Comparative Analysis of the Core Proteomes among the Pseudomonas Major Evolutionary Groups Reveals Species-Specific Adaptations for Pseudomonas aeruginosa and Pseudomonas chlororaphis




Abstract
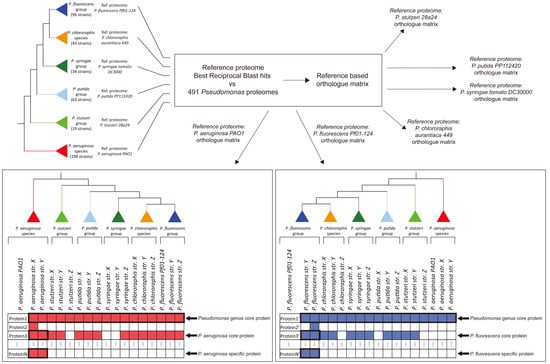
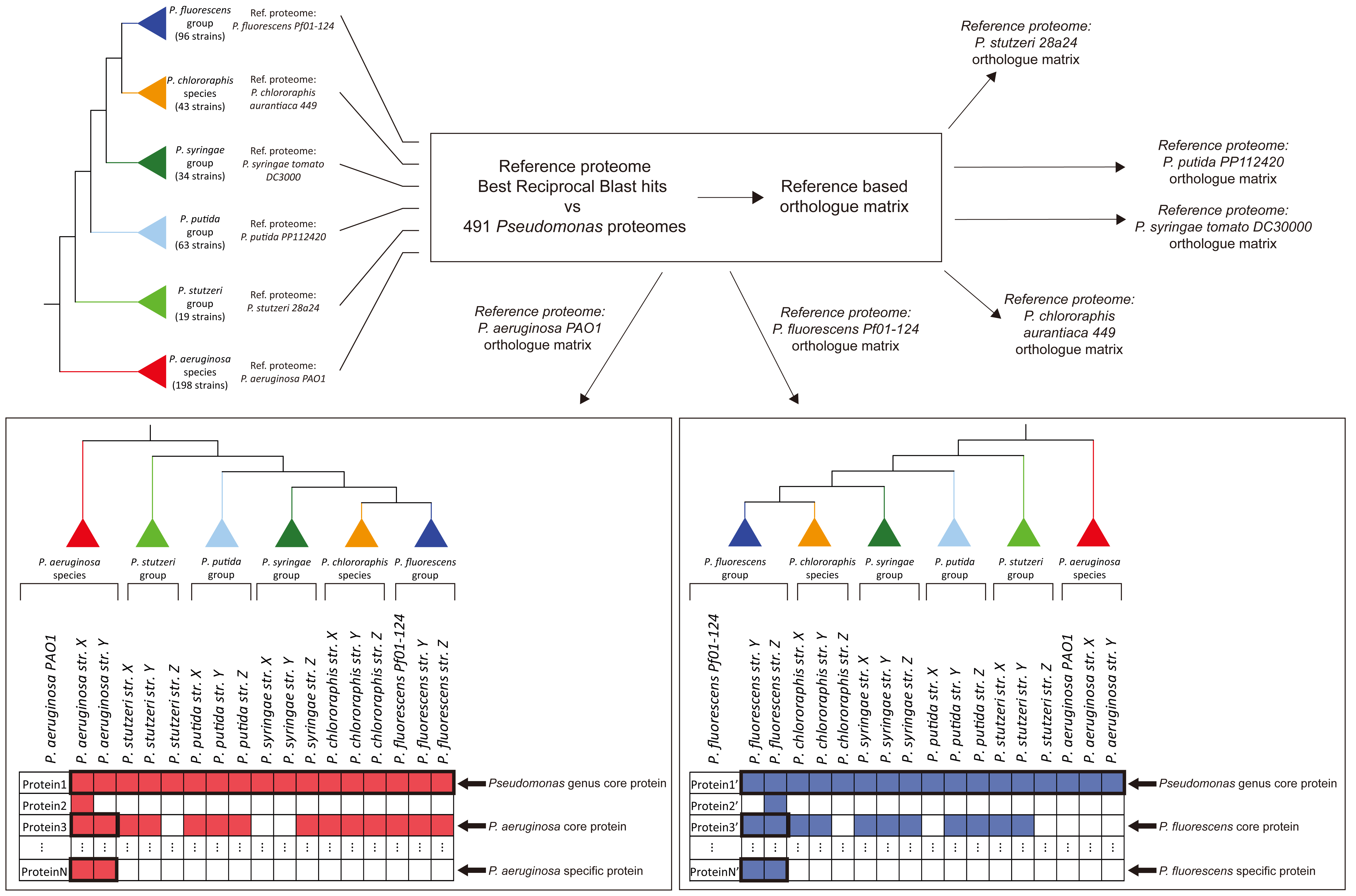
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
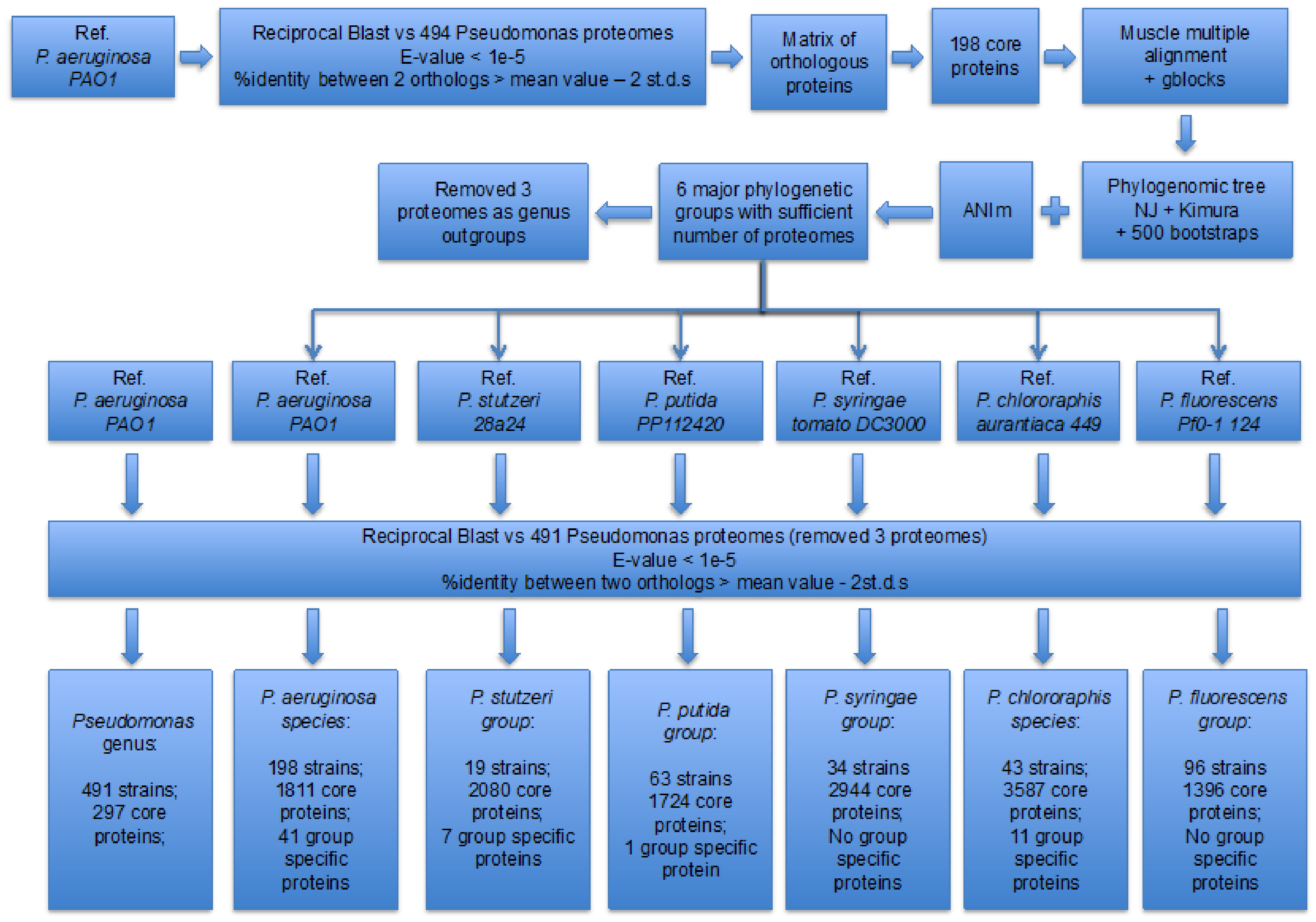
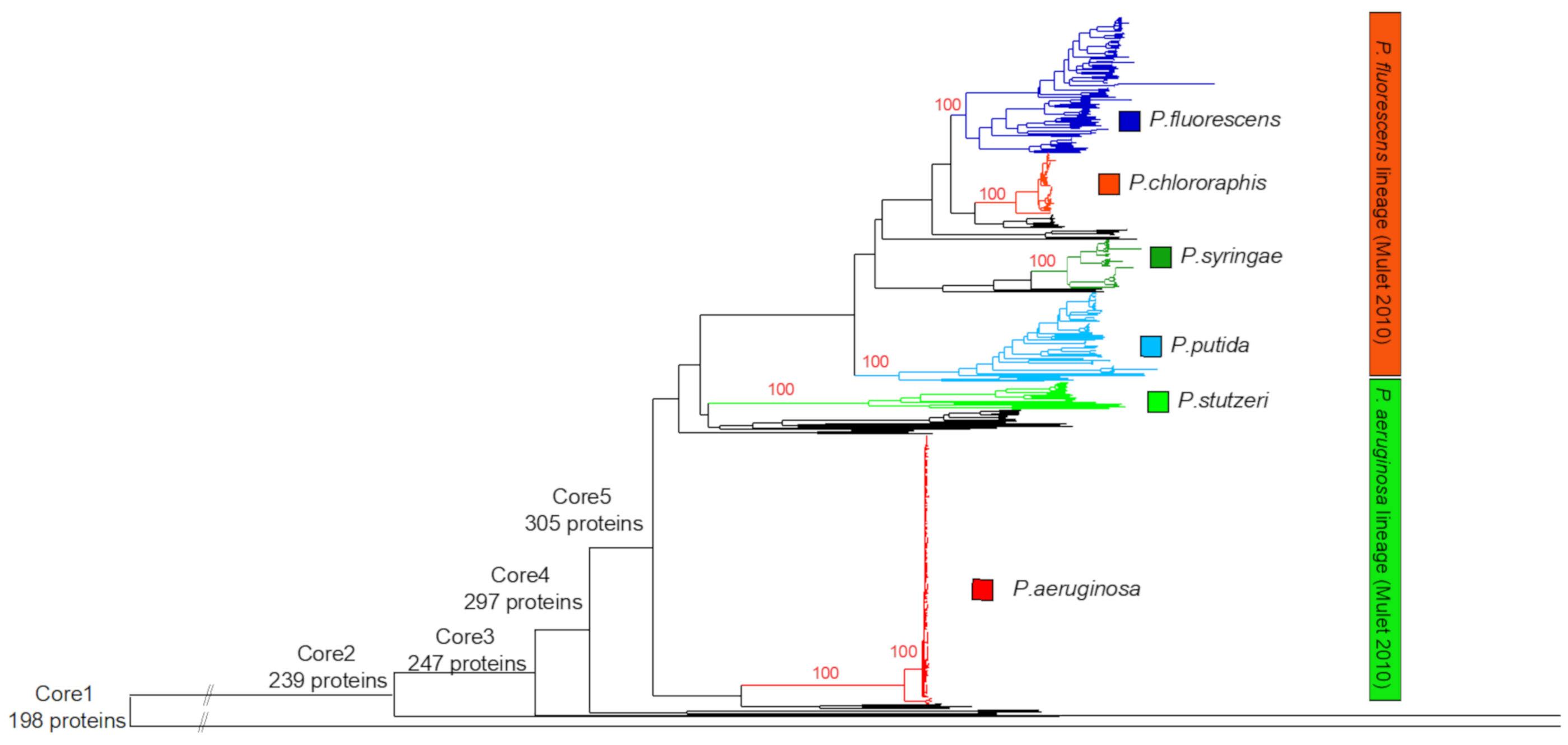
3.1. Identification of the Major Phylogenetic Groups and Species
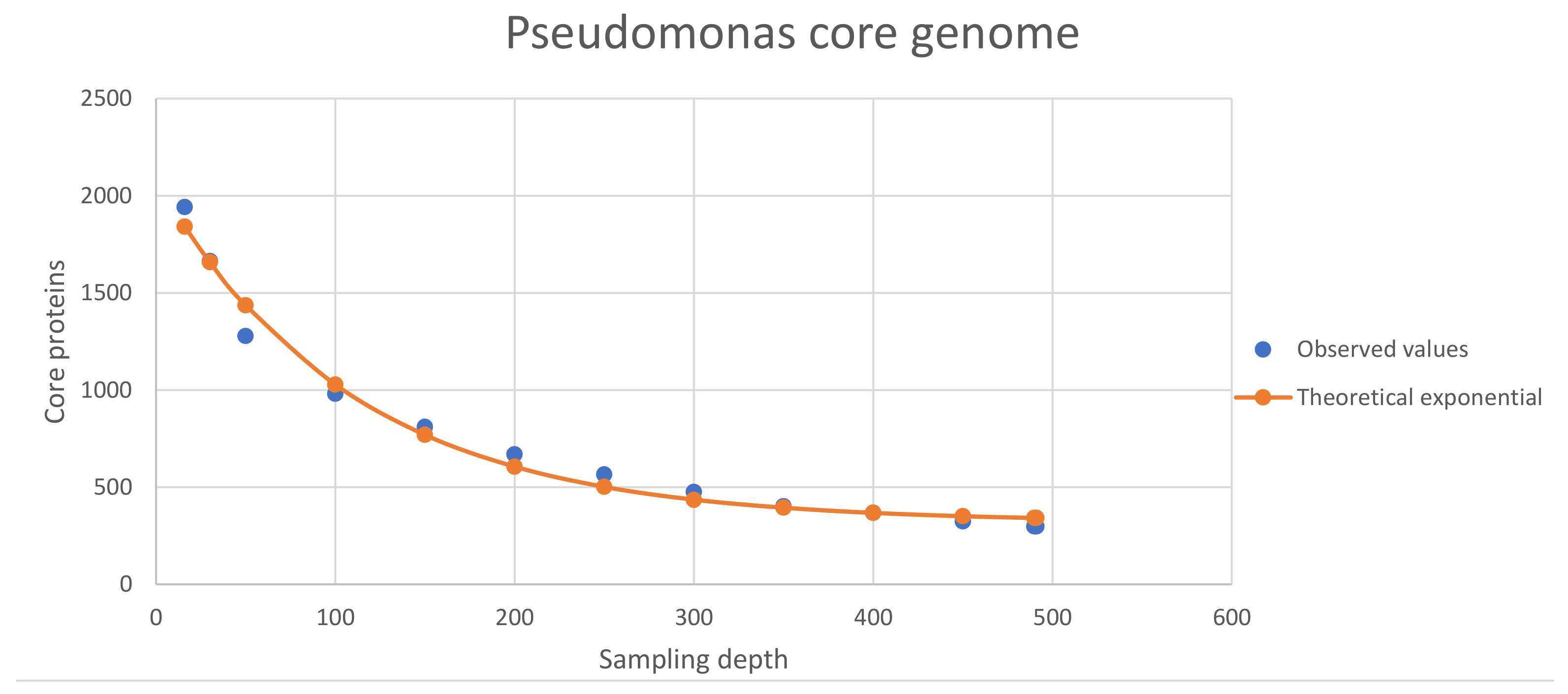
3.2. Properties of the Core Proteomes
3.3. Many P. aeruginosa-Specific Core Proteins Contribute to Pathogenicity
3.4. Pseudomonas chlororaphis-Specific Core Proteins with an Important Role in Niche Adaptation
4. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.C.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peix, A.; Ramírez-Bahena, M.-H.; Velázquez, E. The current status on the taxonomy of Pseudomonas revisited: An update. Infect. Genet. Evol. 2018, 57, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Moradali, M.F.; Ghods, S.; Rehm, B.H.A. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalucat, J.; Bennasar, A.; Bosch, R.; García-Valdés, E.; Palleroni, N.J. Biology of Pseudomonas stutzeri. Microbiol. Mol. Biol. Rev. 2006, 70, 510–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, X.-F.; Kvitko, B.; He, S.Y. Pseudomonas syringae: What it takes to be a pathogen. Nat. Rev. Microbiol. 2018, 16, 316–328. [Google Scholar] [CrossRef]
- Mulet, M.; Lalucat, J.; García-Valdés, E. DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 2010, 12, 1513–1530. [Google Scholar] [CrossRef] [Green Version]
- Scales, B.S.; Dickson, R.P.; LiPuma, J.J.; Huffnagle, G.B. Microbiology, genomics, and clinical significance of the Pseudomonas fluorescens species complex, an unappreciated colonizer of humans. Clin. Microbiol. Rev. 2014, 27, 927–948. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.A.; Staley, J.; Challender, M.; Heuton, J. Safety of Pseudomonas chlororaphis as a gene source for genetically modified crops. Transgenic Res. 2018, 27, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.; Damania, A. Draft Genome Sequence of Pseudomonas putida CBF10-2, a Soil Isolate with Bioremediation Potential in Agricultural and Industrial Environmental Settings. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulou, E.S.; Perruchon, C.; Vasileiadis, S.; Rousidou, C.; Tanou, G.; Samiotaki, M.; Molassiotis, A.; Karpouzas, D.G. Metabolic and Evolutionary Insights in the Transformation of Diphenylamine by a Pseudomonas putida Strain Unravelled by Genomic, Proteomic, and Transcription Analysis. Front. Microbiol. 2018, 9, 676. [Google Scholar] [CrossRef]
- Anzai, Y.; Kim, H.; Park, J.Y.; Wakabayashi, H.; Oyaizu, H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol. 2000, 50, 1563–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woese, C.R.; Stackebrandt, E.; Weisburg, W.G.; Paster, B.J.; Madigan, M.T.; Fowler, V.J.; Hahn, C.M.; Blanz, P.; Gupta, R.; Nealson, K.H.; et al. The phylogeny of purple bacteria: The alpha subdivision. Syst. Appl. Microbiol. 1984, 5, 315–326. [Google Scholar] [CrossRef]
