
Incorporating Topological and Age Uncertainty into Event-Based Biogeography of Sand Spiders Supports Paleo-Islands in Galapagos and Ancient Connections among Neotropical Dry Forests




Abstract
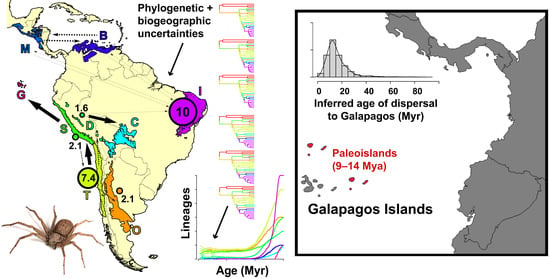
:
1. Introduction
2. Material and Methods
2.1. Summaries of Biogeographic Inferences in Face of Uncertainty
2.2. Model Selection
2.3. Phylogeny and Distribution Data
2.4. BioGeoBEARS Parameters and Time Stratification
3. Results
3.1. Model Selection and Estimates of Ancestral Ranges
3.2. Lineages through Time by Area
3.3. Comparing Time-Stratified vs. Unconstrained Models in the Face of Age Uncertainty
3.4. Ancestral Range Estimates of Particular Nodes in the Face of Topological Uncertainty
3.5. Summary of Biogeographic Events in the Face of Uncertainty
4. Discussion
4.1. Inference of Biogeographic Events in the Face of Uncertainty
4.2. Cladogenetic Events, Founder-Event Speciation, and Uncertainty
4.3. Lineages through Time by Area
4.4. Sand Spiders Dispersed to Galapagos Paleo-Islands
4.5. Ancient Connections among Neotropical Dry Forests
4.6. Script Availability
4.7. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huelsenbeck, J.P.; Imennov, N.S. Geographic origin of human mitochondrial DNA: Accommodating phylogenetic uncertainty and model comparison. Syst. Biol. 2002, 51, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Dupin, J.; Matzke, N.J.; Särkinen, T.; Knapp, S.; Olmstead, R.G.; Bohs, L.; Smith, S.D. Bayesian estimation of the global biogeographical history of the Solanaceae. J. Biogeogr. 2017, 44, 887–899. [Google Scholar] [CrossRef]
- Ronquist, F. Dispersal-vicariance analysis: A new approach to the quantification of historical biogeography. Syst. Biol. 1997, 46, 195–203. [Google Scholar] [CrossRef]
- Bremer, K. Ancestral areas: A cladistic reinterpretation of the center of origin concept. Syst. Biol. 1992, 41, 436–445. [Google Scholar] [CrossRef]
- Ree, R.H.; Sanmartín, I. Conceptual and statistical problems with the DEC+J model of founder-event speciation and its comparison with DEC via model selection. J. Biogeogr. 2018, 45, 741–749. [Google Scholar] [CrossRef]
- Ree, R.H.; Smith, S.A. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst. Biol. 2008, 57, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Ree, R.H.; Moore, B.R.; Webb, C.O.; Donoghue, M.J. A likelihood framework for inferring the evolution of geographic range on phylogenetic trees. Evolution 2005, 59, 2299–2311. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Harris, A.J.; Blair, C.; He, X. RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography. Mol. Phylogenet. Evol. 2015, 87, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Matzke, N.J. BioGeoBEARS: BioGeography with Bayesian (and Likelihood) Evolutionary Analysis in R Scripts. 2013. Available online: https://github.com/nmatzke/BioGeoBEARS (accessed on 13 October 2020).
