansola, G. High-throughput sequencing of 16S RNA genes of soil bacterial communities from a naturally occurring CO2 gas vent. Int. J. Greenh. Gas Control 2014, 29, 176–184. [Google Scholar] [CrossRef]
- Ko, D.; Yoo, G.; Yun, S.-T.; Chung, H. Impacts of CO2 leakage on plants and microorganisms: A review of results from CO2 release experiments and storage sites. Greenh. Gases Sci. Technol. 2016, 6, 319–338. [Google Scholar] [CrossRef]
- Li, Q.; Liu, G. Risk Assessment of the Geological Storage of CO2: A Review. In Geologic Carbon Sequestration: Understanding Reservoir Behavior; Vishal, V., Singh, T.N., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 249–284. [Google Scholar]
- Li, Q.; Liu, G.; Cai, B.; Leamon, G.; Liu, L.-C.; Chen, Z.-A.; Li, X. Public awareness of the environmental impact and management of carbon dioxide capture, utilization and storage technology: The views of educated people in China. Clean Technol. Environ. Policy 2017, 19, 2041–2056. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, W.; Zhang, S.; Zhu, Q.; Feng, Q.; Chen, F. Short-term effects of CO2 leakage on the soil bacterial community in a simulated gas leakage scenario. PeerJ 2017, 5, e4024. [Google Scholar] [CrossRef]
- Drigo, B.; Kowalchuk, G.A.; van Veen, J.A. Climate change goes underground: Effects of elevated atmospheric CO2 on microbial community structure and activities in the rhizosphere. Biol. Fertil. Soils 2008, 44, 667–679. [Google Scholar] [CrossRef]
- Eisenhauer, N.; Cesarz, S.; Koller, R.; Worm, K.; Reich, P.B. Global change belowground: Impacts of elevated CO2, nitrogen, and summer drought on soil food webs and biodiversity. Glob. Chang. Biol. 2012, 18, 435–447. [Google Scholar] [CrossRef]
- Banerjee, S.; Kirkby, C.A.; Schmutter, D.; Bissett, A.; Kirkegaard, J.A.; Richardson, A.E. Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 2016, 97, 188–198. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef]
- Ruan, Q.; Dutta, D.; Sun, F.; Fuhrman, J.A.; Steele, J.A.; Schwalbach, M.S. Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics 2006, 22, 2532–2538. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Tu, Q.; Zhi, X. Functional Molecular Ecological Networks. mBio 2010, 1, e00169-10. [Google Scholar] [CrossRef]
- Krueger, M.; Jones, D.; Frerichs, J.; Oppermann, B.I.; West, J.; Coombs, P.; Green, K.; Barlow, T.; Lister, R.; Shaw, R.; et al. Effects of elevated CO2 concentrations on the vegetation and microbial populations at a terrestrial CO2 vent at Laacher See, Germany. Int. J. Greenh. Gas Control 2011, 5, 1093–1098. [Google Scholar] [CrossRef]
- Beaubien, S.E.; Ciotoli, G.; Coombs, P.; Dictor, M.C.; Krueger, M.; Lombardi, S.; Pearce, J.M.; West, J.M. The impact of a naturally occurring CO2 gas vent on the shallow ecosystem and soil chemistry of a Mediterranean pasture (Latera, Italy). Int. J. Greenh. Gas Control 2008, 2, 373–387. [Google Scholar] [CrossRef]
- Oesterreicher-Cunha, P.; Molinaro, B.S.; Feijo, I.V.A.; Vargas, E.A., Jr.; Guimaraes, J.R.D. Experimental evaluation of CO2 percolation effects on subsurface soil microbiota. Int. J. Greenh. Gas Control 2015, 32, 135–146. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, H.; Wang, J.; Liu, X.; Cheng, K.; Li, L.; Zheng, J.; Zhang, X.; Zheng, J.; Pan, G. Short-term response of nitrifier communities and potential nitrification activity to elevated CO2 and temperature interaction in a Chinese paddy field. Appl. Soil Ecol. 2015, 96, 88–98. [Google Scholar] [CrossRef]
