Using ISSR Genomic Fingerprinting to Study the Genetic Differentiation of Artemia Leach, 1819 (Crustacea: Anostraca) from Iran and Neighbor Regions with the Focus on the Invasive American Artemia franciscana



Abstract
:1. Introduction
2. Material and Methods
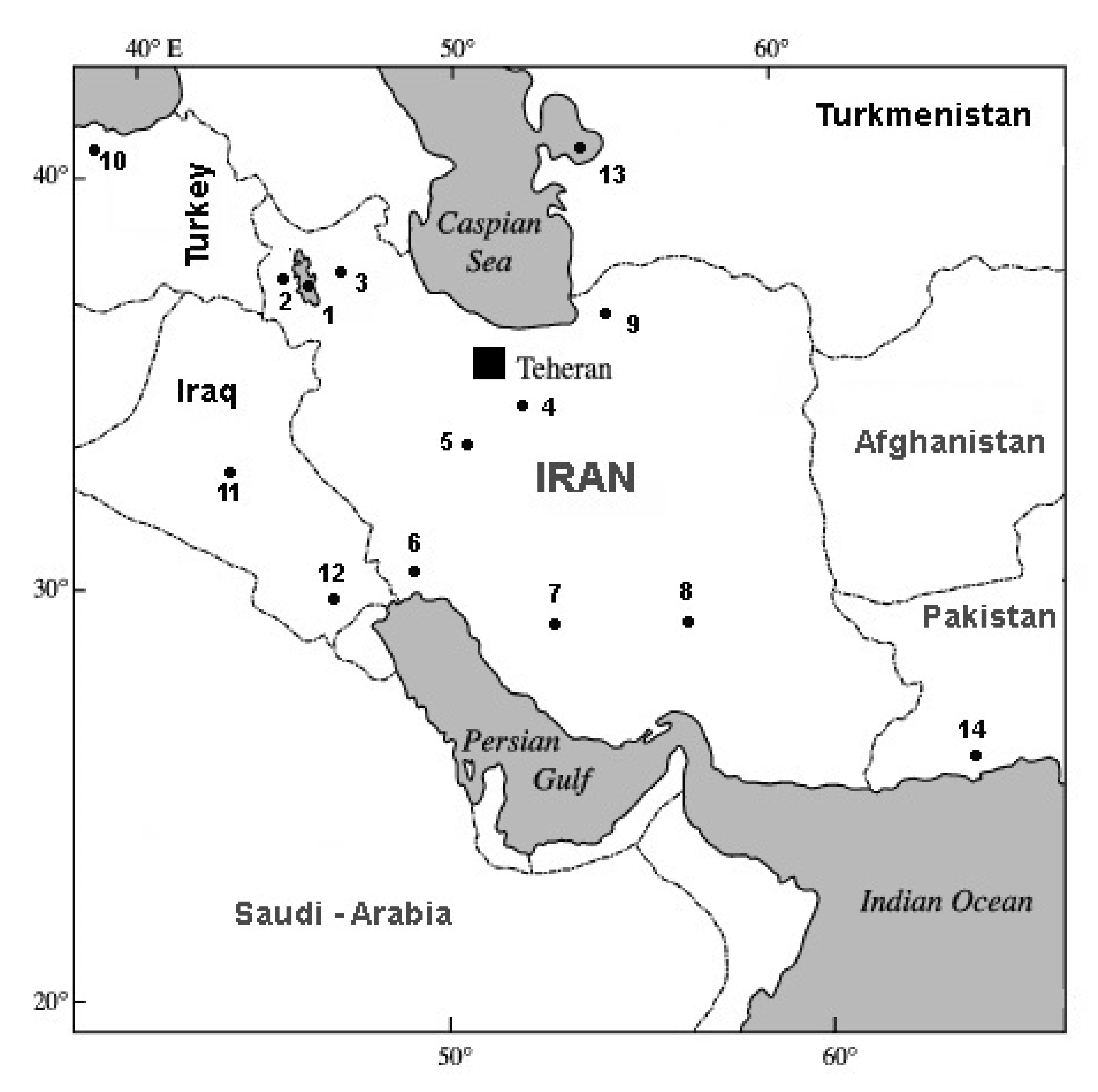
2.1. Sample Collection and DNA Extraction
2.2. Population Identification and Phylogenetic Analyses
2.3. Genomic Fingerprinting by ISSR-PCR
2.4. ISSR Statistics
3. Results
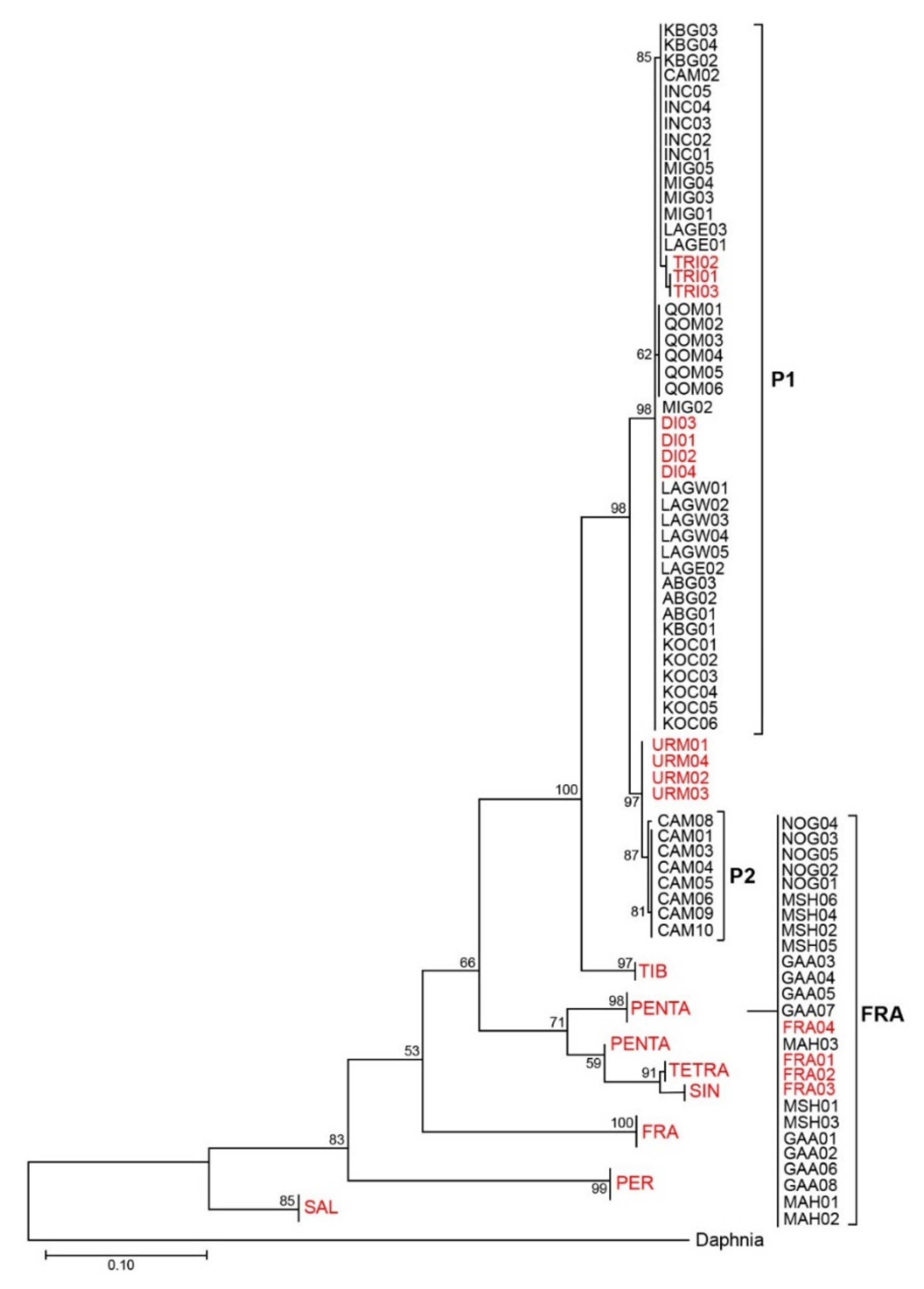
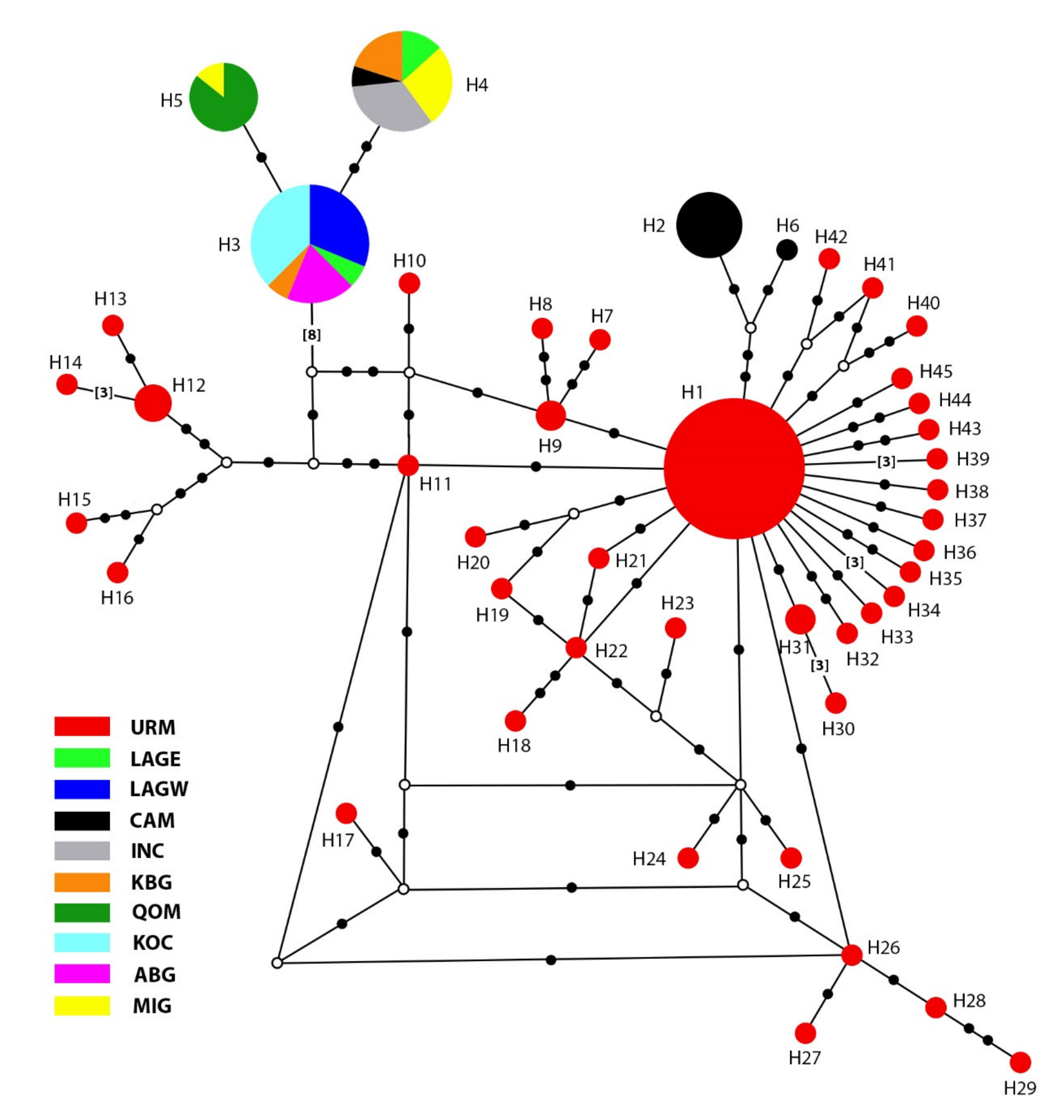
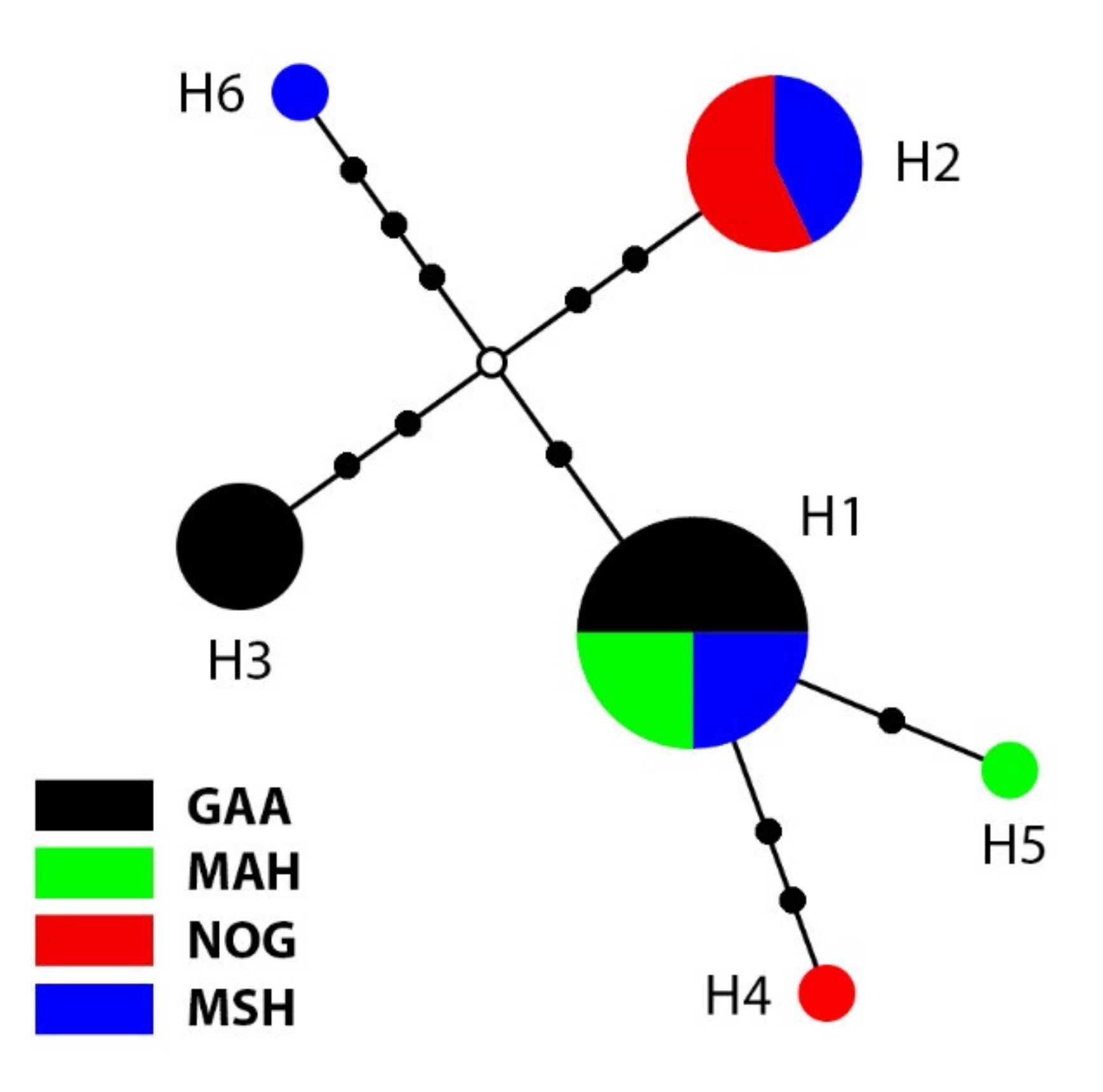
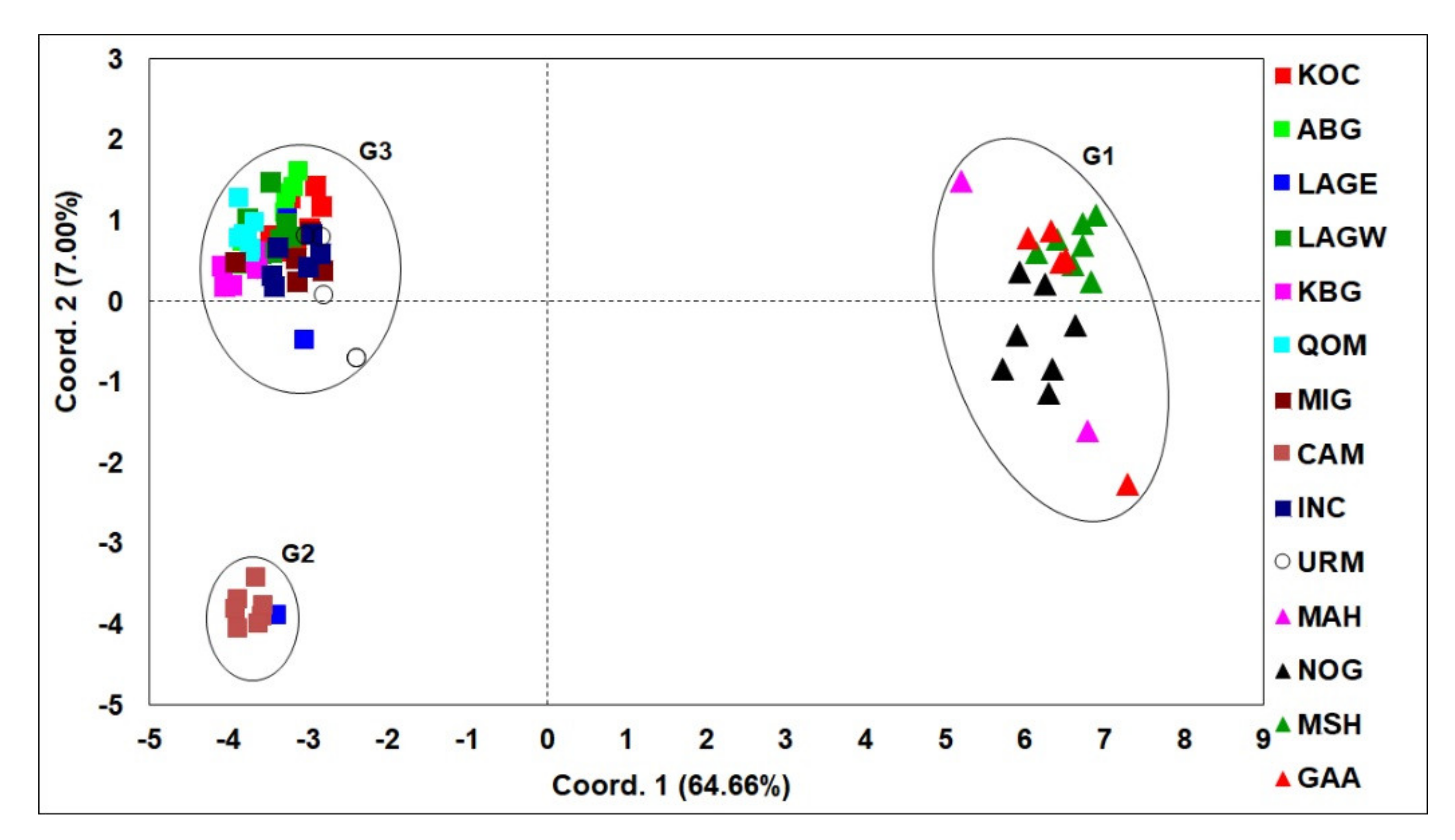
3.1. Phylogenetic Analyses and Haplotype Distribution
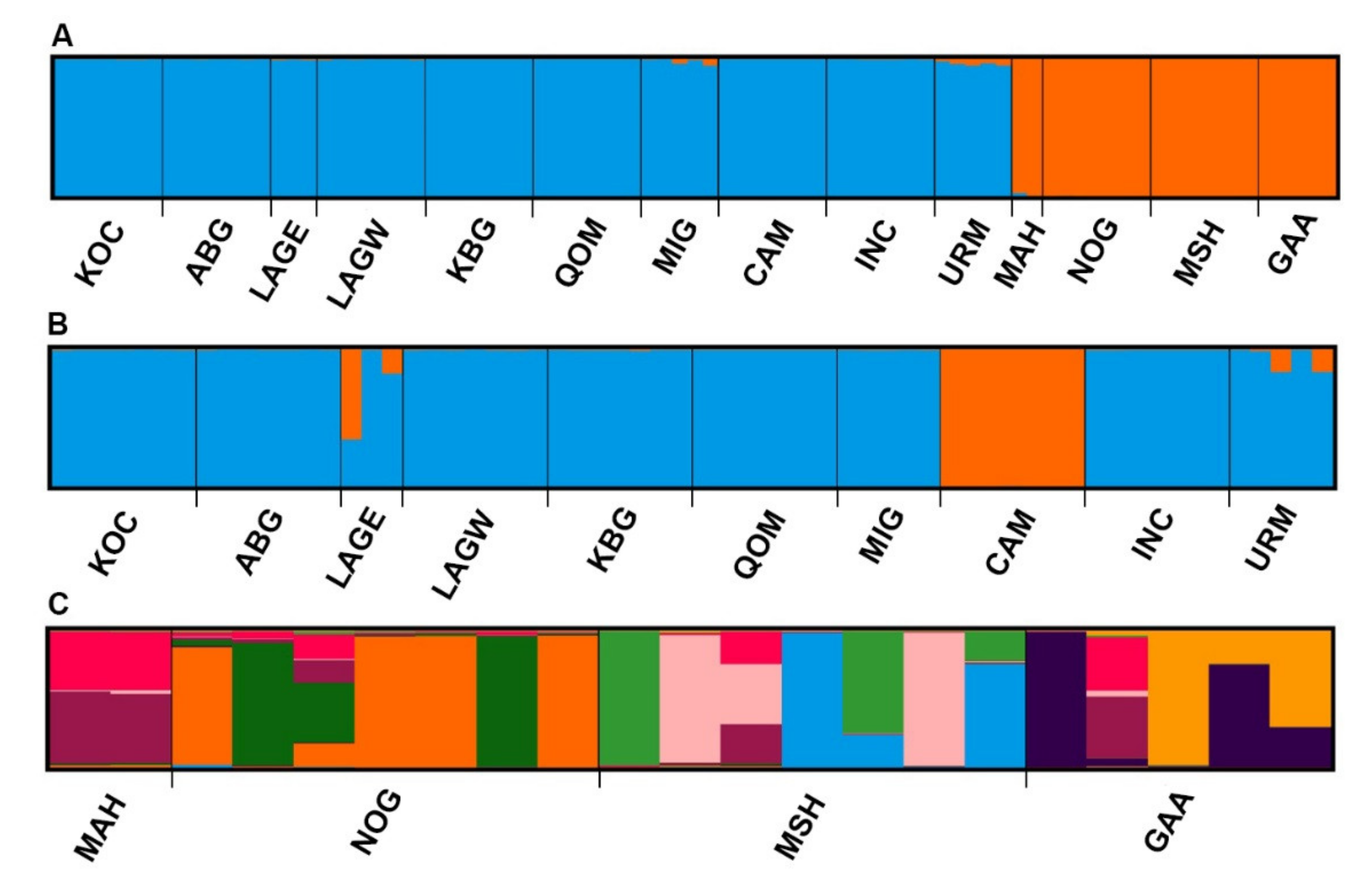
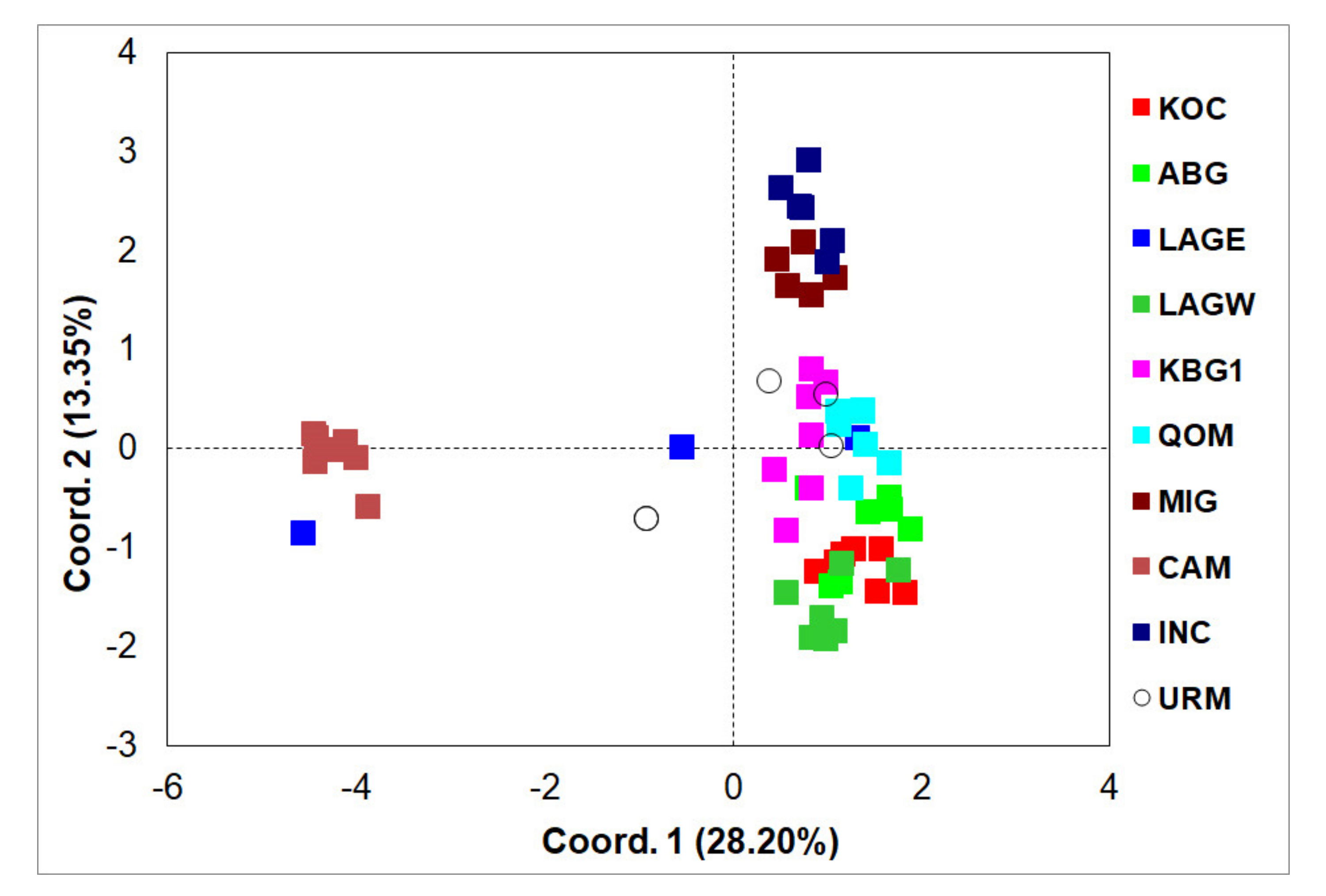
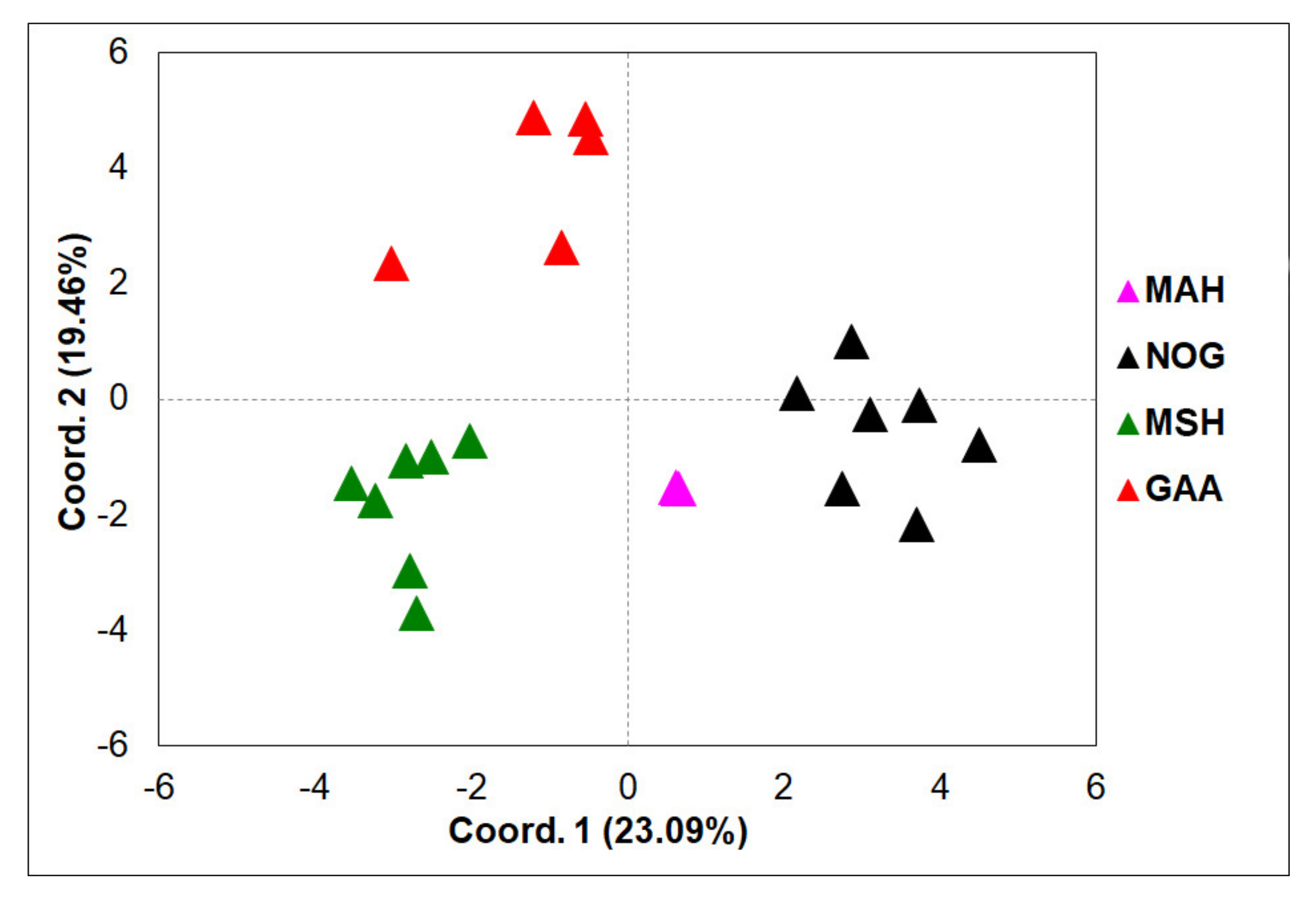
3.2. ISSR Profiling
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Haplotype | Ind. | Pop. (Ind.) | Haplotype | Ind. | Pop. (Ind.) |
|---|---|---|---|---|---|
| H1 | 18 | URM (18) | H24 | 1 | URM (1) |
| H2 | 7 | CAM (7) | H25 | 1 | URM (1) |
| H3 | 16 | KOC (6), LAGW (5)ABG (3), LAGE (1), KBG (1) | H26 | 1 | URM (1) |
| H4 | 15 | ING (5), MIG (4), KBG (3), LAGE (2) | H27 | 1 | URM (1) |
| H5 | 7 | QOM (6), MIG (1) | H28 | 1 | URM (1) |
| H6 | 1 | CAM (1) | H29 | 1 | URM (1) |
| H7 | 1 | URM (1) | H30 | 1 | URM (1) |
| H8 | 1 | URM (1) | H31 | 2 | URM (2) |
| H9 | 2 | URM (2) | H32 | 1 | URM (1) |
| H10 | 1 | URM (1) | H33 | 1 | URM (1) |
| H11 | 1 | URM (1) | H34 | 1 | URM (1) |
| H12 | 3 | URM (3) | H35 | 1 | URM (1) |
| H13 | 1 | URM (1) | H36 | 1 | URM (1) |
| H14 | 1 | URM (1) | H37 | 1 | URM (1) |
| H15 | 1 | URM (1) | H38 | 1 | URM (1) |
| H16 | 1 | URM (1) | H39 | 1 | URM (1) |
| H17 | 1 | URM (1) | H40 | 1 | URM (1) |
| H18 | 1 | URM (1) | H41 | 1 | URM (1) |
| H19 | 1 | URM (1) | H42 | 1 | URM (1) |
| H20 | 1 | URM (1) | H43 | 1 | URM (1) |
| H21 | 1 | URM (1) | H44 | 1 | URM (1) |
| H22 | 1 | URM (1) | H45 | 1 | URM (1) |
| H23 | 1 | URM (1) | - | - | - |
| Haplotype | Ind. | Pop. (Ind.) |
|---|---|---|
| H1 | 8 | GAA (4), MAH (2), MSH (2) |
| H2 | 7 | NOG (4), MSH (3) |
| H3 | 4 | GAA (4) |
| H4 | 1 | NOG (1) |
| H5 | 1 | MAH (1) |
| H6 | 1 | MSH (1) |
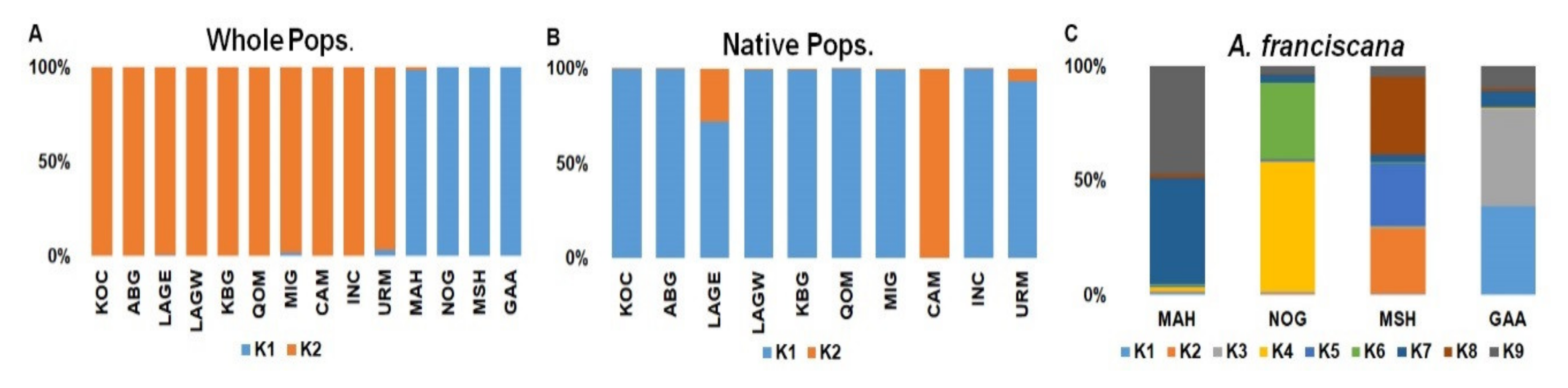
| Population | K1 (%) | K2 (%) |
|---|---|---|
| KOC | 0.1 | 99.9 |
| ABG | 0.2 | 99.8 |
| LAGE | 0.7 | 99.3 |
| LAGW | 0.3 | 99.7 |
| KBG | 0.1 | 99.9 |
| QOM | 0.1 | 99.9 |
| MIG | 1.9 | 98.1 |
| CAM | 0.1 | 99.9 |
| INC | 0.2 | 99.8 |
| URM | 3.6 | 96.4 |
| MAH | 98.7 | 1.3 |
| NOG | 99.9 | 0.1 |
| MSH | 99.9 | 0.1 |
| GAA | 99.9 | 0.1 |
| Population | K1 (%) | K2 (%) |
|---|---|---|
| KOC | 000099.7 | 0.3 |
| ABG | 99.8 | 0.2 |
| LAGE | 72.1 | 27.9 |
| LAGW | 99.6 | 0.4 |
| KBG | 99.5 | 0.5 |
| QOM | 99.8 | 0.2 |
| MIG | 99.6 | 0.4 |
| CAM | 0.2 | 99.8 |
| INC | 99.7 | 0.3 |