- Matzke, N.J. Probabilistic historical biogeography: New models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing. Front. Biogeogr. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Matzke, N.J. Model selection in historical biogeography reveals that founder-event speciation is a crucial process in island clades. Syst. Biol. 2014, 63, 951–970. [Google Scholar] [CrossRef]
- Turk, E.; Bond, J.; Cheng, R.-C.; Čandek, K.; Hamilton, C.A.; Kralj-Fišer, S.; Kuntner, M. A natural colonization of Asia: Phylogenomic and biogeographic history of coin spiders (Araneae: Nephilidae: Herennia). Diversity 2021, in press. [Google Scholar]
- Hortal, J.; De Bello, F.; Diniz-Filho, J.A.F.; Lewinsohn, T.M.; Lobo, J.M.; Ladle, R.J. Seven Shortfalls that Beset Large-Scale Knowledge of Biodiversity. Annu. Rev. Ecol. Evol. Syst. 2015, 46, 523–549. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, J.A.; Sharma, P.P. A critical appraisal of the placement of Xiphosura (Chelicerata) with account of known sources of phylogenetic error. Syst. Biol. 2019, 68, 896–917. [Google Scholar] [CrossRef] [PubMed]
- Suh, A. The phylogenomic forest of bird trees contains a hard polytomy at the root of Neoaves. Zool. Scr. 2016, 45, 50–62. [Google Scholar] [CrossRef]
- Ho, S.Y.W.; Duchêne, S. Molecular-clock methods for estimating evolutionary rates and timescales. Mol. Ecol. 2014, 23, 5947–5965. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Bromham, L. Six impossible things before breakfast: Assumptions, models, and belief in molecular dating. Trends Ecol. Evol. 2019, 34, 474–486. [Google Scholar] [CrossRef]
- Parham, J.F.; Donoghue, P.C.J.; Bell, C.J.; Calway, T.D.; Head, J.J.; Holroyd, P.A.; Inoue, J.G.; Irmis, R.B.; Joyce, W.G.; Ksepka, D.T.; et al. Best practices for justifying fossil calibrations. Syst. Biol. 2012, 61, 346–359. [Google Scholar] [CrossRef]
- Warnock, R.C.M.; Parham, J.F.; Donoghue, P.C.J.; Joyce, W.G.; Lyson, T.R. Calibration uncertainty in molecular dating analyses: There is no substitute for the prior evaluation of time priors. Proc. R. Soc. B Biol. Sci. 2015, 282, 20141013. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P.; Rannala, B.; Masly, J.P. Accommodating phylogenetic uncertainty in evolutionary studies. Science 2000, 288, 2349–2350. [Google Scholar] [CrossRef] [Green Version]
- Nylander, J.A.A.; Olsson, U.; Alström, P.; Sanmartín, I. Accounting for phylogenetic uncertainty in biogeography: A Bayesian approach to dispersal-vicariance analysis of the thrushes (Aves: Turdus). Syst. Biol. 2008, 57, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Harris, A.J.; He, X. S-DIVA (Statistical Dispersal-Vicariance Analysis): A tool for inferring biogeographic histories. Mol. Phylogenet. Evol. 2010, 56, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.M.; Boyer, S.L.; Giribet, G. A well-resolved transcriptomic phylogeny of the mite harvestman family Pettalidae (Arachnida, Opiliones, Cyphophthalmi) reveals signatures of Gondwanan vicariance. J. Biogeogr. 2020, 47, 1345–1361. [Google Scholar] [CrossRef]
- Santaquiteria, A.; Siqueira, A.C.; Duarte-Ribeiro, E.; Carnevale, G.; White, W.; Pogonoski, J.; Baldwin, C.C.; Ortí, G.; Arcila, D.; Betancur, R. Phylogenomics and historical biogeography of seahorses, dragonets, goatfishes, and allies (Teleostei: Syngnatharia): Assessing the factors driving uncertainty in biogeographic inferences. Syst. Biol. 2021, in press. [Google Scholar] [CrossRef]
