
Transcriptome Analysis of Cambium Tissue of Paulownia Collected during Winter and Spring

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Sampling and RNA Isolation
2.2. cDNA Synthesis and RNA Sequencing
2.3. Assembly and Annotation
2.4. Gene Ontology and Coding Sequences
2.5. Gene Expression Analysis
2.6. Simple Sequence Repeats Analyses
2.7. Validation of Wood-Forming Candidate Genes with RT-qPCR
3. Results and Discussion
3.1. RNA-Seq and Transcriptome Assembly of Paulownia Cambial Tissue
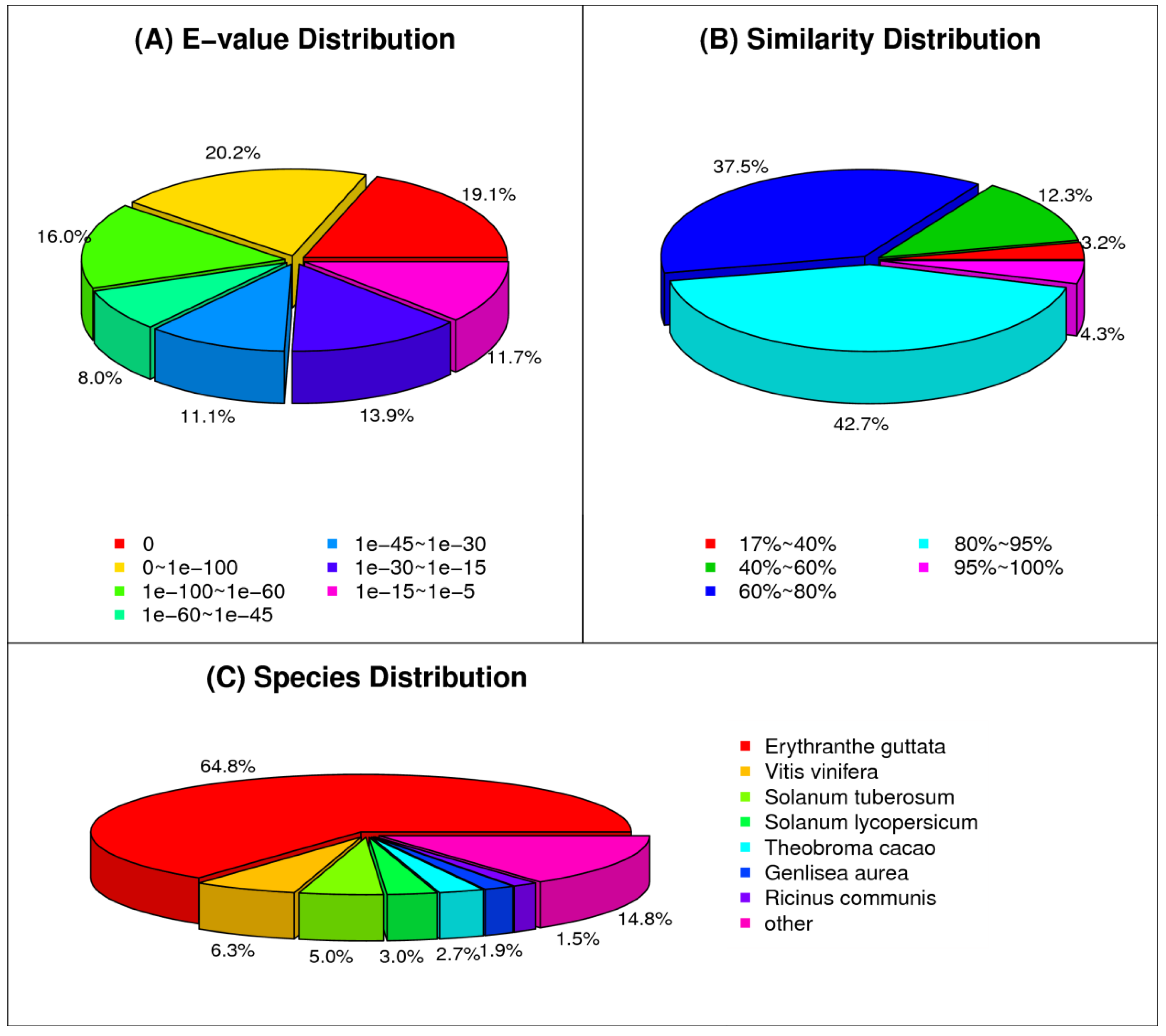
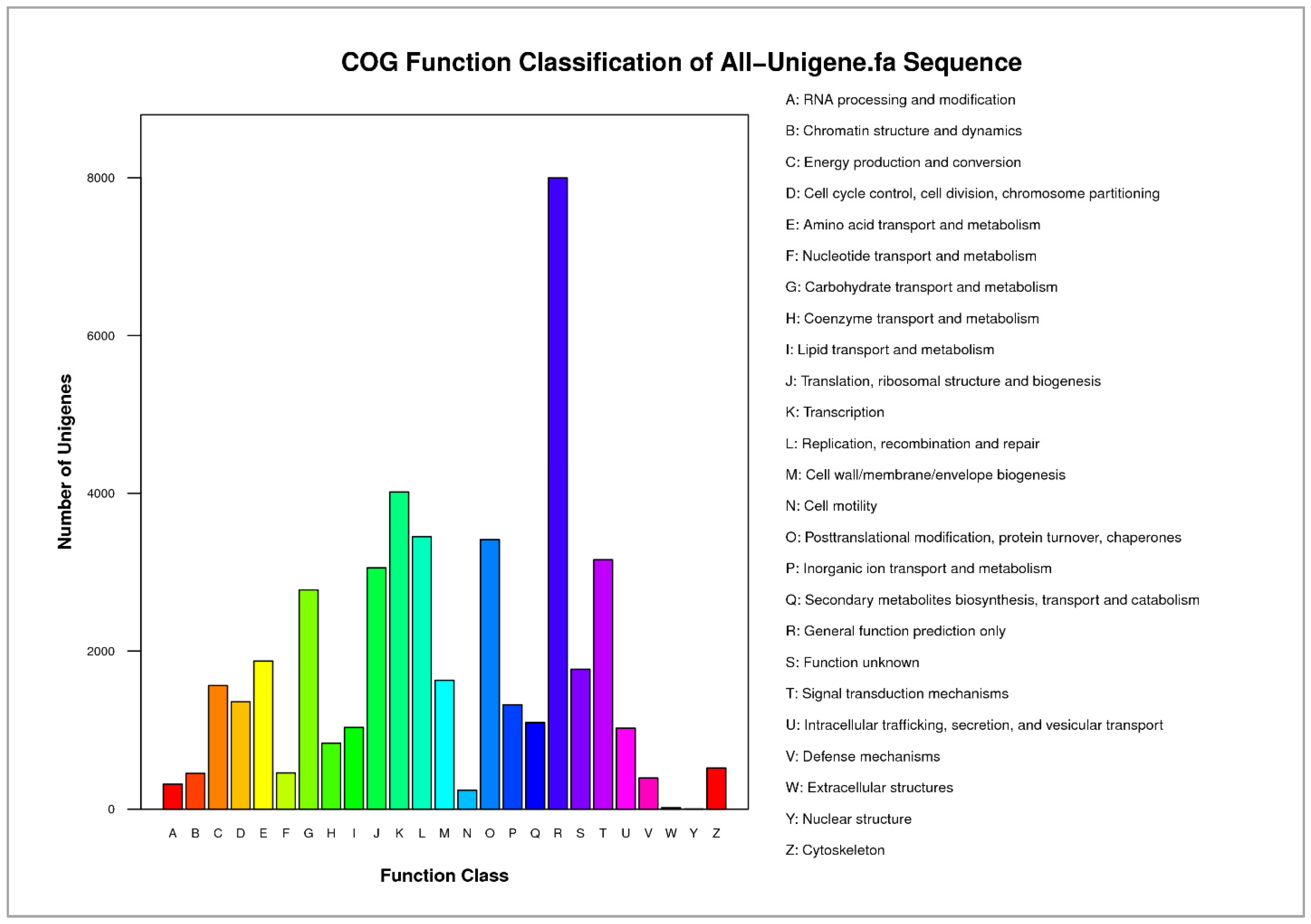