| URM | 93.1 | 6.9 |
| Population | K1 (%) | K2 (%) | K3 (%) | K4 (%) | K5 (%) | K6 (%) | K7 (%) | K8 (%) | K9 (%) |
|---|---|---|---|---|---|---|---|---|---|
| MAH | 1.1 | 0.4 | 0.3 | 1.7 | 0.4 | 1.1 | 46 | 1.8 | 47.1 |
| NOG | 0.6 | 0.5 | 0.4 | 57 | 0.6 | 33.6 | 3.6 | 0.3 | 3.4 |
| MSH | 0.5 | 28.6 | 0.3 | 0.4 | 27.9 | 0.4 | 3.4 | 33.9 | 4.6 |
| GAA | 38.8 | 0.2 | 42.3 | 0.3 | 0.4 | 0.2 | 7 | 1.1 | 9.7 |
References
- Van Stappen, G. Zoogeography. In Artemia: Basic and Applied Biology; Abatzopoulos, T.J., Beardmore, J.A., Clegg, J.S., Sorgeloos., P., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2002; pp. 171–224. [Google Scholar]
- Eimanifar, A.; Van Stappen, G.; Marden, B.; Wink, M. Artemia biodiversity in Asia with the focus on the phylogeography of the introduced American species Artemia franciscana Kellogg, 1906. Mol. Phylogenetics Evol. 2014, 79, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Asem, A.; Eimanifar, A.; Sun, S.C. Genetic variation and evolutionary origins of parthenogenetic Artemia (Crustacea: Anostraca) with different ploidies. Zool. Scr. 2016, 45, 421–436. [Google Scholar] [CrossRef]
- Van Stappen, G. Artemia biodiversity in Central and Eastern Asia. Ph.D. Thesis, Ghent University, Ghent, Belgium, 2008. [Google Scholar]
- Jones, A.G.; Ewing, C.M.; Melvin, M.V. Biotechnology of solar saltfields. Hydrobiologia 1981, 81, 391–406. [Google Scholar] [CrossRef]
- Ruebhart, D.R.; Cock, I.E.; Shaw, G.R. Invasive character of the brine shrimp Artemia franciscana Kellogg 1906 (Branchiopoda: Anostraca) and its potential impact on Australian inland hypersalinewaters. Mar. Freshw. Res. 2008, 59, 587–595. [Google Scholar] [CrossRef]
- Li, D.R.; Ye, H.L.; Yang, J.S.; Yang, F.; Wang, M.R.; De Vos, S.; Vuylsteke, M.; Sorgeloos, P.; Van Stappen, G.; Bossier, P.; et al. Identification and characterization of a Masculinizer (Masc) gene involved in sex differentiation in Artemia. Gene 2017, 614, 56–64. [Google Scholar] [CrossRef]
- Kappas, I.; Baxevanis, D.; Abatzopoulos, T.J. Phylogeographic patterns in Artemia: A model organism for hypersaline crustaceans. In Phylogeography and Population Genetics in Crustacea; Koenemann, S., Schubart, C., Held, C., Eds.; CRC Press: Boca Raton, FL, USA, 2011; pp. 233–255. [Google Scholar]
- Vazquez-Silva, G.; Aguirre-Garrido, J.F.; Ramirez-Saad, H.C.; Mayorga-Reyes, L.; Azaola-Espinosa, A.; Morales-Jiménez, J. Effect of bacterial probiotics bioencapsulated in Artemia franciscana on weight and length of the short fin silverside (Chirostoma humboldtianum) and the characterization of its intestinal bacterial community by DGGE. Lat. Am. J. Aquat. Res. 2018, 45, 1031–1043. [Google Scholar]
- Rajabi, S.; Ramazani, A.; Hamidi, M.; Naji, T. Artemia salina as a model organism in toxicity assessment of nanoparticles. J. Pharm. Sci. 2015, 23, 20. [Google Scholar] [CrossRef] [Green Version]