- Özen, A.I.; Ussery, D.W. Defining the Pseudomonas genus: Where do we draw the line with Azotobacter? Microb. Ecol. 2012, 63, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tindall, B.J.; Rosselló-Móra, R.; Busse, H.-J.; Ludwig, W.; Kämpfer, P. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 2010, 60, 249–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palleroni, N.J. Pseudomonas. In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Rainey, F., Kämpfer, P., Trujillo, M., Chun, J., DeVos, P., Hedlund, B., Dedysh, S., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2015; p. 1. ISBN 978-1-118-96060-8. [Google Scholar]
- Bennasar, A.; Mulet, M.; Lalucat, J.; García-Valdés, E. PseudoMLSA: A database for multigenic sequence analysis of Pseudomonas species. BMC Microbiol. 2010, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- García-Valdés, E.; Lalucat, J. Pseudomonas: Molecular Phylogeny and Current Taxonomy. In Pseudomonas: Molecular and Applied Biology; Kahlon, R.S., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–23. ISBN 978-3-319-31197-5. [Google Scholar]
- Lalucat, J.; Mulet, M.; Gomila, M.; García-Valdés, E. Genomics in Bacterial Taxonomy: Impact on the Genus Pseudomonas. Genes 2020, 11, 139. [Google Scholar] [CrossRef] [Green Version]
- Jolley, K.A.; Bliss, C.M.; Bennett, J.S.; Bratcher, H.B.; Brehony, C.; Colles, F.M.; Wimalarathna, H.; Harrison, O.B.; Sheppard, S.K.; Cody, A.J.; et al. Ribosomal multilocus sequence typing: Universal characterization of bacteria from domain to strain. Microbiology 2012, 158, 1005–1015. [Google Scholar] [CrossRef]
- Kyrpides, N.C.; Hugenholtz, P.; Eisen, J.A.; Woyke, T.; Göker, M.; Parker, C.T.; Amann, R.; Beck, B.J.; Chain, P.S.G.; Chun, J.; et al. Genomic encyclopedia of bacteria and archaea: Sequencing a myriad of type strains. PLoS Biol. 2014, 12, e1001920. [Google Scholar] [CrossRef] [Green Version]
- Whitman, W.B.; Woyke, T.; Klenk, H.-P.; Zhou, Y.; Lilburn, T.G.; Beck, B.J.; De Vos, P.; Vandamme, P.; Eisen, J.A.; Garrity, G.; et al. Genomic Encyclopedia of Bacterial and Archaeal Type Strains, Phase III: The genomes of soil and plant-associated and newly described type strains. Stand. Genom. Sci. 2015, 10, 26. [Google Scholar] [CrossRef]
- Wu, D.; Hugenholtz, P.; Mavromatis, K.; Pukall, R.; Dalin, E.; Ivanova, N.N.; Kunin, V.; Goodwin, L.; Wu, M.; Tindall, B.J.; et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009, 462, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Gogarten, J.P.; Townsend, J.P. Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 2005, 3, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Ouzounis, C.A. The balance of driving forces during genome evolution in prokaryotes. Genome Res. 2003, 13, 1589–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunin, V.; Goldovsky, L.; Darzentas, N.; Ouzounis, C.A. The net of life: Reconstructing the microbial phylogenetic network. Genome Res. 2005, 15, 954–959. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [Green Version]
- Jun, S.-R.; Wassenaar, T.M.; Nookaew, I.; Hauser, L.; Wanchai, V.; Land, M.; Timm, C.M.; Lu, T.-Y.S.; Schadt, C.W.; Doktycz, M.J.; et al. Diversity of Pseudomonas Genomes, Including Populus-Associated Isolates, as Revealed by Comparative Genome Analysis. Appl. Environ. Microbiol. 2016, 82, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Winsor, G.L.; Griffiths, E.J.; Lo, R.; Dhillon, B.K.; Shay, J.A.; Brinkman, F.S.L. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016, 44, D646–D653. [Google Scholar] [CrossRef] [Green Version]
