
16S rRNA–Based Analysis Reveals Differences in the Bacterial Community Present in Tissues of Choromytilus chorus (Mytilidae, Bivalvia) Grown in an Estuary and a Bay in Southern Chile

 , ,
, ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Material and Methods
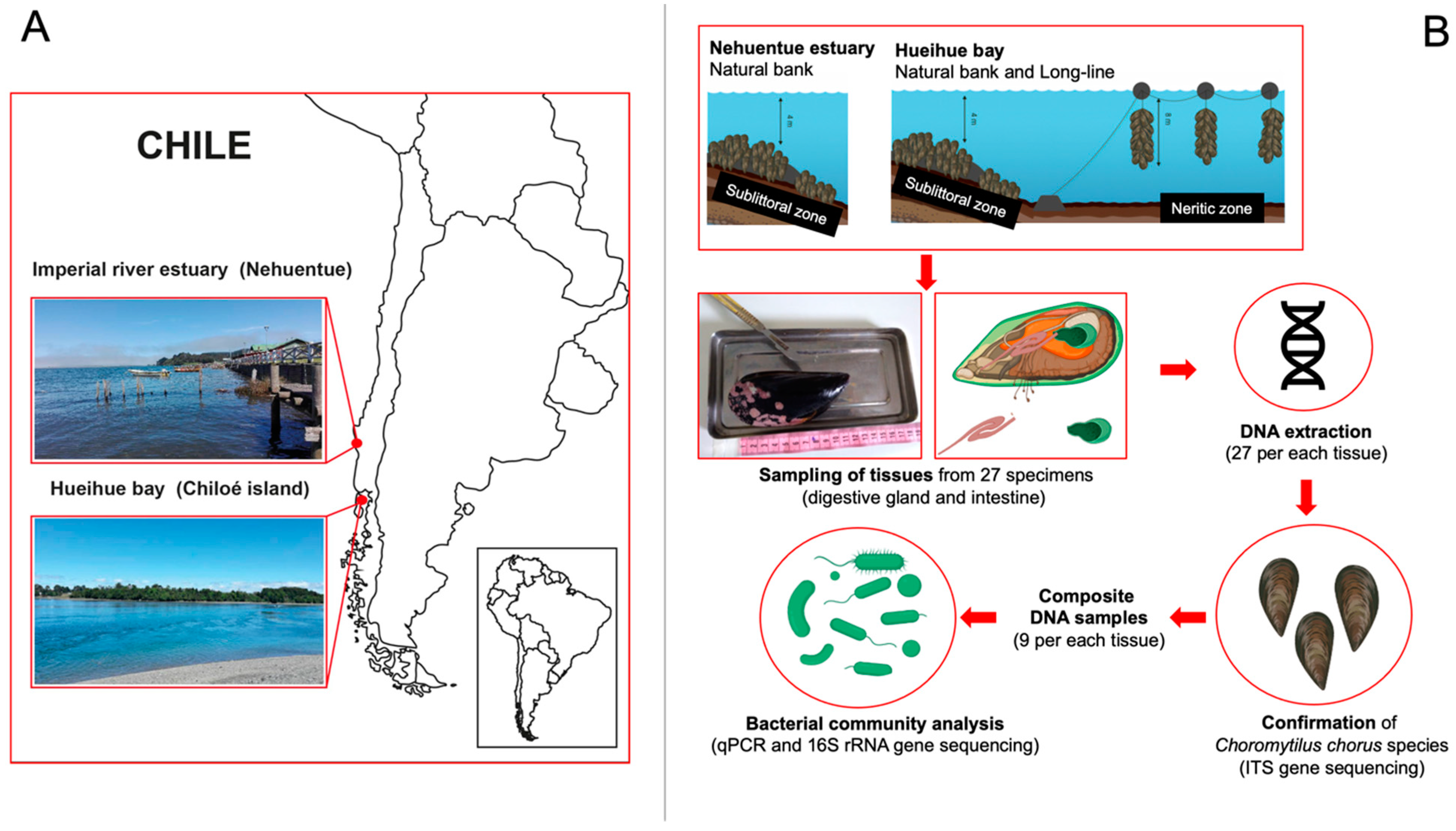
2.1. Collection of Specimens
2.2. Samples
2.3. DNA Extraction
2.4. Collection of Choromytilus Chorus
2.5. Bacterial Abundance in Digestive Gland and Intestinal Tissues