- Asem, A.; Rastegar-Pouyani, N.; De los Rios, P. The genus Artemia Leach, 1819 (Crustacea: Branchiopoda): True and false taxonomical descriptions. Lat. Am. J. Aquat. Res. 2010, 38, 501–506. [Google Scholar]
- Asem, A.; Eimanifar, A.; Rastegar-Pouyani, N.; Hontoria, F.; De Vos, S.; Van Stappen, G.; Sun, S.C. An overview on the nomenclatural and phylogenetic problems of native Asian brine shrimps of the genus Artemia Leach, 1819 (Crustacea: Anostraca). Zookeys 2020, 902, 1. [Google Scholar] [CrossRef]
- Naganawa, H.; Mura, G. Two new cryptic species of Artemia (Branchiopoda, Anostraca) from Mongolia and the possibility of invasion and disturbance by the aquaculture industry in East Asia. Crustaceana 2017, 90, 1679–1698. [Google Scholar] [CrossRef]
- Wang, W.; Luo, Q.; Guo, H.; Bossier, P.; Van Stappen, G.; Sorgeloos, P.; Xin, N.; Sun, Q.; Hu, S.; Yu, J. Phylogenetic analysis of brine shrimp (Artemia) in China using DNA barcoding. Genom. Proteom. Bioinform. 2008, 6, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, M.I.; Paredes, I.; Lebouvier, M.; Green, A.J. Functional role of native and invasive filter-feeders, and the effect of parasites: Learning from hypersaline ecosystems. PLoS ONE 2016, 11, e0161478. [Google Scholar] [CrossRef] [PubMed]
- Horvath, Z.; Lejeusne, C.; Amat, F.; Sanchez-Fontenla, J.; Vad, C.F.; Green, A.J. Eastern spread of the invasive Artemia franciscana in the Mediterranean Basin, with the first record from the Balkan Peninsula. Hydrobiologia 2018, 822, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Asem, A.; Eimanifar, A.; Li, W.; Wang, P.; Brooks, S.A.; Wink, M. Phylogeography and population genetic structure of an exotic invasive brine shrimp Artemia Leach, 1819 (Crustacea: Anostraca) in Australia. Aust. J. Zool. 2018, 66, 307–316. [Google Scholar] [CrossRef]
- Saji, A.; Eimanifar, A.; Soorae, P.S.; Al Dhaheri, S.H.; Asem, A. Phylogenetic Analysis of exotic invasive species of Brine Shrimp Artemia Leach, 1819 (Branchiopoda, Anostraca) in Al Wathba Wetland Reserve (UAE.; Abu Dhabi). Crustacean 2019, 92, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Amat, F.; Hontoria, F.; Navarro, J.C.; Vieira, N.; Mura, G. Biodiversity loss in the genus Artemia in the Western Mediterranean region. Limnetica 2007, 26, 387–404. [Google Scholar]
- Lee, C.E. Evolutionary genetics of invasive species. Trends Ecol. Evol. 2002, 17, 386–391. [Google Scholar] [CrossRef]
- Lavergne, S.; Molofsky, J. Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc. Natl. Acad. Sci. USA 2007, 104, 3883–3888. [Google Scholar] [CrossRef] [Green Version]
- Kappas, I.; Abatzopoulos, T.J.; Van Hoa, N.; Sorgeloos, P.; Beardmore, J.A. Genetic and reproductive differentiation of Artemia franciscana in a new environment. Mar. Biol. 2004, 146, 103–117. [Google Scholar] [CrossRef]