- Freschi, L.; Jeukens, J.; Kukavica-Ibrulj, I.; Boyle, B.; Dupont, M.-J.; Laroche, J.; Larose, S.; Maaroufi, H.; Fothergill, J.L.; Moore, M.; et al. Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front. Microbiol. 2015, 6, 1036. [Google Scholar] [CrossRef] [Green Version]
- Jeukens, J.; Emond-Rheault, J.-G.; Freschi, L.; Kukavica-Ibrulj, I.; Levesque, R.C. Major Release of 161 Whole-Genome Sequences from the International Pseudomonas Consortium Database. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, P.N.; Savka, M.A.; Gan, H.M. In-silico Taxonomic Classification of 373 Genomes Reveals Species Misidentification and New Genospecies within the Genus Pseudomonas. Front. Microbiol. 2017, 8, 1296. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.D.; Vernikos, G.S.; Snyder, L.A.S.; Churcher, C.; Arrowsmith, C.; Chillingworth, T.; Cronin, A.; Davis, P.H.; Holroyd, N.E.; Jagels, K.; et al. Meningococcal genetic variation mechanisms viewed through comparative analysis of serogroup C strain FAM18. PLoS Genet. 2007, 3, e23. [Google Scholar] [CrossRef] [Green Version]
- Hiller, N.L.; Janto, B.; Hogg, J.S.; Boissy, R.; Yu, S.; Powell, E.; Keefe, R.; Ehrlich, N.E.; Shen, K.; Hayes, J.; et al. Comparative genomic analyses of seventeen Streptococcus pneumoniae strains: Insights into the pneumococcal supragenome. J. Bacteriol. 2007, 189, 8186–8195. [Google Scholar] [CrossRef] [Green Version]
- Méric, G.; Yahara, K.; Mageiros, L.; Pascoe, B.; Maiden, M.C.J.; Jolley, K.A.; Sheppard, S.K. A reference pan-genome approach to comparative bacterial genomics: Identification of novel epidemiological markers in pathogenic Campylobacter. PLoS ONE 2014, 9, e92798. [Google Scholar] [CrossRef]
- Rasko, D.A.; Rosovitz, M.J.; Myers, G.S.A.; Mongodin, E.F.; Fricke, W.F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N.R.; Chaudhuri, R.; et al. The pangenome structure of Escherichia coli: Comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 2008, 190, 6881–6893. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- Loveridge, E.J.; Jones, C.; Bull, M.J.; Moody, S.C.; Kahl, M.W.; Khan, Z.; Neilson, L.; Tomeva, M.; Adams, S.E.; Wood, A.C.; et al. Reclassification of the Specialized Metabolite Producer Pseudomonas mesoacidophila ATCC 31433 as a Member of the Burkholderia cepacia Complex. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Hesse, C.; Schulz, F.; Bull, C.T.; Shaffer, B.T.; Yan, Q.; Shapiro, N.; Hassan, K.A.; Varghese, N.; Elbourne, L.D.H.; Paulsen, I.T.; et al. Genome-based evolutionary history of Pseudomonas spp. Environ. Microbiol. 2018, 20, 2142–2159. [Google Scholar] [CrossRef]
- Gomila, M.; Peña, A.; Mulet, M.; Lalucat, J.; García-Valdés, E. Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 2015, 6, 214. [Google Scholar] [CrossRef] [Green Version]
- Ozer, E.A.; Allen, J.P.; Hauser, A.R. Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genom. 2014, 15, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, D.; Vijay, A.K.; Kohli, G.S.; Rice, S.A.; Willcox, M. Comparative genomics of clinical strains of Pseudomonas aeruginosa strains isolated from different geographic sites. Sci. Rep. 2018, 8, 15668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freschi, L.; Vincent, A.T.; Jeukens, J.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Dupont, M.-J.; Charette, S.J.; Boyle, B.; Levesque, R.C. The Pseudomonas aeruginosa Pan-Genome Provides New Insights on Its Population Structure, Horizontal Gene Transfer, and Pathogenicity. Genome Biol. Evol. 2019, 11, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Valot, B.; Guyeux, C.; Rolland, J.Y.; Mazouzi, K.; Bertrand, X.; Hocquet, D. What It Takes to Be a Pseudomonas aeruginosa? The Core Genome of the Opportunistic Pathogen Updated. PLoS ONE 2015, 10, e0126468. [Google Scholar] [CrossRef] [PubMed]