2.6. Bacterial Community in Digestive Gland and Intestinal Tissues
2.7. Bioinformatics and Statistical Analyses
3. Results
3.1. Collection of Choromytilus Chorus
3.2. Bacterial Abundances in Digestive Gland and Intestinal Tissues
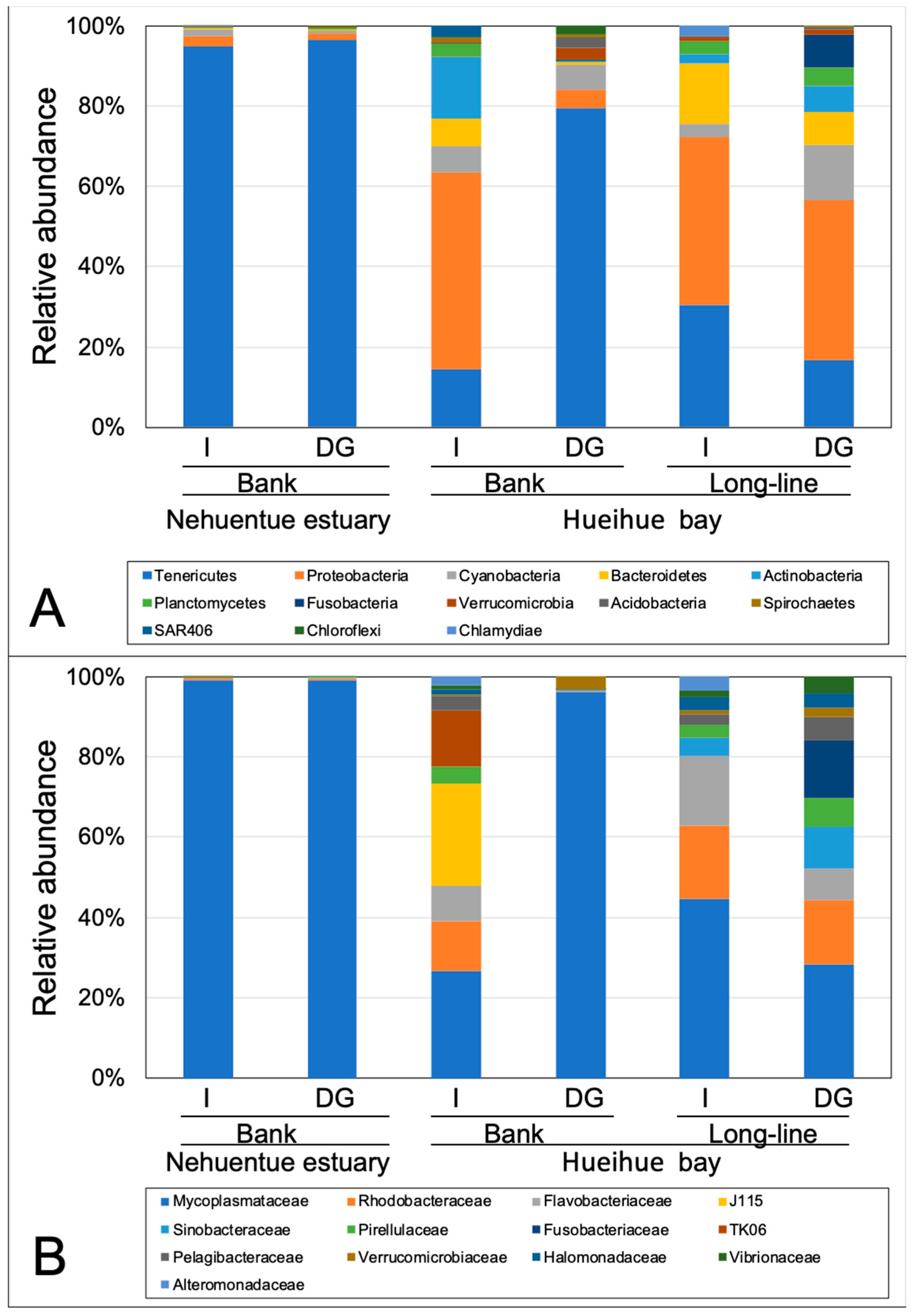
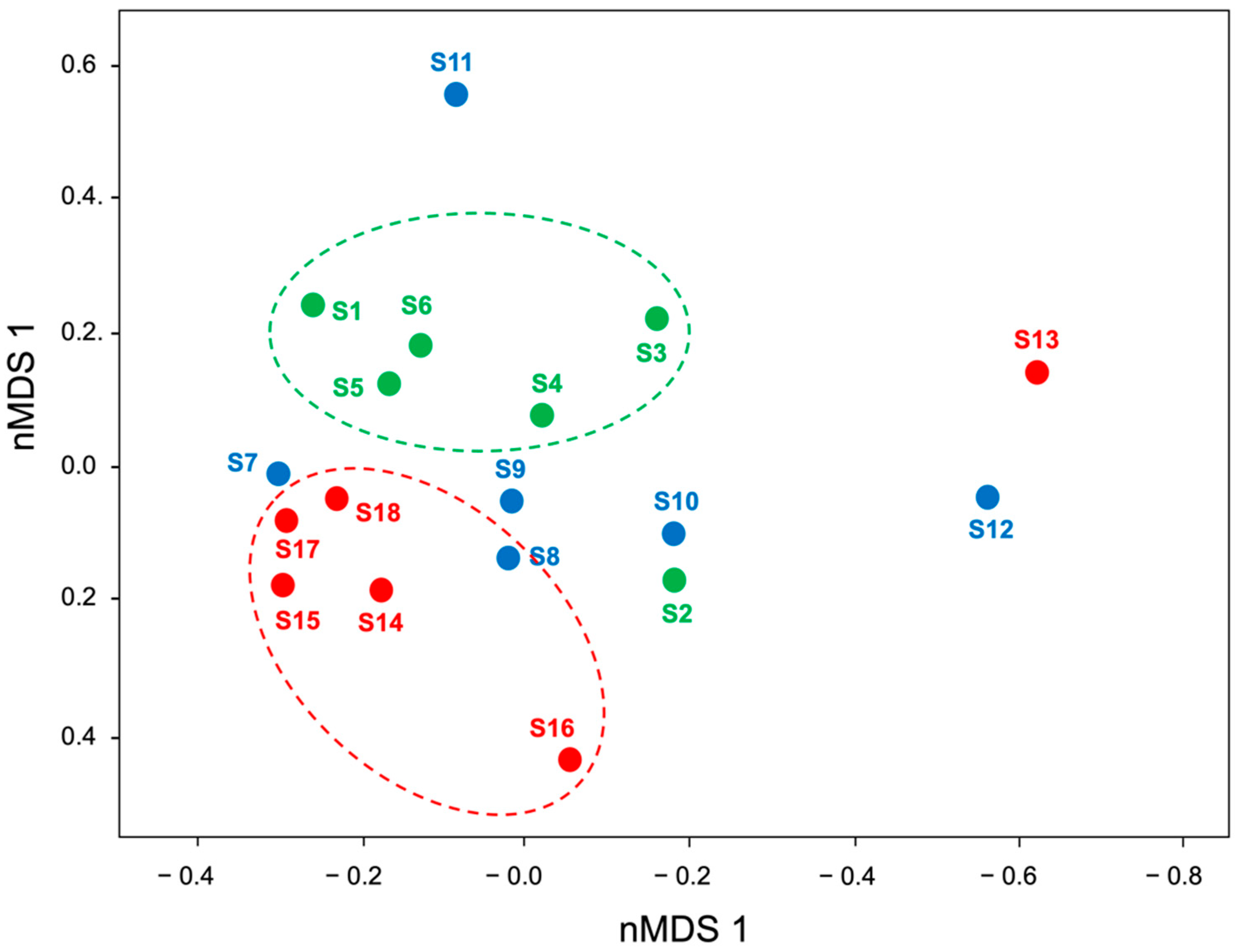
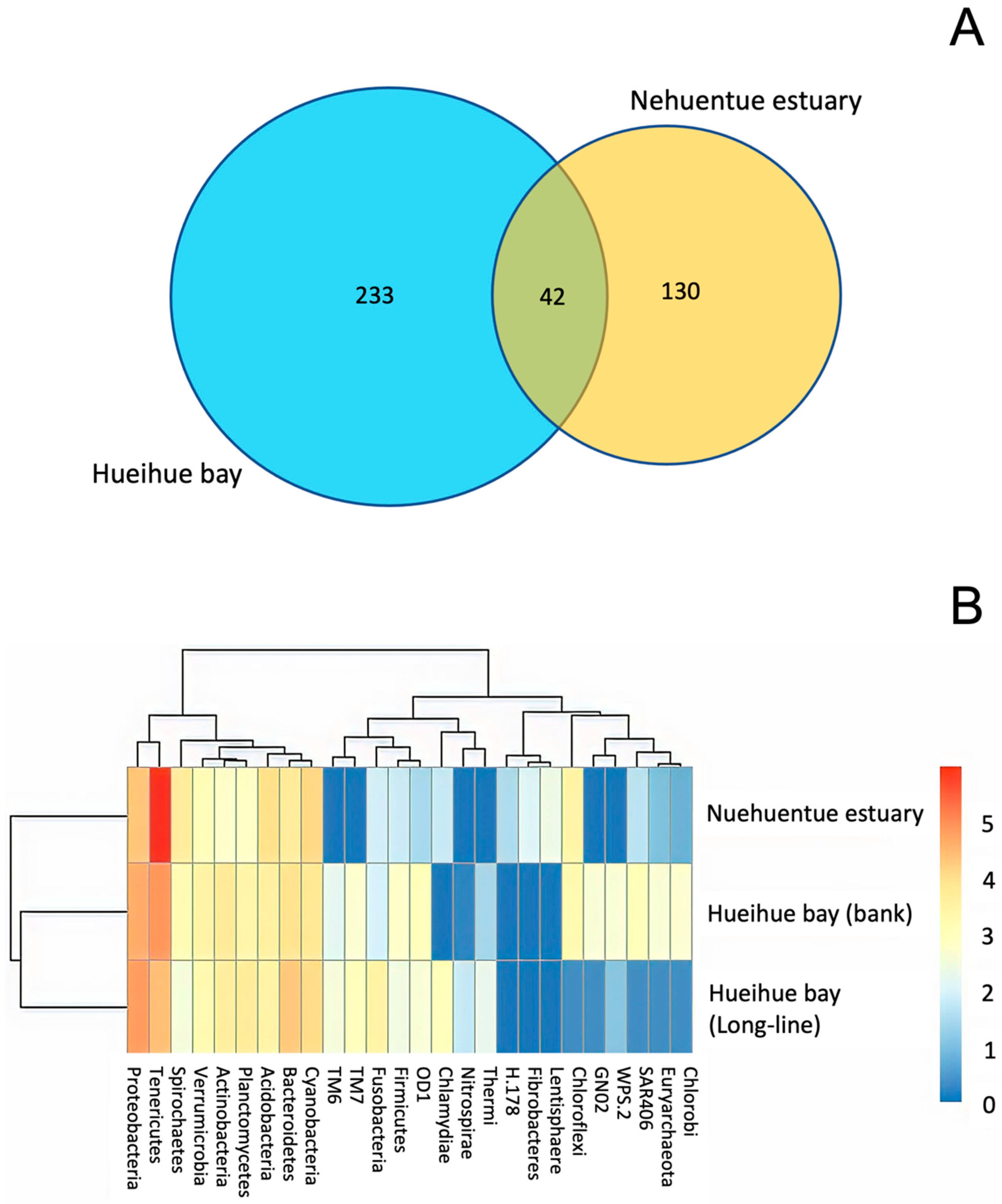
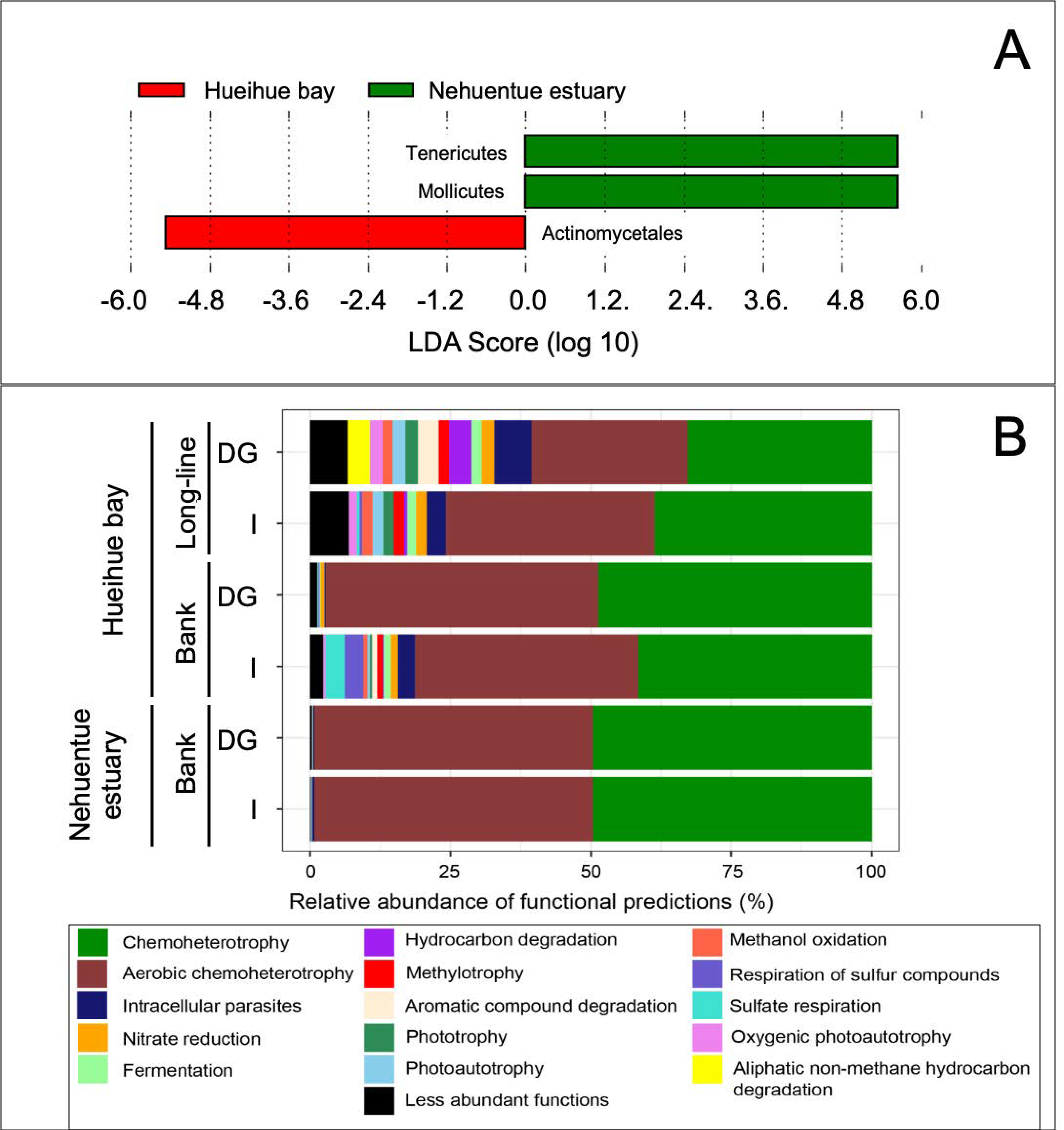
3.3. Bacterial Community in Digestive Gland and Intestinal Tissues
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wijsman, J.W.M.; Troost, K.; Fang, J.; Roncarati, A. Global production of marine bivalves. Trends and challenges. In Goods and Services of Marine Bivalves; Smaal, A., Ferreira, J., Grant, J., Petersen, J., Strand, Ø., Eds.; Springer: Cham, Switzerland, 2019; Volume 2, pp. 7–26. [Google Scholar] [CrossRef] [Green Version]
- FAO. El estado mundial de la pesca y la acuicultura. In Food and Agriculture Organization of the United Nations; FAO: Rome, Italy, 2018; Available online: http://www.fao.org/3/I9540ES/i9540es.pdf (accessed on 12 May 2021).
- Reid, D.G.; Osorio, C. The shallow-water marine mollusca of the estero elefantes and laguna. san rafael, southern Chile. Bull. Br. Mus. Nat. Hist. London 2000, 66, 109–146. Available online: https://www.biodiversitylibrary.org/page/41005314#page/3/mode/1up (accessed on 12 May 2021).