- Beulig, F.; Urich, T.; Nowak, M.; Trumbore, S.E.; Gleixner, G.; Gilfillan, G.D.; Fjelland, K.E.; Küsel, K. Altered carbon turnover processes and microbiomes in soils under long-term extremely high CO2 exposure. Nat. Microbiol. 2016, 1, 15025. [Google Scholar] [CrossRef]
- Fernández-Montiel, I.; Touceda, M.; Pedescoll, A.; Gabilondo, R.; Prieto-Fernández, A.; Bécares, E. Short-term effects of simulated below-ground carbon dioxide leakage on a soil microbial community. Int. J. Greenh. Gas Control 2015, 36, 51–59. [Google Scholar] [CrossRef]
- Sáenz de Miera, L.E.; Arroyo, P.; de Luis Calabuig, E.; Ansola, G. Effects of varying CO2 flows on bacterial communities in mesocosms created from two soils. Int. J. Greenh. Gas Control 2016, 46, 205–214. [Google Scholar] [CrossRef]
- Tait, K.; Stahl, H.; Taylor, P.; Widdicombe, S. Rapid response of the active microbial community to CO2 exposure from a controlled sub-seabed CO2 leak in Ardmucknish Bay (Oban, Scotland). Int. J. Greenh. Gas Control 2015, 38, 171–181. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2011, 6, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic Molecular Ecological Network of Soil Microbial Communities in Response to Elevated CO2. MBio 2011, 2, e00122-11. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.E.; Ward, C.S. A network-based approach to disturbance transmission through microbial interactions. Front. Microbiol. 2015, 6, 1182. [Google Scholar] [CrossRef]
- Morriën, E.; Hannula, S.E.; Snoek, L.B.; Helmsing, N.R.; Zweers, H.; de Hollander, M.; Soto, R.L.; Bouffaud, M.-L.; Buée, M.; Dimmers, W.; et al. Soil networks become more connected and take up more carbon as nature restoration progresses. Nat. Commun. 2017, 8, 14349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Zhang, P.; Qin, Y.; Tu, Q.; Yang, Y.; He, Z.; Schadt, C.W.; Zhou, J. Network succession reveals the importance of competition in response to emulsified vegetable oil amendment for uranium bioremediation. Environ. Microbiol. 2016, 18, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, X.; Han, X.; Deng, Y. Higher precipitation strengthens the microbial interactions in semi-arid grassland soils. Glob. Ecol. Biogeogr. 2018, 27, 570–580. [Google Scholar] [CrossRef]
- Jiang, X.; Takacs-Vesbach, C.D. Microbial community analysis of pH 4 thermal springs in Yellowstone National Park. Extremophiles 2017, 21, 135–152. [Google Scholar] [CrossRef]
- Van Horn, D.J.; Wolf, C.R.; Colman, D.R.; Jiang, X.; Kohler, T.J.; McKnight, D.M.; Stanish, L.F.; Yazzie, T.; Takacs-Vesbach, C.D. Patterns of bacterial biodiversity in the glacial meltwater streams of the McMurdo Dry Valleys, Antarctica. FEMS Microbiol. Ecol. 2016, 92, fiw148. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef]
- Killcoyne, S.; Carter, G.W.; Smith, J.; Boyle, J. Cytoscape: A community-based framework for network modeling. Methods Mol. Biol. (Clifton N.J.) 2009, 563, 219–239. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of ‘small-world’ networks. Nature 1998, 393, 440–442. [Google Scholar] [CrossRef]
- Alon, U. Biological Networks: The Tinkerer as an Engineer. Science 2003, 301, 1866–1867. [Google Scholar] [CrossRef]
- Olesen, J.M.; Bascompte, J.; Dupont, Y.L.; Jordano, P. The modularity of pollination networks. Proc. Natl. Acad. Sci. USA 2007, 104, 19891–19896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling Interactions in the Microbiome: A Network Perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Scherer-Lorenzen, M.; Bauhus, J. Belowground facilitation and competition in young tree species mixtures. For. Ecol. Manag. 2012, 265, 191–200. [Google Scholar] [CrossRef]