- Landis, M.J.; Freyman, W.A.; Baldwin, B.G. Retracing the Hawaiian silversword radiation despite phylogenetic, biogeographic, and paleogeographic uncertainty. Evolution 2018, 72, 2343–2359. [Google Scholar] [CrossRef]
- Landis, M.J.; Eaton, D.A.R.; Clement, W.L.; Park, B.; Spriggs, E.L.; Sweeney, P.W.; Edwards, E.J.; Donoghue, M.J. Joint phylogenetic estimation of geographic movements and biome shifts during the global diversification of Viburnum. Syst. Biol. 2021, 70, 67–85. [Google Scholar] [CrossRef]
- Matos-Maraví, P.; Wahlberg, N.; Freitas, A.V.L.; Devries, P.; Antonelli, A.; Penz, C.M. Mesoamerica is a cradle and the Atlantic Forest is a museum of Neotropical butterfly diversity: Insights from the evolution and biogeography of Brassolini (Lepidoptera: Nymphalidae). Biol. J. Linn. Soc. 2021, 133, 704–724. [Google Scholar] [CrossRef]
- Yan, Y.; Davis, C.C.; Dimitrov, D.; Wang, Z.; Rahbek, C.; Borregaard, M.K. Phytogeographic history of the tea family inferred through high-resolution phylogeny and fossils. Syst. Biol. 2021, in press. [Google Scholar] [CrossRef]
- Magalhaes, I.L.F.; Neves, D.M.; Santos, F.R.; Vidigal, T.H.D.A.; Brescovit, A.D.; Santos, A.J. Phylogeny of Neotropical Sicarius sand spiders suggests frequent transitions from deserts to dry forests despite antique, broad-scale niche conservatism. Mol. Phylogenet. Evol. 2019, 140, 106569. [Google Scholar] [CrossRef]
- Binford, G.J.; Callahan, M.S.; Bodner, M.R.; Rynerson, M.R.; Núñez, P.B.; Ellison, C.E.; Duncan, R.P. Phylogenetic relationships of Loxosceles and Sicarius spiders are consistent with Western Gondwanan vicariance. Mol. Phylogenet. Evol. 2008, 49, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, I.L.F.; Oliveira, U.; Santos, F.R.; Vidigal, T.H.D.A.; Brescovit, A.D.; Santos, A.J. Strong spatial structure, Pliocene diversification and cryptic diversity in the Neotropical dry forest spider Sicarius cariri. Mol. Ecol. 2014, 23, 5323–5336. [Google Scholar] [CrossRef]
- Magalhaes, I.L.F.; Brescovit, A.D.; Santos, A.J. Phylogeny of Sicariidae spiders (Araneae: Haplogynae), with a monograph on Neotropical Sicarius. Zool. J. Linn. Soc. 2017, 179, 767–864. [Google Scholar]
- White, W.M.; McBirney, A.R.; Duncan, R.A. Petrology and geochemistry of the Galápagos Islands: Portrait of a pathological mantle plume. J. Geophys. Res. 1993, 98, 533–563. [Google Scholar] [CrossRef]
- Christie, D.M.; Duncan, R.A.; McBirney, A.R.; Richards, M.A.; White, W.M.; Harpp, K.S.; Fox, C.G. Drowned islands downstream from the Galapagos hotspot imply extended speciation times. Nature 1992, 355, 246–248. [Google Scholar] [CrossRef]
- Werner, R.; Hoernle, K.; Van Den Bogaard, P.; Ranero, C.; Von Huene, R.; Korich, D. Drowned 14-m.y.-old Galapagos archipelago off the coast of Costa Rica: Implications for tectonic and evolutionary models. Geology 1999, 27, 499–502. [Google Scholar] [CrossRef]
- DRYFLOR. Plant diversity patterns in neotropical dry forests and their conservation implications. Science 2016, 353, 1383–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, R.T.; Prado, D.E.; Pendry, C.A. Neotropical seasonally dry forests and Quaternary vegetation changes. J. Biogeogr. 2000, 27, 261–273. [Google Scholar] [CrossRef]