- Weiser, R.; Green, A.E.; Bull, M.J.; Cunningham-Oakes, E.; Jolley, K.A.; Maiden, M.C.J.; Hall, A.J.; Winstanley, C.; Weightman, A.J.; Donoghue, D.; et al. Not all Pseudomonas aeruginosa are equal: Strains from industrial sources possess uniquely large multireplicon genomes. Microb. Genom. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, B.E.; Yang, R.; Clatworthy, A.E.; White, T.; Osmulski, S.J.; Li, L.; Penaranda, C.; Lander, E.S.; Shoresh, N.; Hung, D.T. Defining the core essential genome of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2019, 116, 10072–10080. [Google Scholar] [CrossRef] [Green Version]
- Liberati, N.T.; Urbach, J.M.; Miyata, S.; Lee, D.G.; Drenkard, E.; Wu, G.; Villanueva, J.; Wei, T.; Ausubel, F.M. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. USA 2006, 103, 2833–2838. [Google Scholar] [CrossRef] [Green Version]
- Skurnik, D.; Roux, D.; Aschard, H.; Cattoir, V.; Yoder-Himes, D.; Lory, S.; Pier, G.B. A comprehensive analysis of In Vitro and in vivo genetic fitness of Pseudomonas aeruginosa using high-throughput sequencing of transposon libraries. PLoS Pathog. 2013, 9, e1003582. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.H.; Wessel, A.K.; Palmer, G.C.; Murray, J.L.; Whiteley, M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 2015, 112, 4110–4115. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Gallagher, L.A.; Thongdee, M.; Staudinger, B.J.; Lippman, S.; Singh, P.K.; Manoil, C. General and condition-specific essential functions of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2015, 112, 5189–5194. [Google Scholar] [CrossRef] [Green Version]
- Silby, M.W.; Cerdeño-Tárraga, A.M.; Vernikos, G.S.; Giddens, S.R.; Jackson, R.W.; Preston, G.M.; Zhang, X.-X.; Moon, C.D.; Gehrig, S.M.; Godfrey, S.A.C.; et al. Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol. 2009, 10, R51. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Salmerón, J.E.; Moreno-Hagelsieb, G.; Santoyo, G. Genome Comparison of Pseudomonas fluorescens UM270 with Related Fluorescent Strains Unveils Genes Involved in Rhizosphere Competence and Colonization. J. Genom. 2017, 5, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Sanz, D.; Arrebola, E.; Martínez-Granero, F.; García-Méndez, S.; Muriel, C.; Blanco-Romero, E.; Martín, M.; Rivilla, R.; Redondo-Nieto, M. Classification of Isolates from the Pseudomonas fluorescens Complex into Phylogenomic Groups Based in Group-Specific Markers. Front. Microbiol. 2017, 8, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udaondo, Z.; Molina, L.; Segura, A.; Duque, E.; Ramos, J.L. Analysis of the core genome and pangenome of Pseudomonas putida. Environ. Microbiol. 2016, 18, 3268–3283. [Google Scholar] [CrossRef]
- Koehorst, J.J.; van Dam, J.C.J.; van Heck, R.G.A.; Saccenti, E.; Dos Santos, V.A.P.M.; Suarez-Diez, M.; Schaap, P.J. Comparison of 432 Pseudomonas strains through integration of genomic, functional, metabolic and expression data. Sci. Rep. 2016, 6, 38699. [Google Scholar] [CrossRef] [Green Version]