- Hontoria, F.; Redón, S.; Maccari, M.; Varó, I.; Vavarro, C.J.; Ballell, L.; Amat, F. A revision of Artemia biodiversity in Macaronesia. Aquat. Biosyst. 2012, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, J.; Gómez, A.; Figuerola, J.; Amat, F.; Rico, C.; Green, A.J. Colonization and dispersal patterns of the invasive American brine shrimp Artemia franciscana (Branchiopoda: Anostraca) in the Mediterranean region. Hydrobiologia 2014, 726, 25–41. [Google Scholar] [CrossRef]
- Dlugosch, M.K.; Parker, M.I. Founding events in species invasions: Genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 2008, 17, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Vikas, P.A.; Sajeshkumar, N.K.; Thomas, P.C.; Chakraborty, K.; Vijayan, K.K. Aquaculture related invasion of the exotic Artemia franciscana and displacement of the autochthonous Artemia populations from the hypersaline habitats of India. Hydrobiologia 2012, 684, 129–142. [Google Scholar] [CrossRef]
- Hou, L.; Li, H.; Zou, X.; Yao, F.; Bi, X.; He, C. Population genetic structure and genetic differentiation of Artemia parthenogenetica in China. J. Shellfish Res. 2006, 25, 999–1005. [Google Scholar]
- Eimanifar, A.; Wink, M. Fine-scale population genetic structure in Artemia urmiana (Günther, 1890) based on mtDNA sequences and ISSR genomic fingerprinting. Org. Divers. Evol. 2013, 13, 531–543. [Google Scholar] [CrossRef]
- Eimanifar, A.; Marden, B.; Braun, M.S.; Wink, M. Analysis of the genetic variability of Artemia franciscana Kellogg, 1906 from the Great Salt Lake (USA) based on mtDNA sequences, ISSR genomic fingerprinting and biometry. Mar. Biodivers. 2015, 45, 311–319. [Google Scholar] [CrossRef]
- Asem, A.; Eimanifar, A.; Van Stappen, G.; Sun, S.C. The impact of one-decade ecological disturbance on genetic changes: A study on the brine shrimp Artemia urmiana from Urmia Lake, Iran. PeerJ 2019, 7, e7190. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Maniatsi, S.; Baxevanis, A.D.; Kappas, I.; Deligiannidis, P.; Triantafyllidis, A.; Papakostas, S.; Bougiouklis, D.; Abatzopoulos, T.J. Is polyploidy a persevering accident or an adaptive evolutionary pattern? The case of the brine shrimp Artemia. Mol. Phylogenetics Evol. 2011, 58, 353–364. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; von Holdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software Structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Sayg, Y. Characterization of parthenogenetic Artemia populations from Camalti (Izmir, Turkey) and Kalloni (Lesbos, Greece): Survival, growth, maturation, biometrics, fatty acid profiles and hatching characteristics. Hydrobiologia 2004, 527, 227–239. [Google Scholar] [CrossRef]
- Kelts, K.; Shahrabi, M. Holocene sedimentalogy of hypersaline Lake Urmia, northwestern Iran. Paleogeography. Paleoclimatol. Paleoecol. 1986, 54, 105–130. [Google Scholar] [CrossRef]