- Navarro, J.M. The effects of salinity on the physiological ecology of Choromytilus chorus (Molina, 1782) (Bivalvia: Mytilidae). J. Exp. Mar. Biol. Ecol. 1988, 122, 19–33. [Google Scholar] [CrossRef]
- Campos, B.; Landaeta, M.F. Moluscos planctónicos entre el fiordo Reloncaví y el golfo Corcovado, sur de Chile: Ocurrencia, distribución y abundancia en invierno. Rev. Biol. Mar. Oceanogr. 2016, 51, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Hazen, T.H.; Pan, L.; Gu, J.-D.; Sobecky, P.A. The contribution of mobile genetic elements to the evolution and ecology of Vibrios. FEMS Microbiol. Ecol. 2010, 74, 485–499. [Google Scholar] [CrossRef] [Green Version]
- La Valley, K.J.; Jones, S.; Gómez-Chiarri, M.; DeAlteris, J.; Rice, M. Bacterial Community Profiling of the Eastern oyster (Crassostrea virginica): Comparison of Culture-Dependent and Culture-Independent Outcomes. J. Shellfish. Res. 2009, 28, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Pierce, M.L.; Ward, J.E.; Holohan, B.A.; Zhao, X.; Hicks, R.E. The influence of site and season on the gut and pallial fluid microbial communities of the eastern oyster, Crassostrea virginica (Bivalvia, Ostreidae): Community-level physiological profiling and genetic structure. Hydrobiol. 2016, 765, 97–113. [Google Scholar] [CrossRef]
- Rubiolo, J.A.; Botana, L.M.; Martínez, P. Insights into mussel microbiome. Microb. Communities Aquac. Ecosyst. 2019, 3, 95–120. [Google Scholar] [CrossRef]
- Austin, B. The bacterial microflora of fish, revised. Sci. World J. 2006, 6, 931–945. [Google Scholar] [CrossRef] [Green Version]
- Sweet, M.J.; Bulling, M.T. On the importance of the microbiome and pathobiome in coral health and disease. Front. Mar. Sci. 2017, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Flores, R.; Cáceres-Martínez, J. Rickettsiales-like organisms in bivalves and marine gastropods: A review. Rev. Aquac. 2020, 12, 1–17. [Google Scholar] [CrossRef]
- Carella, F.; Carrasco, N.; Andree, K.B.; Lacuesta, B.; Furones, D.; De Vico, G. Nocardiosis in Mediterranean bivalves: First detection of Nocardia crassostreae in a new host Mytilus galloprovincialis and in Ostrea edulis from the gulf of Naples (Italy). J. Invertebr. Pathol. 2013, 114, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.J.G.; Acheson, D. Cholera and other types of vibriosis: A story of human pandemics and oysters on the half shell. Clin. Infect. Dis. 2003, 37, 272–280. [Google Scholar] [CrossRef]
- Travers, M.-A.; Miller, K.B.; Roque, A.; Friedman, C.S. Bacterial diseases in marine bivalves. J. Invertebr. Pathol. 2015, 131, 11–31. [Google Scholar] [CrossRef] [Green Version]
- Clerissi, C.; De Lorgeril, J.; Petton, B.; Lucasson, A.; Escoubas, J.-M.; Gueguen, Y.; Dégremont, L.; Mitta, G.; Toulza, E. Microbiota composition and evenness predict survival rate of oysters confronted to pacific oyster mortality syndrome. Front. Microbiol. 2020, 11, 311. [Google Scholar] [CrossRef] [Green Version]
- Milan, M.; Smits, M.; Rovere, G.D.; Iori, S.; Zampieri, A.; Carraro, L.; Martino, C.; Papetti, C.; Ianni, A.; Ferri, N.; et al. Host-microbiota interactions shed light on mortality events in the striped venus clam Chamelea gallina. Mol. Ecol. 2019, 28, 4486–4499. [Google Scholar] [CrossRef] [PubMed]
- Lasa, A.; Di Cesare, A.; Tassistro, G.; Borello, A.; Gualdi, S.; Furones, D.; Carrasco, N.; Cheslett, D.; Brechon, A.; Paillard, C.; et al. Dynamics of the Pacific oyster pathobiota during mortality episodes in Europe assessed by 16S rRNA gene profiling and a new target enrichment next-generation sequencing strategy. Environ. Microbiol. 2019, 21, 4548–4562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musella, M.; Wathsala, R.; Tavella, T.; Rampelli, S.; Barone, M.; Palladino, G.; Biagi, E.; Brigidi, P.; Turroni, S.; Franzellitti, S.; et al. Tissue-scale microbiota of the Mediterranean mussel (Mytilus galloprovincialis) and its relationship with the environment. Sci. Total. Environ. 2020, 717, 137209. [Google Scholar] [CrossRef]
- De La Fuente, M.; Miranda, C.; Faundez, V. Bacteriología asociada al cultivo de moluscos en Chile: Avances y perspectivas. Rev. Biol. Mar. Oceanogr. 2015, 50, 1–12. [Google Scholar] [CrossRef]
- Tello, M.; Valdes, N.; Vargas, R.; Rojas, J.; Parra, M.; Gajardo, M.; Gonzalez, A. Metagenomics, Basics, Methods and Applications; IntechOpen: Santiago, Chile, 2019; Volume 8, pp. 286–302. [Google Scholar] [CrossRef]
- Jorquera, M.; Silva, F.; Riquelme, C. Bacteria in the culture of the scallop Argopecten purpuratus (Lamarck, 1819). Aquac. Int. 2001, 9, 285–303. [Google Scholar] [CrossRef]
- Rojas, R.; Miranda, C.D.; Opazo, R.; Romero, J. Characterization and pathogenicity of Vibrio splendidus strains associated with massive mortalities of commercial hatchery-reared larvae of scallop Argopecten purpuratus (Lamarck, 1819). J. Invertebr. Pathol. 2015, 124, 61–69. [Google Scholar] [CrossRef]
- Romero, J.; Gonzalez, N.; Espejo, R. Marine Pseudoalteromonas sp. Composes most of the bacterial population developed in oysters spoiled during storage. J. Food Sci. 2002, 67, 2300–2303. [Google Scholar] [CrossRef]
- Trabal, N.; Mazón-Suástegui, J.M.; Vázquez-Juárez, R.; Asencio-Valle, F.; Morales-Bojórquez, E.; Romero, J. Molecular analysis of bacterial microbiota associated with oysters (Crassostrea gigas and Crassostrea corteziensis) in different growth phases at two cultivation sites. Microb. Ecol. 2012, 64, 555–569. [Google Scholar] [CrossRef]
- Aranda, C.P.; Yevenes, M.; Rodriguez-Benito, C.; Godoy, F.A.; Ruiz, M.; Cachicas, V. Distribution and growth of Vibrio parahaemolyticus in Southern Chilean clams (Venus antiqua) and Blue Mussels (Mytilus chilensis). Foodborne Pathog. Dis. 2015, 12, 1–7. [Google Scholar] [CrossRef]
- Larenas, J.; Perez, M.; Moraleda, C.; Godoy, M.; Larenas, C.; Acuña-Retamar, M. In vitro adhesion and infectiveness of Piscirickettsia salmonis on mussel shells Mytilus chilensis. Bull. Eur. Ass. Fish Pathol. 2019, 39, 114–121. Available online: https://eafp.org/download/2019-volume39/issue_3/39-3-114-larenas.pdf (accessed on 12 May 2021).