- Prado, D.E.; Gibbs, P.E. Patterns of species distributions in the dry seasonal forests of South America. Ann. Missouri Bot. Gard. 1993, 80, 902–927. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Lüdecke, D. sjmisc: Data and variable transformation functions. J. Open Source Softw. 2018, 3, 754. [Google Scholar] [CrossRef]
- Akaike, H. Information theory and an extension of the maximum likelihood principle. In Proceedings of the Second International Symposium on Information Theory, Tsahkadsor, Armenia, 2–8 September 1971; Petrov, B.N., Caski, F., Eds.; Akademiai Kiado: Budapest, Hungary, 1973; pp. 267–281. [Google Scholar]
- Wagenmakers, E.J.; Farrell, S. AIC model selection using Akaike weights. Psychon. Bull. Rev. 2004, 11, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Cala-Riquelme, F.; Gutiérrez-Estrada, M.; Florez-Daza, A.E.; Agnarsson, I. A new six-eyed sand spider Sicarius Walckenaer, 1847 (Araneae: Haplogynae: Sicariidae) from Colombia, with information on its natural history. Arachnology 2017, 17, 176–182. [Google Scholar] [CrossRef]
- Echeverría-Londoño, S.; Enquist, B.J.; Neves, D.M.; Violle, C.; Boyle, B.; Kraft, N.J.B.; Maitner, B.S.; McGill, B.; Peet, R.K.; Sandel, B.; et al. Plant functional diversity and the biogeography of biomes in North and South America. Front. Ecol. Evol. 2018, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Nash, J.C. On best practice optimization methods in R. J. Stat. Softw. 2014, 60, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Matzke, N.J. Statistical Comparison of DEC and DEC+J Is Identical to Comparison of Two ClaSSE Submodels, and Is Therefore Valid. OSF Prepr. 2021, 1–40. [Google Scholar] [CrossRef]
- Renner, S.S. Available data point to a 4-km-high Tibetan Plateau by 40 Ma, but 100 molecular-clock papers have linked supposed recent uplift to young node ages. J. Biogeogr. 2016, 43, 1479–1487. [Google Scholar] [CrossRef]
- Spriggs, E.L.; Clement, W.L.; Sweeney, P.W.; Madriñán, S.; Edwards, E.J.; Donoghue, M.J. Temperate radiations and dying embers of a tropical past: The diversification of Viburnum. New Phytol. 2015, 207, 340–354. [Google Scholar] [CrossRef]
- Ceccarelli, F.S.; Koch, N.M.; Soto, E.M.; Barone, M.L.; Arnedo, M.A.; Ramírez, M.J. The grass was greener: Repeated Evolution of specialized morphologies and habitat shifts in ghost spiders following grassland expansion in South America. Syst. Biol. 2019, 68, 63–77. [Google Scholar] [CrossRef]
- Skeels, A. Lineages through space and time plots: Visualising spatial and temporal changes in diversity. Front. Biogeogr. 2019, 11, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lotz, L.N. An update on the spider genus Hexophthalma (Araneae: Sicariidae) in the Afrotropical region, with descriptions of new species. Eur. J. Taxon. 2018, 2018, 475–494. [Google Scholar] [CrossRef] [Green Version]
- Rabosky, D.L. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS ONE 2014, 9, e89543. [Google Scholar] [CrossRef] [Green Version]
- Grehan, J. Biogeography and evolution of the Galapagos: Integration of the biological and geological evidence. Biol. J. Linn. Soc. 2001, 74, 267–287. [Google Scholar] [CrossRef]
- Heads, M.; Grehan, J.R. The Galápagos Islands: Biogeographic patterns and geology. Biol. Rev. 2021, 96, 1160–1185. [Google Scholar] [CrossRef] [PubMed]
- Heads, M. Metapopulation vicariance explains old endemics on young volcanic islands. Cladistics 2018, 34, 292–311. [Google Scholar] [CrossRef]