- Djamali, M.; Kürschner, H.; Akhani, H.; De Beaulieu, J.L.; Amini, A.; Andrieu-Ponel, V.; Ponel, P.; Stevens, L. Palaeoecological significance of the spores of the liverwort Riella (Riellaceae) in a late Pleistocene long pollen record from the hypersaline Lake Urmia, NW Iran. Rev. Palaeobot. Palynol. 2008, 152, 66–73. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Ratnasingham, S.; Waard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proceedings of the Royal Society of London. Ser. B Biol. Sci. 2003, 270, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Golani, D.G.; Azzurro, E.; Corsini-Foka, M.; Falautana, M.; Andaloro, F.; Bernardi, G. Genetic bottlenecks and successful biological invasions: The case of a recent Lessepsian migrant. Biol. Lett. 2007, 3, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Rattanawannee, A.; Duangphakdee, O.; Chanchao, C.; Teerapakpinyo, C.; Warrit, N.; Wongsiri, S.; Oldroyd, B.P. Genetic Characterization of Exotic Commercial Honey Bee (Hymenoptera: Apidae) Populations in Thailand Reveals High Genetic Diversity and Low Population Substructure. J. Econ. Entomol. 2020, 113, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Asem, A.; Sun, S.C. Morphological differentiation of seven parthenogenetic Artemia (Crustacea: Branchiopoda) populations from China, with special emphasis on ploidy degrees. Microsc. Res. Tech. 2016, 79, 258–266. [Google Scholar] [CrossRef] [PubMed]










| No. | Voucher Number (IPMB) | Abb. | Species/Population | Locality | Country | Geographic Coordinates | COI Accession Numbers |
|---|---|---|---|---|---|---|---|
| 1 | 57211 | URM | A. urmiana | Urmia Lake | Iran | 37°20′ E–45°40′ N | JX512748-808 [28] |
| 2 | 57223 | LAGW | Parthenogenetic | Western Lagoon around Urmia Lake | Iran | 37°15′ E–45°85′ N | KF691338-342 [2] |
| 3 | 57224 | LAGE | Parthenogenetic | Eastern Lagoon around Urmia Lake | Iran | 37°50′ E–46°40′ N | KF691343-345 [2] |
| 4 | 57225 | QOM | Parthenogenetic | Qom Salt Lake | Iran | 34°40′ E–51°80′ N | KF691367-372 [2] |
| 5 | 57226 | MIG | Parthenogenetic | Mighan Salt Lake | Iran | 34°20′ E–49°80′ N | KF691357-361 [2] |
| 6 | 57230 | MSH | A. franciscana | Mahshar port | Iran | 49°11′ E–30°33′ N | KF691351-356 [2] |
| 7 | 57228 | MAHR | A. franciscana | Maharlu Lake | Iran | 29°57′ E–52°14′ N | KF691347, 349-350 [2] |
| 8 | 57229 | NOG | A. franciscana | Nough Catchment | Iran | 30°60′ E–56°50′ N | KF691362-366 [2] |
| 9 | 57227 | INC | Parthenogenetic | Incheh Lake | Iran | 37°24′ E–54°36′ N | KF691333-337 [2] |
| 10 | 57292 | CAM | Parthenogenetic | Camalti Lake | Turkey | 27°08′ E–38°25′ N | KF691520-525; 527-529 [2] |
| 11 | 57255 | ABG | Parthenogenetic | Abu-Ghraib | Iraq | 44°30′ E–33°20′ N | KF691373-375 [2] |