- Cremonte, F.; Puebla, C.; Tilleria, J.; Videla, V. Histopathological survey of the mussel Mytilus chilensis (Mytilidae) and the clam gari solida (psammobiidae) from southern Chile. Lat. Am. J. Aquat. Res. 2015, 43, 248–254. [Google Scholar] [CrossRef]
- Faggio, C.; Tsarpali, V.; Dailianis, S. Mussel digestive gland as a model tissue for assessing xenobiotics: An overview. Sci. Total. Environ. 2018, 636, 220–229. [Google Scholar] [CrossRef]
- Meyer, A. DNA technology and Phylogeny of fish. Gen. Evol. Aqua. Org. 1994, 5, 219–290. [Google Scholar]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Shade, A.; McManus, P.S.; Handelsman, J. Unexpected diversity during community succession in the apple flower microbiome. mBio 2013, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Acuña, J.J.; Inostroza, N.G.; Mora, M.L.; Radic, S.; Sadowsky, M.J.; Jorquera, M.A. Endophytic bacterial communities associated with roots and leaves of plants growing in chilean extreme environments. Sci. Rep. 2019, 9, 4950. [Google Scholar] [CrossRef] [Green Version]
- Gohl, D.M.; Vangay, P.; Garbe, J.; MacLean, A.; Hauge, A.; Becker, A.; Gould, T.J.; Clayton, J.B.; Johnson, J.B.C.T.J.; Hunter, R.; et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 2016, 34, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. 2011, 36, 10.7.1–10.7.20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.K.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.J.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2011, 6, 610–618. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D. Vegan: Community Ecology Package v.2. Available online: https://cran.rproject.org/package=vegan (accessed on 12 May 2021).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- SantaClara, F.J.; Espiñeira, M.; Cabado, A.G.; Aldasoro, A.; Gonzalez-Lavín, N.; Vieites, J.M. Development of a method for the genetic identification of mussel species belonging to mytilus, perna, aulacomya, and other genera. J. Agric. Food Chem. 2006, 54, 8461–8470. [Google Scholar] [CrossRef]
- Aguilera-Muñoz, F.; Valenzuela-Muñoz, V.; Gallardo-Escárate, C. Authentication of commercial chilean mollusks using ribosomal internal transcribed spacer (its) as specie-specific DNA marker. Gayana (Concepción) 2008, 72, 178–187. [Google Scholar] [CrossRef]
- Olafsen, J.A.; Mikkelsen, H.V.; Giæver, H.M.; Hansen, G.H. Indigenous bacteria in hemolymph and tissues of marine bivalves at low temperatures. Appl. Environ. Microbiol. 1993, 59, 1848–1854. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Xue, C.-H.; Liu, Q. Changes in microbial flora of Pacific oysters (Crassostrea gigas) during refrigerated storage and its shelf-life extension by chitosan. Int. J. Food Microbiol. 2009, 131, 272–276. [Google Scholar] [CrossRef]
- Hatha, A.A.M.; Christi, K.S.; Singh, R.; Kumar, S. Bacteriology of the fresh water bivalve clam Batissa violacea (Kai) sold in the Suva market. S. Pac. J. Nat. Appl. Sci. 2005, 23, 48–50. [Google Scholar] [CrossRef] [Green Version]
- Rao, K.V.S.; Natarajan, K.; Kumar, L.V.; Manivel, V.; Ahmad, J.; Chatterji, A.; Ansari, Z.A. Extract from an Indian Green Mussel (Perna viridis) for Differentiation and Maturation of Dendric Cells. U.S. Patent 20030401622, 28 March 2003. Available online: https://europepmc.org/article/pat/us2004009596 (accessed on 12 May 2021).
- Moriarty, D.J. The role of microorganisms in aquaculture ponds. Aquaculture 1997, 151, 333–349. [Google Scholar] [CrossRef]
- Westerdahl, A.; Olsson, J.C.; Kjelleberg, S.; Conway, P.L. Isolation and characterization of turbot (scophtalmus maximus)-associated bacteria with inhibitory effects against Vibrio anguillarum. Appl. Environ. Microbiol. 1991, 57, 2223–2228. [Google Scholar] [CrossRef] [Green Version]
- Aceves, A.K.; Johnson, P.; Bullard, S.A.; LaFrentz, S.; Arias, C.R. Description and characterization of the digestive gland microbiome in the freshwater mussel villosa nebulosa (Bivalvia: Unionidae). J. Molluscan Stud. 2018, 84, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Green, T.; Barnes, A. Bacterial diversity of the digestive gland of Sydney rock oysters, Saccostrea glomeratainfected with the paramyxean parasite, Marteilia sydneyi. J. Appl. Microbiol. 2010, 109, 613–622. [Google Scholar] [CrossRef]
- Lokmer, A.; Kuenzel, S.; Baines, J.F.; Wegner, K.M. The role of tissue-specific microbiota in initial establishment success of pacific oysters. Environ. Microbiol. 2016, 18, 970–987. [Google Scholar] [CrossRef] [Green Version]