- Arnedo, M.A.; Hormiga, G. Repeated colonization, adaptive radiation and convergent evolution in the sheet-weaving spiders (Linyphiidae) of the south Pacific Archipelago of Juan Fernandez. Cladistics 2021, 37, 317–342. [Google Scholar] [CrossRef]
- Corbett, E.C.; Bravo, G.A.; Schunck, F.; Naka, L.N.; Silveira, L.F.; Edwards, S.V. Evidence for the Pleistocene Arc Hypothesis from genome-wide SNPs in a Neotropical dry forest specialist, the Rufous-fronted Thornbird (Furnariidae: Phacellodomus rufifrons). Mol. Ecol. 2020, 29, 4457–4472. [Google Scholar] [CrossRef]
- Werneck, F.P.; Gamble, T.; Colli, G.R.; Rodrigues, M.T.; Sites, J.W. Deep diversification and long-term persistence in the south american “dry diagonal”: Integrating continent-wide phylogeography and distribution modeling of geckos. Evolution 2012, 66, 3014–3034. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source Trees (each with 100 BSM) | Biogeographic Model | Caribbean (B) | Chiquitano (C) | Andes (D) | Mesoamerica (M) | Monte (O) | Atacama (T) | Caribbean + Andes | Andes + Mesoamerica | Caribbean + Mesoamerica | Caribbean + Monte | Andes + Monte | Monte + Mesoamerica |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MCCT | DVL + J | 25 | 0 | 13 | 30 | 15 | 15 | 0 | 0 | 2 | 0 | 0 | 0 |
| MCCT | DVL | 22 | 0 | 9 | 19 | 19 | 19 | 0 | 1 | 2 | 2 | 4 | 3 |
| 100 trees | DVL + J | 22 | 2 | 24 | 22 | 16 | 11 | 0 | 0 | 2 | 0 | 0 | 0 |
| 100 trees | DVL | 16.2 | 1.2 | 26.6 | 15.3 | 12.5 | 15.5 | 1.3 | 1.3 | 2.7 | 1.9 | 1.7 | 2 |
| Starting Range | Ending Range | Average Events | Event Type |
|---|---|---|---|
| I | I | 10.0948 | In-situ speciation |
| T | T | 7.5704 | In-situ speciation |
| F | F | 3.9997 | In-situ speciation |
| S | S | 2.5332 | In-situ speciation |
| O | O | 2.168 | In-situ speciation |
| D | D | 1.6566 | In-situ speciation |
| T | TS | 1.0111 | Dispersal |
| MB | M | 0.9475 | Vicariance |
| MB | B | 0.9475 | Vicariance |
| D | DC | 0.8726 | Dispersal |
| SG | S | 0.8004 | Vicariance |
| SG | G | 0.8004 | Vicariance |
| S | SG | 0.7997 | Dispersal |
| OT | O | 0.6377 | Vicariance |
| OT | T | 0.636 | Vicariance |
| TS | S | 0.5241 | Vicariance |
| TS | T | 0.5228 | Vicariance |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhaes, I.L.F.; Santos, A.J.; Ramírez, M.J. Incorporating Topological and Age Uncertainty into Event-Based Biogeography of Sand Spiders Supports Paleo-Islands in Galapagos and Ancient Connections among Neotropical Dry Forests. Diversity 2021, 13, 418. https://doi.org/10.3390/d13090418
Magalhaes ILF, Santos AJ, Ramírez MJ. Incorporating Topological and Age Uncertainty into Event-Based Biogeography of Sand Spiders Supports Paleo-Islands in Galapagos and Ancient Connections among Neotropical Dry Forests. Diversity. 2021; 13(9):418. https://doi.org/10.3390/d13090418
Chicago/Turabian StyleMagalhaes, Ivan L. F., Adalberto J. Santos, and Martín J. Ramírez. 2021. "Incorporating Topological and Age Uncertainty into Event-Based Biogeography of Sand Spiders Supports Paleo-Islands in Galapagos and Ancient Connections among Neotropical Dry Forests" Diversity 13, no. 9: 418. https://doi.org/10.3390/d13090418
APA StyleMagalhaes, I. L. F., Santos, A. J., & Ramírez, M. J. (2021). Incorporating Topological and Age Uncertainty into Event-Based Biogeography of Sand Spiders Supports Paleo-Islands in Galapagos and Ancient Connections among Neotropical Dry Forests. Diversity, 13(9), 418. https://doi.org/10.3390/d13090418