| 12 | 57256 | GAA | A. franciscana | Garmat Ali | Iraq | 47°49′ E–30°30′ N | KF691376-383 [2] |
| 13 | 57258 | KBG | Parthenogenetic | Kara Bogaz Gol | Turkmenistan | 53°33′ E–41°17′ N | KF691530-532,534 [2] |
| 14 | 57257 | KOC | Parthenogenetic | Korangi Creek (Karachi coast) | Pakistan | 67°10′ E–24°48′ N | KF691442-445; 447-448 JX512748 [2] |
| Species/Population | Abbreviation | Individual | Accession Numbers | Ref. |
|---|---|---|---|---|
| A. urmiana | URM | 4 | JX512748-751 | [28] |
| A. sinica | SIN | 4 | KF691298-301 | [2] |
| A. tibetiana | TIB | 4 | KF691215-218 | [2] |
| A. salina | SAL | 4 | KF691512-515 | [2] |
| A. persimilis | PER | 4 | DQ119647 HM998992 EF615594 EF615593 | [27] [33] [14] [14] |
| A. franciscana | FRA | 4 | KJ863440-443 | [2] |
| Diploid Pop. | DI | 4 | KU183949-952 | [3] |
| Triploid Pop. | TRE | 3 | HM998997-999 | [33] |
| Tetraploid Pop. | TETR | 4 | KU183954-957 | [3] |
| Pentaploid Pop. | PEN | 4 | KU183968-971 | [3] |
| Primer | Sequence | GC (%) | Annealing Temperature (°C) | Amplification Pattern |
|---|---|---|---|---|
| ISSR1 | (AC)8T | 47.1 | 48–54 | Smear |
| ISSR2 | (CAC)5 | 66.7 | 48–54 | Smear |
| ISSR3 | (GACA)4 | 50 | 48–54 | Smear |
| ISSR4 | (AG)12 | 50 | 48–54 | Poor |
| ISSR5 | (TC)9 | 50 | 48–54 | Poor |
| ISSR6 | (GT)10 | 50 | 48–54 | Smear |
| ISSR7 | (CA)10A | 47.6 | 48–54 | Poor |
| ISSR8 | (GAA)5 | 33.3 | 48–54 | No amplification |
| ISSR9 | (CAG)6 | 66.7 | 48–54 | No amplification |
| ISSR10 | (GCCG)4 | 100 | 48–54 | No amplification |
| ISSR11 | (AG)8C | 52.9 | 48 | Good and sharp |
| ISSR12 | (AG)8YTa | 50 | 48 | Good and sharp |
| ISSR13 | (GA)9T | 47.4 | 50 | Good and sharp |
| ISSR14 | (TG)8G | 52.9 | 50 | Good and sharp |
| ISSR15 | (AC)8C | 52.9 | 49 | Good and sharp |
| Pop. | N | Na | Ne | I | He | uHe | PPL | NB | NPB |
|---|---|---|---|---|---|---|---|---|---|
| KOC | 7 | 0.645 (0.062) | 1.102 (0.021) | 0.090 (0.017) | 0.059 (0.012) | 0.064 (0.012) | 17.76 | 71 | 0 |
| ABG | 7 | 0.743 (0.066) | 1.149 (0.024) | 0.131 (0.020) | 0.088 (0.013) | 0.095 (0.014) | 23.68 | 77 | 0 |
| LAGE | 3 | 0.914 (0.072) | 1.256 (0.031) | 0.208 (0.024) | 0.143 (0.017) | 0.171 (0.020) | 34.87 | 86 | 0 |
| LAGW | 7 | 0.829 (0.066) | 1.157 (0.024) | 0.140 (0.020) | 0.093 (0.014) | 0.100 (0.015) | 25.66 | 87 | 0 |
| KBG | 7 | 0.743 (0.068) | 1.161 (0.025) | 0.139 (0.020) | 0.094 (0.014) | 0.101 (0.015) | 25.00 | 75 | 0 |
| QOM | 7 | 0.638 (0.062) | 1.097 (0.020) | 0.087 (0.016) | 0.058 (0.011) | 0.062 (0.012) | 17.11 | 71 | 0 |
| MIG | 5 | 0.678 (0.067) | 1.150 (0.025) | 0.128 (0.020) | 0.087 (0.014) | 0.096 (0.015) | 23.03 | 68 | 0 |
| CAM | 7 | 0.559 (0.051) | 1.041 (0.014) | 0.036 (0.011) | 0.024 (0.008) | 0.026 (0.008) | 7.24 | 74 | 0 |
| INC | 7 | 0.658 (0.064) | 1.125 (0.022) | 0.110 (0.018) | 0.074 (0.013) | 0.080 (0.014) | 19.74 | 70 | 0 |