- Pierce, M.L.; Ward, J.E. Gut microbiomes of the eastern oyster (Crassostrea virginica) and the Blue Mussel (Mytilus edulis): Temporal variation and the influence of marine aggregate-associated microbial communities. mSphere 2019, 4, e00730-19. [Google Scholar] [CrossRef] [Green Version]
- Cleary, D.F.R.; Becking, L.E.; Polónia, A.R.M.; Freitas, R.M.; Gomes, N.C.M. Composition and predicted functional ecology of mussel-associated bacteria in Indonesian marine lakes. Anton. Leeuw. Int. J G. 2015, 107, 821–834. [Google Scholar] [CrossRef]
- Weingarten, E.A.; Atkinson, C.L.; Jackson, C.R. The gut microbiome of freshwater unionidae mussels is determined by host species and is selectively retained from filtered seston. PLoS ONE 2019, 14, e0224796. [Google Scholar] [CrossRef] [PubMed]
- Offret, C.; Paulino, S.; Gauthier, O.; Château, K.; Bidault, A.; Corporeau, C.; Miner, P.; Petton, B.; Pernet, F.; Fabioux, C.; et al. The marine intertidal zone shapes oyster and clam digestive bacterial microbiota. FEMS Microbiol. Ecol. 2020, 96, fiaa078. [Google Scholar] [CrossRef] [PubMed]
- Dubé, C.E.; Ky, C.-L.; Planes, S. Microbiome of the black-lipped pearl oyster pinctada margaritifera, a multi-tissue description with functional profiling. Front. Microbiol. 2019, 10, 1548. [Google Scholar] [CrossRef] [PubMed]
- Schill, W.B.; Iwanowicz, D.; Adams, C. Endozoicomonas dominates the gill and intestinal content microbiomes of Mytilus edulis from Barnegat Bay, New Jersey. J. Shellfish. Res. 2017, 36, 391–401. [Google Scholar] [CrossRef]
- King, G.M.; Judd, C.; Kuske, C.R.; Smith, C. Analysis of stomach and gut microbiomes of the Eastern oyster (Crassostrea virginica) from Coastal Louisiana, USA. PLoS ONE 2012, 7, e51475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, N.T.; Mazón-Suástegui, J.M.; Vázquez-Juárez, R.; Ascencio-Valle, F.; Romero, J. Changes in the composition and diversity of the bacterial microbiota associated with oysters (Crassostrea corteziensis, Crassostrea gigas and Crassostrea sikamea) during commercial production. FEMS Microbiol. Ecol. 2014, 88, 69–83. [Google Scholar] [CrossRef] [Green Version]
- Arfken, A.; Song, B.; Bowman, J.S.; Piehler, M. Denitrification potential of the eastern oyster microbiome using a 16S rRNA gene based metabolic inference approach. PLoS ONE 2017, 12, e0185071. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; Van Der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Li, Y.-F.; Xu, J.-K.; Chen, Y.-W.; Ding, W.-Y.; Shao, A.-Q.; Liang, X.; Zhu, Y.-T.; Yang, J.-L. Characterization of gut microbiome in the mussel Mytilus galloprovincialis in response to thermal stress. Front. Physiol. 2019, 10, 1086–1118. [Google Scholar] [CrossRef] [Green Version]
- Harshbarger, J.; Chang, S. Chlamydiae (with phages), mycoplasmas, and richettsiae in Chesapeake Bay bivalves. Science 1977, 196, 666–668. [Google Scholar] [CrossRef]
- Sun, F.; Wang, Y.; Wang, C.; Zhang, L.; Tu, K.; Zheng, Z. Insights into the intestinal microbiota of several aquatic organisms and association with the surrounding environment. Aquaculture 2019, 507, 196–202. [Google Scholar] [CrossRef]
- Sun, F.; Xu, Z. Significant differences in intestinal microbial communities in aquatic animals from an aquaculture area. J. Mar. Sci. Eng. 2021, 9, 104. [Google Scholar] [CrossRef]
- Cavallo, R.A.; Acquaviva, M.I.; Stabili, L. Culturable heterotrophic bacteria in seawater and Mytilus galloprovincialis from a mediterranean area (Northern Ionian Sea—Italy). Environ. Monit. Assess. 2009, 149, 465–475. [Google Scholar] [CrossRef]
- Distel, D.L.; Altamia, M.A.; Lin, Z.; Shipway, J.R.; Han, A.; Forteza, I.; Antemano, R.; Limbaco, M.G.J.P.; Tebo, A.G.; Dechavez, R.; et al. Discovery of chemoautotrophic symbiosis in the giant shipwormKuphus polythalamia(Bivalvia: Teredinidae) extends wooden-steps theory. Proc. Natl. Acad. Sci. USA 2017, 114, E3652–E3658. [Google Scholar] [CrossRef] [Green Version]
- Zaccone, R.; Caruso, G.; Calì, C. Heterotrophic bacteria in the northern Adriatic Sea: Seasonal changes and ectoenzyme profile. Mar. Environ. Res. 2002, 54, 1–19. [Google Scholar] [CrossRef]
- Zheng, Z.; Liao, Y.; Ye, J.; Yang, C.; Adzigbli, L.; Wang, Q.; Du, X.; Deng, Y. Microbiota diversity in pearl oyster pinctada fucata martensii intestine and its aquaculture environment. Front. Mar. Sci. 2021, 8, 271. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Site | Tissue | 16 rRNA Gene (Copy No. g−1 Tissue) |