- Lhospice, S.; Gomez, N.O.; Ouerdane, L.; Brutesco, C.; Ghssein, G.; Hajjar, C.; Liratni, A.; Wang, S.; Richaud, P.; Bleves, S.; et al. Pseudomonas aeruginosa zinc uptake in chelating environment is primarily mediated by the metallophore pseudopaline. Sci. Rep. 2017, 7, 17132. [Google Scholar] [CrossRef] [Green Version]
- McFarlane, J.S.; Lamb, A.L. Biosynthesis of an Opine Metallophore by Pseudomonas aeruginosa. Biochemistry 2017, 56, 5967–5971. [Google Scholar] [CrossRef]
- Ghssein, G.; Brutesco, C.; Ouerdane, L.; Fojcik, C.; Izaute, A.; Wang, S.; Hajjar, C.; Lobinski, R.; Lemaire, D.; Richaud, P.; et al. Biosynthesis of a broad-spectrum nicotianamine-like metallophore in Staphylococcus aureus. Science 2016, 352, 1105–1109. [Google Scholar] [CrossRef]
- Crespo, A.; Pedraz, L.; Astola, J.; Torrents, E. Pseudomonas aeruginosa Exhibits Deficient Biofilm Formation in the Absence of Class II and III Ribonucleotide Reductases Due to Hindered Anaerobic Growth. Front. Microbiol. 2016, 7, 688. [Google Scholar] [CrossRef] [Green Version]
- Mossialos, D.; Amoutzias, G.D. Role of siderophores in cystic fibrosis pathogenesis: Foes or friends? Int. J. Med. Microbiol. 2009, 299, 87–98. [Google Scholar] [CrossRef]
- Mossialos, D.; Amoutzias, G.D. Siderophores in fluorescent pseudomonads: New tricks from an old dog. Future Microbiol. 2007, 2, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Amoutzias, G.D.; Van de Peer, Y.; Mossialos, D. Evolution and taxonomic distribution of nonribosomal peptide and polyketide synthases. Future Microbiol. 2008, 3, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Amoutzias, G.D.; Chaliotis, A.; Mossialos, D. Discovery Strategies of Bioactive Compounds Synthesized by Nonribosomal Peptide Synthetases and Type-I Polyketide Synthases Derived from Marine Microbiomes. Mar. Drugs 2016, 14, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossialos, D.; Ochsner, U.; Baysse, C.; Chablain, P.; Pirnay, J.-P.; Koedam, N.; Budzikiewicz, H.; Fernández, D.U.; Schäfer, M.; Ravel, J.; et al. Identification of new, conserved, non-ribosomal peptide synthetases from fluorescent pseudomonads involved in the biosynthesis of the siderophore pyoverdine. Mol. Microbiol. 2002, 45, 1673–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, A.P.; Vasil, A.I.; Filloux, A.; Ball, G.; Wilderman, P.J.; Vasil, M.L. A novel extracellular phospholipase C of Pseudomonas aeruginosa is required for phospholipid chemotaxis. Mol. Microbiol. 2004, 53, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Wagner, V.E.; Bushnell, D.; Passador, L.; Brooks, A.I.; Iglewski, B.H. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: Effects of growth phase and environment. J. Bacteriol. 2003, 185, 2080–2095. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.C.; Sharma, A.; Brown, R.; Weatherly, M.; Moya, F.R.; Lewandoski, J.; Zimmerman, J.J. Function and composition of pulmonary surfactant and surfactant-derived fatty acid profiles are altered in young adults with cystic fibrosis. Chest 2000, 118, 164–174. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.-C.; Wu, Y.-Q.; Cao, D.; Zhang, W.-B.; Wang, H.-H. Only Acyl Carrier Protein 1 (AcpP1) Functions in Pseudomonas aeruginosa Fatty Acid Synthesis. Front. Microbiol 2017, 8, 2186. [Google Scholar] [CrossRef] [Green Version]