| URM | 5 | 0.783 (0.072) | 1.200 (0.027) | 0.173 (0.022) | 0.117 (0.015) | 0.130 (0.017) | 30.26 | 73 | 0 |
| MAH | 2 | 0.822 (0.066) | 1.181 (0.025) | 0.155 (0.021) | 0.106 (0.015) | 0.142 (0.020) | 25.66 | 86 | 1 |
| NOG | 7 | 1.013 (0.071) | 1.244 (0.030) | 0.208 (0.023) | 0.140 (0.016) | 0.151 (0.017) | 38.82 | 95 | 0 |
| MSH | 7 | 1.066 (0.066) | 1.223 (0.028) | 0.193 (0.022) | 0.129 (0.015) | 0.139 (0.016) | 36.84 | 106 | 0 |
| GAA | 5 | 0.921 (0.067) | 1.206 (0.028) | 0.172 (0.022) | 0.117 (0.015) | 0.130 (0.017) | 30.26 | 94 | 0 |
| Mean | 5.9 (0.035) | 0.787 (0.018) | 1.164 (0.007) | 0.141 (0.005) | 0.095 (0.004) | 0.106 (0.004) | 25.42 (2.28%) | - | - |
| Population | N | Na | Ne | I | He | uHe | PPL | NB | NPB |
|---|---|---|---|---|---|---|---|---|---|
| KOC | 7.000 | 0.731 (0.067) | 1.115 (0.023) | 0.102 (0.019) | 0.067 (0.013) | 0.073 (0.014) | 20.15 | 71 | 2 |
| ABG | 7.000 | 0.843 (0.071) | 1.169 (0.027) | 0.148 (0.022) | 0.100 (0.015) | 0.107 (0.016) | 26.87 | 77 | 4 |
| LAGE | 3.000 | 1.037 (0.075) | 1.291 (0.034) | 0.236 (0.026) | 0.162 (0.018) | 0.195 (0.022) | 39.55 | 86 | 1 |
| LAGW | 7.000 | 0.940 (0.069) | 1.178 (0.027) | 0.158 (0.022) | 0.106 (0.015) | 0.114 (0.016) | 29.10 | 87 | 4 |
| KBG | 7.000 | 0.843 (0.073) | 1.183 (0.028) | 0.158 (0.023) | 0.107 (0.015) | 0.115 (0.017) | 28.36 | 75 | 0 |
| QOM | 7.000 | 0.724 (0.066) | 1.110 (0.023) | 0.099 (0.018) | 0.065 (0.013) | 0.070 (0.014) | 19.40 | 71 | 0 |
| MIG | 5.000 | 0.769 (0.073) | 1.170 (0.027) | 0.145 (0.022) | 0.098 (0.015) | 0.109 (0.017) | 26.12 | 68 | 3 |
| CAM | 7.000 | 0.634 (0.055) | 1.046 (0.016) | 0.041 (0.013) | 0.027 (0.009) | 0.029 (0.009) | 8.21 | 74 | 0 |
| INC | 7.000 | 0.746 (0.069) | 1.142 (0.025) | 0.125 (0.021) | 0.084 (0.014) | 0.091 (0.015) | 22.39 | 70 | 1 |
| URM | 5.000 | 0.888 (0.077) | 1.227 (0.029) | 0.196 (0.024) | 0.133 (0.017) | 0.148 (0.018) | 34.33 | 73 | 3 |
| Mean | 6.200 (0.036) | 0.816 (0.022) | 1.163 (0.008) | 0.141 (0.007) | 0.095 (0.005) | 0.105 (0.005) | 25.45 (2.74) | - | - |
| Population | N | Na | Ne | I | He | uHe | PPL | NB | NPB |
|---|---|---|---|---|---|---|---|---|---|
| MAH | 2.000 | 0.992 (0.071) | 1.219 (0.029) | 0.187 (0.025) | 0.128 (0.017) | 0.171 (0.023) | 30.95 | 86 | 3 |
| NOG | 7.000 | 1.222 (0.073) | 1.295 (0.034) | 0.251 (0.026) | 0.169 (0.018) | 0.182 (0.019) | 46.83 | 95 | 6 |
| MSH | 7.000 | 1.286 (0.065) | 1.269 (0.033) | 0.233 (0.025) | 0.156 (0.018) | 0.168 (0.019) | 44.44 | 106 | 8 |
| GAA | 5.000 | 1.111 (0.070) | 1.249 (0.033) | 0.207 (0.025) | 0.141 (0.018) | 0.157 (0.020) | 36.51 | 94 | 5 |
| Mean | 5.250 (0.091) | 1.153 (0.035) | 1.258 (0.016) | 0.220 (0.013) | 0.149 (0.009) | 0.169 (0.010) | 39.68 (3.65) | - | - |
| Population | KOC | ABG | LAGE | LAGW | KBG | QOM | MIG | CAM | INC |
|---|---|---|---|---|---|---|---|---|---|