|---|---|---|
| Estuary (bank) | Digestive gland | 1.32 ± 2.61 × 103 † b * |
| Intestine | 5.25 ± 2.49 × 105 a | |
| Bay (bank) | Digestive gland | 1.49 ± 2.32 × 103 b |
| Intestine | 6.82 ± 1.52 × 104 ab | |
| Bay (long–line) | Digestive gland | 1.95 ± 1.19 × 103 b |
| Intestine | 1.85 ± 5.61 × 103 b |
| Collection Site | Tissue | ID Sample | Input Numeric | Filtered Numeric | Percentage of Input Passed Filter Numeric | Denoised Numeric | Non-Chimeric Numeric | Percentage of Input |
|---|---|---|---|---|---|---|---|---|
| Non-Chimeric Numeric | ||||||||
| Nehuentue estuary (bank) | Digestive gland | S4 | 111,820 | 92,771 | 82.96% | 92,384 | 84,149 | 75.25% |
| S5 | 1,021,727 | 841,987 | 82.41% | 840,357 | 646,711 | 63.30% | ||
| S6 | 128,850 | 107,349 | 83.31% | 106,665 | 90,197 | 70.00% | ||
| Intestine | S1 | 153,833 | 128,787 | 83.72% | 128,153 | 115,093 | 74.82% | |
| S2 | 62,996 | 52,256 | 82.95% | 51,940 | 46,339 | 73.56% | ||
| S3 | 30,677 | 23,610 | 76.96% | 23,000 | 19,253 | 62.76% | ||
| Hueihue bay (bank) | Digestive gland | S10 | 66,618 | 55,223 | 82.90% | 54,831 | 48,429 | 72.70% |
| S11 | 49,621 | 39,265 | 79.13% | 39,063 | 26,974 | 54.36% | ||
| S12 | 32,893 | 22,963 | 69.81% | 22,794 | 12,728 | 38.70% | ||
| Intestine | S7 | 102,545 | 83,973 | 81.89% | 83,332 | 49,164 | 47.94% | |
| S8 | 29,508 | 21,780 | 73.81% | 21,594 | 12,745 | 43.19% | ||
| S9 | 22,699 | 15,600 | 68.73% | 15,506 | 10,297 | 45.36% | ||
| Hueihue bay (long-line) | Digestive gland | S16 | 61,463 | 48,887 | 79.54% | 48,649 | 31,910 | 51.92% |
| S17 | 23,182 | 17,896 | 77.20% | 17,810 | 10,993 | 47.42% | ||
| S18 | 65,698 | 53,659 | 81.68% | 53,478 | 30,905 | 47.04% | ||
| Intestine | S13 | 83,007 | 29,294 | 35.29% | 28,803 | 15,723 | 18.94% | |
| S14 | 54,765 | 42,006 | 76.70% | 41,693 | 25,168 | 45.96% | ||
| S15 | 55,987 | 45,461 | 81.20% | 45,239 | 25,795 | 46.07% |
| Collection Site | Tissue | Sobs † | Shannon Index H’ | Inverse Simpson Index 1/λ | Pielous’s Evenness | Faith’s Phylogenetic Diversity |
|---|---|---|---|---|---|---|
| Estuary (bank) | Digestive gland | 90.66 ± 13.61 ‡ a* | 2.77 ± 0.25 a | 5.04 ± 0.028 a | 0.43 ± 0.03 b | 14.05 ± 3.12 a |
| Intestine | 113.66 ± 38.12 a | 2.99 ± 0.34 a | 15.20 ± 5.07 b | 0.45 ± 0.01 b | 11.21 ± 3.71 a | |
| Bay (bank) | Digestive gland | 82.00 ± 44.83 a | 4.89 ± 0.94 a | 4.83 ± 0.17 a | 0.81 ± 0.05 ab | 8.77 ± 4.29 a |
| Intestine | 41.33 ± 15.05 a | 2.92 ± 0.58 a | 5.00 ± 0.08 a | 0.56 ± 0.08 a | 6.86 ± 0.34 a | |
| Bay (long–line) | Digestive gland | 82.66 ± 26.40 a | 5.10 ± 1.23 a | 4.98 ± 0.01 a | 0.80 ± 0.13 a | 12.27 ± 1.51 a |
| Intestine | 55.66 ± 17.74 a | 4.94 ± 0.58 a | 4.48 ± 0.58 a | 0.87 ± 0.01 a | 7.94 ± 2.85 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenzuela, T.; Rilling, J.I.; Larama, G.; Acuña, J.J.; Campos, M.; Inostroza, N.G.; Araya, M.; Altamirano, K.; Fujiyoshi, S.; Yarimizu, K.; et al. 16S rRNA–Based Analysis Reveals Differences in the Bacterial Community Present in Tissues of Choromytilus chorus (Mytilidae, Bivalvia) Grown in an Estuary and a Bay in Southern Chile. Diversity 2021, 13, 209. https://doi.org/10.3390/d13050209
Valenzuela T, Rilling JI, Larama G, Acuña JJ, Campos M, Inostroza NG, Araya M, Altamirano K, Fujiyoshi S, Yarimizu K, et al. 16S rRNA–Based Analysis Reveals Differences in the Bacterial Community Present in Tissues of Choromytilus chorus (Mytilidae, Bivalvia) Grown in an Estuary and a Bay in Southern Chile. Diversity. 2021; 13(5):209. https://doi.org/10.3390/d13050209
Chicago/Turabian StyleValenzuela, Tamara, Joaquin I. Rilling, Giovanni Larama, Jacquelinne J. Acuña, Marco Campos, Nitza G. Inostroza, Macarena Araya, Katherine Altamirano, So Fujiyoshi, Kyoko Yarimizu, and et al. 2021. "16S rRNA–Based Analysis Reveals Differences in the Bacterial Community Present in Tissues of Choromytilus chorus (Mytilidae, Bivalvia) Grown in an Estuary and a Bay in Southern Chile" Diversity 13, no. 5: 209. https://doi.org/10.3390/d13050209
APA StyleValenzuela, T., Rilling, J. I., Larama, G., Acuña, J. J., Campos, M., Inostroza, N. G., Araya, M., Altamirano, K., Fujiyoshi, S., Yarimizu, K., Maruyama, F., & Jorquera, M. A. (2021). 16S rRNA–Based Analysis Reveals Differences in the Bacterial Community Present in Tissues of Choromytilus chorus (Mytilidae, Bivalvia) Grown in an Estuary and a Bay in Southern Chile. Diversity, 13(5), 209. https://doi.org/10.3390/d13050209