- Qiu, D.; Eisinger, V.M.; Rowen, D.W.; Yu, H.D. Regulated proteolysis controls mucoid conversion in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2007, 104, 8107–8112. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Damron, F.H.; Withers, T.R.; Pritchett, C.L.; Wang, X.; Schurr, M.J.; Yu, H.D. Expression of mucoid induction factor MucE is dependent upon the alternate sigma factor AlgU in Pseudomonas aeruginosa. BMC Microbiol. 2013, 13, 232. [Google Scholar] [CrossRef] [Green Version]
- Wood, L.F.; Ohman, D.E. Cell wall stress activates expression of a novel stress response facilitator (SrfA) under σ22 (AlgT/U) control in Pseudomonas aeruginosa. Microbiology 2015, 161, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, R.D.; Singh, P.; Hsu, F.; Güvener, T.; Carl, M.A.; Trinidad, R.R.S.; Silverman, J.M.; Ohlson, B.B.; Hicks, K.G.; Plemel, R.L.; et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 2010, 7, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, A.B.; Hood, R.D.; Bui, N.K.; LeRoux, M.; Vollmer, W.; Mougous, J.D. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 2011, 475, 343–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purssell, A.; Fruci, M.; Mikalauskas, A.; Gilmour, C.; Poole, K. EsrC, an envelope stress-regulated repressor of the mexCD-oprJ multidrug efflux operon in Pseudomonas aeruginosa. Environ. Microbiol. 2015, 17, 186–198. [Google Scholar] [CrossRef]
- Tielker, D.; Hacker, S.; Loris, R.; Strathmann, M.; Wingender, J.; Wilhelm, S.; Rosenau, F.; Jaeger, K.-E. Pseudomonas aeruginosa lectin LecB is located in the outer membrane and is involved in biofilm formation. Microbiology 2005, 151, 1313–1323. [Google Scholar] [CrossRef]
- Mitchell, E.; Houles, C.; Sudakevitz, D.; Wimmerova, M.; Gautier, C.; Pérez, S.; Wu, A.M.; Gilboa-Garber, N.; Imberty, A. Structural basis for oligosaccharide-mediated adhesion of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Nat. Struct. Mol. Biol. 2002, 9, 918–921. [Google Scholar] [CrossRef]
- Gilboa-Garber, N.; Katcoff, D.J.; Garber, N.C. Identification and characterization of Pseudomonas aeruginosa PA-IIL lectin gene and protein compared to PA-IL. FEMS Immunol. Med. Microbiol. 2000, 29, 53–57. [Google Scholar] [CrossRef]
- Chin-A-Woeng, T.F.; Bloemberg, G.V.; Mulders, I.H.; Dekkers, L.C.; Lugtenberg, B.J. Root colonization by phenazine-1-carboxamide-producing bacterium Pseudomonas chlororaphis PCL1391 is essential for biocontrol of tomato foot and root rot. Mol. Plant Microbe Interact. 2000, 13, 1340–1345. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.N.; Smith, D.L.; Young, R. Holins: The protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 2000, 54, 799–825. [Google Scholar] [CrossRef]
- Dorosky, R.J.; Yu, J.M.; Pierson, L.S.; Pierson, E.A. Pseudomonas chlororaphis Produces Two Distinct R-Tailocins That Contribute to Bacterial Competition in Biofilms and on Roots. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [Green Version]