| ABG | 0.162 | - | - | - | - | - | - | - | - |
| LAGE | 0.167 | 0.175 | - | - | - | - | - | - | - |
| LAGW | 0.193 | 0.127 | 0.190 | - | - | - | - | - | - |
| KBG1 | 0.156 | 0.117 | 0.142 | 0.111 | - | - | - | - | - |
| QOM | 0.182 | 0.131 | 0.171 | 0.135 | 0.099 | - | - | - | - |
| MIG | 0.217 | 0.185 | 0.171 | 0.212 | 0.124 | 0.148 | - | - | - |
| CAM | 0.362 | 0.346 | 0.249 | 0.358 | 0.279 | 0.354 | 0.331 | - | - |
| INC | 0.233 | 0.165 | 0.184 | 0.223 | 0.149 | 0.176 | 0.091 | 0.351 | - |
| URM | 0.219 | 0.194 | 0.165 | 0.190 | 0.141 | 0.174 | 0.194 | 0.351 | 0.211 |
| Population | MAH | NOG | MAH |
|---|---|---|---|
| NOG | 0.143 | - | - |
| MSH | 0.145 | 0.144 | - |
| GAA | 0.198 | 0.170 | 0.152 |
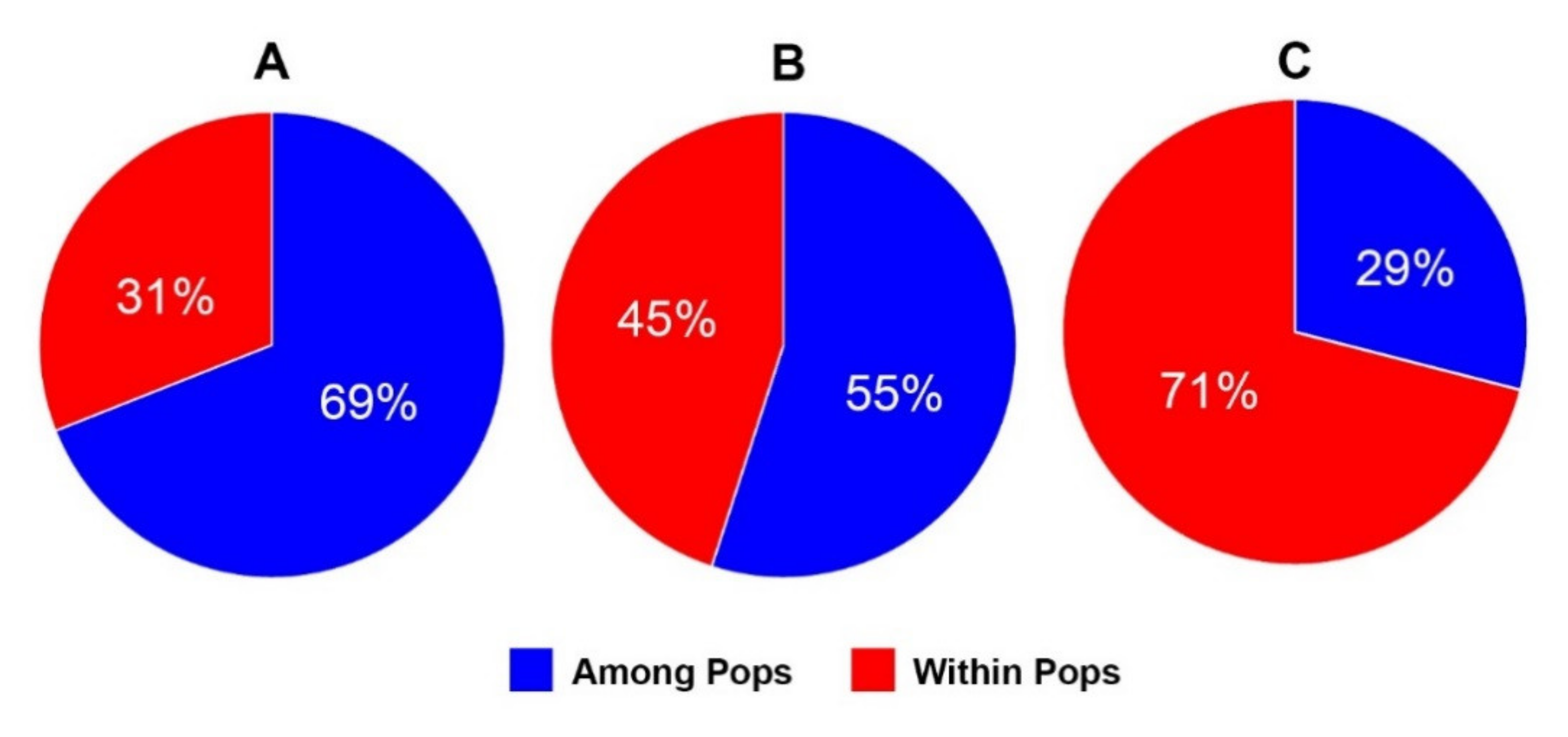
| Whole Populations | |||||
|---|---|---|---|---|---|
| Source | df | SS | MS | Est. Var. | % |
| Among Pops | 13 | 1673.318 | 128.717 | 20.265 | 69% |
| Within Pops | 69 | 639.405 | 9.267 | 9.267 | 31% |
| Total | 82 | 2312.723 | - | 29.532 | 100% |
| Source | df | SS | MS | Est. Var. | % |
| Native Populations | |||||
| Among Pops | 9 | 627.912 | 69.768 | 10.006 | 55% |
| Within Pops | 52 | 418.362 | 8.045 | 8.045 | 45% |
| Total | 61 | 1046.274 | - | 18.052 | 100% |
| A. franciscana | |||||
| Source | df | SS | MS | Est. Var. | % |
| Among Pops | 3 | 117.338 | 39.113 | 5.239 | 29% |
| Within Pops | 17 | 221.043 | 13.003 | 13.003 | 71% |
| Total | 20 | 338.381 | - | 18.241 | 100% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eimanifar, A.; Asem, A.; Wang, P.-Z.; Li, W.; Wink, M. Using ISSR Genomic Fingerprinting to Study the Genetic Differentiation of Artemia Leach, 1819 (Crustacea: Anostraca) from Iran and Neighbor Regions with the Focus on the Invasive American Artemia franciscana. Diversity 2020, 12, 132. https://doi.org/10.3390/d12040132
Eimanifar A, Asem A, Wang P-Z, Li W, Wink M. Using ISSR Genomic Fingerprinting to Study the Genetic Differentiation of Artemia Leach, 1819 (Crustacea: Anostraca) from Iran and Neighbor Regions with the Focus on the Invasive American Artemia franciscana. Diversity. 2020; 12(4):132. https://doi.org/10.3390/d12040132
Chicago/Turabian StyleEimanifar, Amin, Alireza Asem, Pei-Zheng Wang, Weidong Li, and Michael Wink. 2020. "Using ISSR Genomic Fingerprinting to Study the Genetic Differentiation of Artemia Leach, 1819 (Crustacea: Anostraca) from Iran and Neighbor Regions with the Focus on the Invasive American Artemia franciscana" Diversity 12, no. 4: 132. https://doi.org/10.3390/d12040132
APA StyleEimanifar, A., Asem, A., Wang, P. -Z., Li, W., & Wink, M. (2020). Using ISSR Genomic Fingerprinting to Study the Genetic Differentiation of Artemia Leach, 1819 (Crustacea: Anostraca) from Iran and Neighbor Regions with the Focus on the Invasive American Artemia franciscana. Diversity, 12(4), 132. https://doi.org/10.3390/d12040132