- Oliver, T.H.; Heard, M.S.; Isaac, N.J.B.; Roy, D.B.; Procter, D.; Eigenbrod, F.; Freckleton, R.; Hector, A.; Orme, C.D.L.; Petchey, O.L.; et al. Biodiversity and Resilience of Ecosystem Functions. Trends Ecol. Evol. 2015, 30, 673–684. [Google Scholar] [CrossRef] [Green Version]
- van der Plas, F.; Manning, P.; Allan, E.; Scherer-Lorenzen, M.; Verheyen, K.; Wirth, C.; Zavala, M.A.; Hector, A.; Ampoorter, E.; Baeten, L.; et al. Jack-of-all-trades effects drive biodiversity–ecosystem multifunctionality relationships in European forests. Nat. Commun. 2016, 7, 11109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Brose, U. Biodiversity and ecosystem functioning in food webs: The vertical diversity hypothesis. Ecol. Lett. 2018, 21, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Montiel, I.; Pedescoll, A.; Bécares, E. Microbial communities in a range of carbon dioxide fluxes from a natural volcanic vent in Campo de Calatrava, Spain. Int. J. Greenh. Gas Control 2016, 50, 70–79. [Google Scholar] [CrossRef]
- Xiao Fan, W.; Guanrong, C. Complex networks: Small-world, scale-free and beyond. IEEE Circuits Syst. Mag. 2003, 3, 6–20. [Google Scholar] [CrossRef] [Green Version]
- Kitano, H. Biological robustness. Nat. Rev. Genet. 2004, 5, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Chaffron, S.; Rehrauer, H.; Pernthaler, J.; von Mering, C. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 2010, 20, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-Gonzalez, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Harris, J. Soil Microbial Communities and Restoration Ecology: Facilitators or Followers? Science 2009, 325, 573–574. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network | Index | C | L | M | H | E |
|---|---|---|---|---|---|---|
| Empirical networks | Similarity threshold | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 |
| R2 of power law | 0.5 | 0.594 | 0.568 | 0.352 | 0.636 | |
| avgK | 8.077 | 8.026 | 5.521 | 8.553 | 7.387 | |
| avgCC | 0.487 | 0.570 | 0.452 | 0.548 | 0.536 | |
| GD | 2.915 | 2.841 | 3.068 | 2.942 | 2.773 | |
| Modularity | 0.393 | 0.437 | 0.494 | 0.337 | 0.383 | |
| Random networks | avgCC | 0.235 ± 0.02 | 0.223 ± 0.020 | 0.130 ± 0.021 | 0.299 ± 0.021 | 0.266 ± 0.024 |
| GD | 2.410 ± 0.046 | 2.346 ± 0.036 | 2.655 ± 0.044 | 2.287 ± 0.027 | 2.408 ± 0.049 | |
| Modularity | 0.237 ± 0.009 | 0.244 ± 0.010 | 0.327 ± 0.012 | 0.224 ± 0.008 | 0.247 ± 0.010 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Luo, Z.; Chen, F.; Chen, R.; Zhu, Q.; Zhang, S. Impacts of Elevated CO2 Levels on the Soil Bacterial Community in a Natural CO2-Enhanced Oil Recovery Area. Diversity 2019, 11, 77. https://doi.org/10.3390/d11050077
Ma J, Luo Z, Chen F, Chen R, Zhu Q, Zhang S. Impacts of Elevated CO2 Levels on the Soil Bacterial Community in a Natural CO2-Enhanced Oil Recovery Area. Diversity. 2019; 11(5):77. https://doi.org/10.3390/d11050077
Chicago/Turabian StyleMa, Jing, Zhanbin Luo, Fu Chen, Run Chen, Qianlin Zhu, and Shaoliang Zhang. 2019. "Impacts of Elevated CO2 Levels on the Soil Bacterial Community in a Natural CO2-Enhanced Oil Recovery Area" Diversity 11, no. 5: 77. https://doi.org/10.3390/d11050077
APA StyleMa, J., Luo, Z., Chen, F., Chen, R., Zhu, Q., & Zhang, S. (2019). Impacts of Elevated CO2 Levels on the Soil Bacterial Community in a Natural CO2-Enhanced Oil Recovery Area. Diversity, 11(5), 77. https://doi.org/10.3390/d11050077