- Dorosky, R.J.; Pierson, L.S.; Pierson, E.A. Pseudomonas chlororaphis Produces Multiple R-Tailocin Particles That Broaden the Killing Spectrum and Contribute to Persistence in Rhizosphere Communities. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domalaon, R.; Ammeter, D.; Brizuela, M.; Gorityala, B.K.; Zhanel, G.G.; Schweizer, F. Repurposed Antimicrobial Combination Therapy: Tobramycin-Ciprofloxacin Hybrid Augments Activity of the Anticancer Drug Mitomycin C Against Multidrug-Resistant Gram-Negative Bacteria. Front. Microbiol 2019, 10, 1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.-B.; Singh, R.K.; Singh, P.; Song, Q.-Q.; Xing, Y.-X.; Yang, L.-T.; Li, Y.-R. Genetic Diversity of Nitrogen-Fixing and Plant Growth Promoting Pseudomonas Species Isolated from Sugarcane Rhizosphere. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus Core4 | Pseudomonas aeruginosa | Pseudomonas chlororaphis | Pseudomonas stutzeri | Pseudomonas putida | Pseudomonas fluorescens | Pseudomonas syringae | |
|---|---|---|---|---|---|---|---|
| Number_of_Strains | 491 | 189 | 43 | 19 | 63 | 96 | 34 |
| Amino acids in phylogenomic alignment | Core1:27,997 | 31,145 | 103,483 | 193,427 | 155,470 | 115,099 | 110,643 |
| Core proteins | 297 | 1811 | 3587 | 2080 | 1724 | 1396 | 2944 |
| % core proteins with significant presence in other groups (≥90% presence) | 100.0 | 44.3 | 38.7 | 70.1 | 70.5 | 62.7 | 50.1 |
| Group-specific core proteins | - | 41 | 11 | 7 | 1 | 0 | 0 |
| Relaxed group-specific core proteins (10% in others) | - | 84 | 32 | 32 | 4 | 0 | 61 |
| Relaxed group-specific core proteins (20% in others) | - | 116 | 61 | 51 | 4 | 0 | 87 |
| %core—Unknown | 19.9 | 33.4 | 30.4 | 27.1 | 25.4 | 26.6 | 31.3 |
| %core—Other | 24.9 | 16.6 | 20.1 | 19.9 | 17.9 | 17.1 | 18.8 |
| %core—K:Transcription | 4.7 | 7.3 | 7.8 | 4.6 | 5.4 | 6.5 | 5.9 |
| %core—E:Amino acid transport and metabolism | 11.4 | 7.2 | 8.6 | 7.2 | 9.5 | 10.7 | 8.2 |
| %core—P:Inorganic ion transport and metabolism | 4.4 | 5.9 | 5.5 | 4.5 | 5.5 | 4.7 | 5.8 |
| %core—C:Energy production and conversion | 4.4 | 6.5 | 5.8 | 6.3 | 7.0 | 6.1 | 4.8 |
| %core—M:Cell wall/membrane/envelope biogenesis | 6.7 | 4.4 | 5.5 | 6.1 | 5.3 | 5.2 | 5.7 |
| %core—J:Translation, ribosomal structure and biogenesis | 13.1 | 5.6 | 4.2 | 6.9 | 7.1 | 6.0 | 5.2 |
| %core—H:Coenzyme transport and metabolism | 7 | 3.4 | 3.5 | 4.6 | 5.0 | 5.1 | 4.2 |
| %core—L:Replication, recombination and repair | 3.4 | 3.0 | 2.8 | 5.1 | 4.4 | 4.0 | 3.7 |
| Orthologue | Closest Paralogue in Reference Strain | Closest Homologue outside of Group | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Organism | Name | General Function | Presence within Group | Presence outside of Group | Accession | Name | Annotation | % Identity | Organism | Name | Annotation | % Identity |
| P. aeruginosa | cntO | pseudopalin production | 185/189 | 234/305 | PA0151 | - | probable TonB-dependent receptor | 34.84 | Pseudomonas furukawaii | KF707C_RS00865 | Ton-B-dependent receptor | 34.56 |
| P. aeruginosa | cntL | pseudopalin production | 189/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | cntM | pseudopalin production | 189/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | cntI | pseudopalin production | 187/189 | 2/305 | PA2628 | - | hypothetical protein | 34.17 | Pseudomonas mendocina | EL191_RS11685 | DMT family transporter | 34.52 |
| P. aeruginosa | pvdL | pyoverdine biosynthesis | 171/189 | 255/305 | PA2400 | pvdJ | pvdJ | 47.28 | Pseudomonas fulva | CJ462_RS07705 | amino acid adenylation domain-containing protein | 39.98 |
| P. aeruginosa | pchF | pyochelin biosynthesis | 176/189 | 58/305 | PA4226 | pchE | dihydroaeruginoic acid synthetase | 39.57 | Pseudomonas sp. R2-7-07 9503 | C4J86_RS18060 | amino acid adenylation domain-containing protein | 26 |
| P. aeruginosa | plcB | motility | 189/189 | 0/305 | - | - | - | - | - | - | - | |
| P. aeruginosa | acp1 | motility | 189/189 | 0/305 | PA2966 | acpP | acyl carrier protein | 53.4 | Pseudomonas mesoacidophila | B7P44_RS05820 | acyl carrier protein | 61.11 |
| P. aeruginosa | mucE | mucoidy | 189/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | srfA | mucoidy | 189/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | tse1 | toxin/antitoxin operon | 189/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | tsi1 | toxin/antitoxin operon | 189/189 | 1/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | tse2 | toxin/antitoxin operon | 187/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | tsi2 | toxin/antitoxin operon | 189/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | tse3 | toxin/antitoxin operon | 189/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | tsi3 | toxin/antitoxin operon | 186/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. aeruginosa | esrC | multidrug efflux | 189/189 | 0/305 | PA4600 | nfxB | transcriptional regulator NfxB | 60.71 | Pseudomonas chlororaphis | C4K30_RS15575 | TetR/AcrR family transcriptional regulator | 66.47 |
| P. aeruginosa | mexC | multidrug efflux | 186/189 | 240/305 | PA0425 | mexA | resistance-nodulation cell division (RND) multidrug efflux membrane fusion protein MexA precursor | 44.76 | P. sp URMO17WK12 I11 | PSHI_RS20595 | hemolysin secretion protein D | 27.78 |
| P. aeruginosa | mexD | multidrug efflux | 188/189 | 264/305 | PA2018 | mexY | resistance nodulation cell division (RND) multidrug efflux transporter MexY | 52 | Pseudomonas alcaligenes | A0T30_RS19090 | multidrug efflux RND transporter permease subunit | 53.09 |
| P. aeruginosa | lecB | biofilm formation | 188/189 | 0/305 | - | - | - | - | - | - | - | - |
| P. chlororaphis | C4K22 RS23595 | holin family | 43/43 | 0/451 | - | - | - | - | - | - | - | - |
| P. chlororaphis | C4K22 RS23415 | mitomycin biosynthesis | 42/43 | 0/451 | - | - | - | - | - | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolaidis, M.; Mossialos, D.; Oliver, S.G.; Amoutzias, G.D. Comparative Analysis of the Core Proteomes among the Pseudomonas Major Evolutionary Groups Reveals Species-Specific Adaptations for Pseudomonas aeruginosa and Pseudomonas chlororaphis. Diversity 2020, 12, 289. https://doi.org/10.3390/d12080289
Nikolaidis M, Mossialos D, Oliver SG, Amoutzias GD. Comparative Analysis of the Core Proteomes among the Pseudomonas Major Evolutionary Groups Reveals Species-Specific Adaptations for Pseudomonas aeruginosa and Pseudomonas chlororaphis. Diversity. 2020; 12(8):289. https://doi.org/10.3390/d12080289
Chicago/Turabian StyleNikolaidis, Marios, Dimitris Mossialos, Stephen G. Oliver, and Grigorios D. Amoutzias. 2020. "Comparative Analysis of the Core Proteomes among the Pseudomonas Major Evolutionary Groups Reveals Species-Specific Adaptations for Pseudomonas aeruginosa and Pseudomonas chlororaphis" Diversity 12, no. 8: 289. https://doi.org/10.3390/d12080289
APA StyleNikolaidis, M., Mossialos, D., Oliver, S. G., & Amoutzias, G. D. (2020). Comparative Analysis of the Core Proteomes among the Pseudomonas Major Evolutionary Groups Reveals Species-Specific Adaptations for Pseudomonas aeruginosa and Pseudomonas chlororaphis. Diversity, 12(8), 289. https://doi.org/10.3390/d12080289