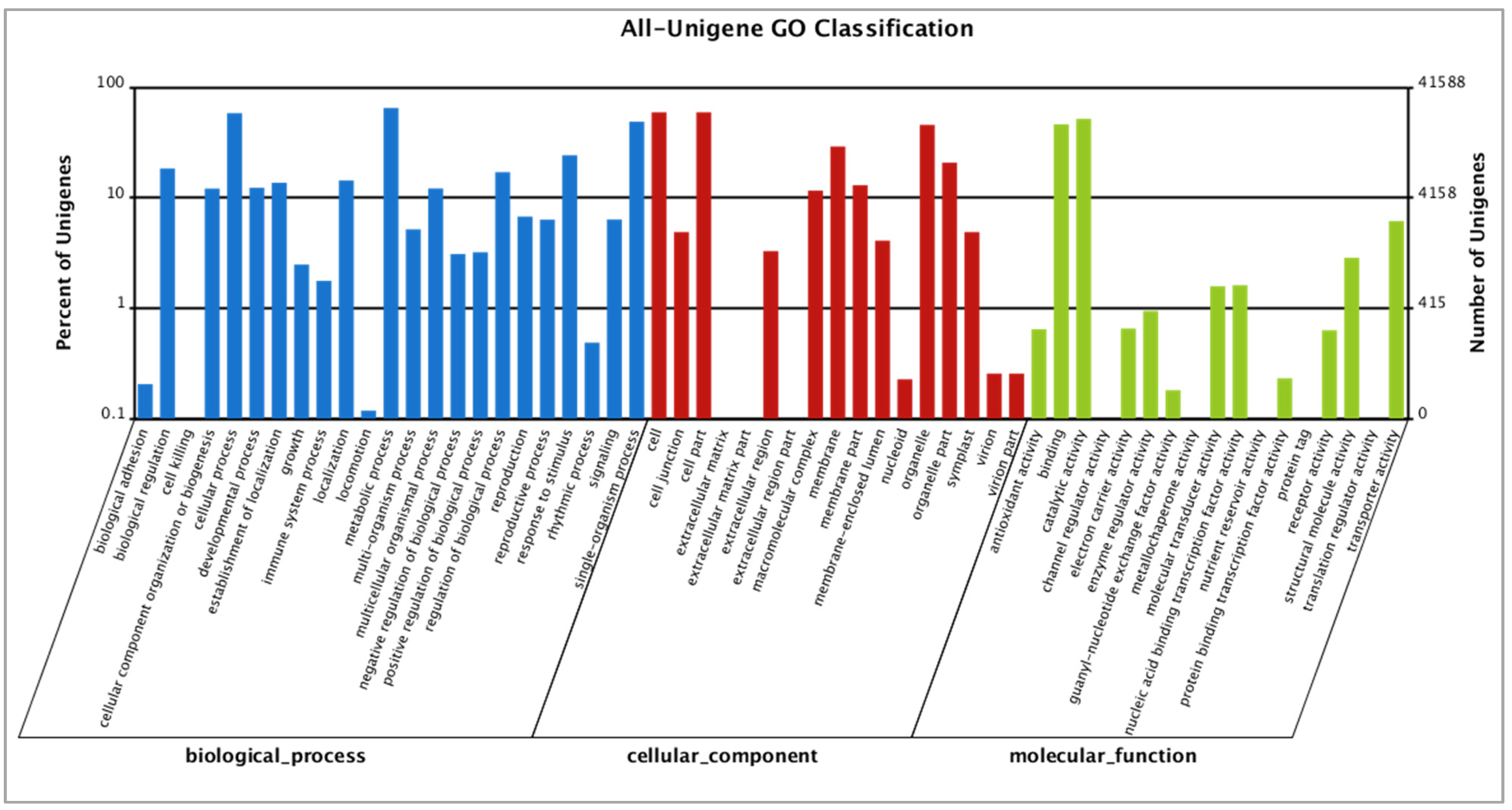
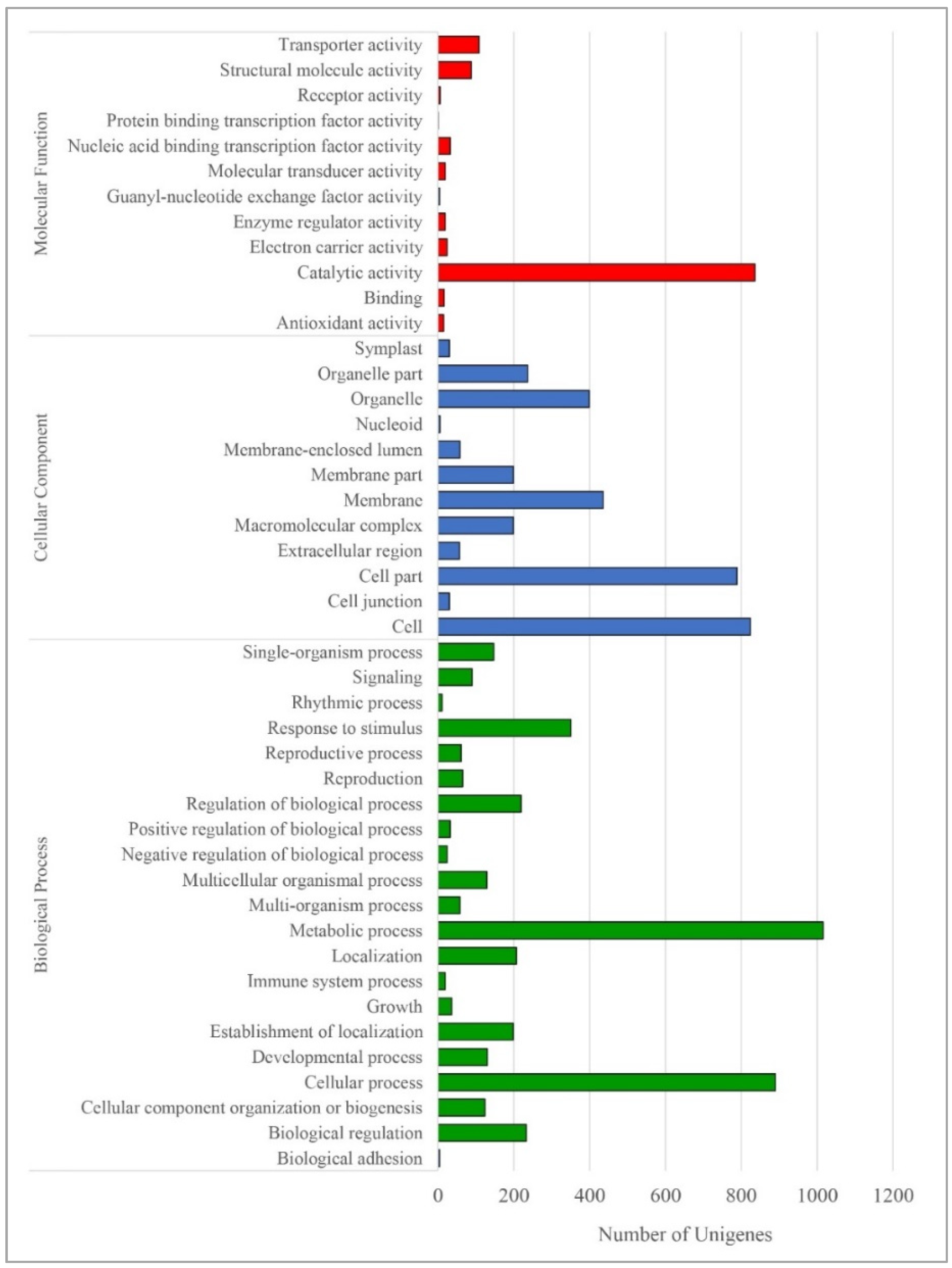
3.2. Functional Annotation of Paulownia Cambial Transcriptome
3.3. Transcriptional Profiling of Cambial Tissues in Winter and Spring
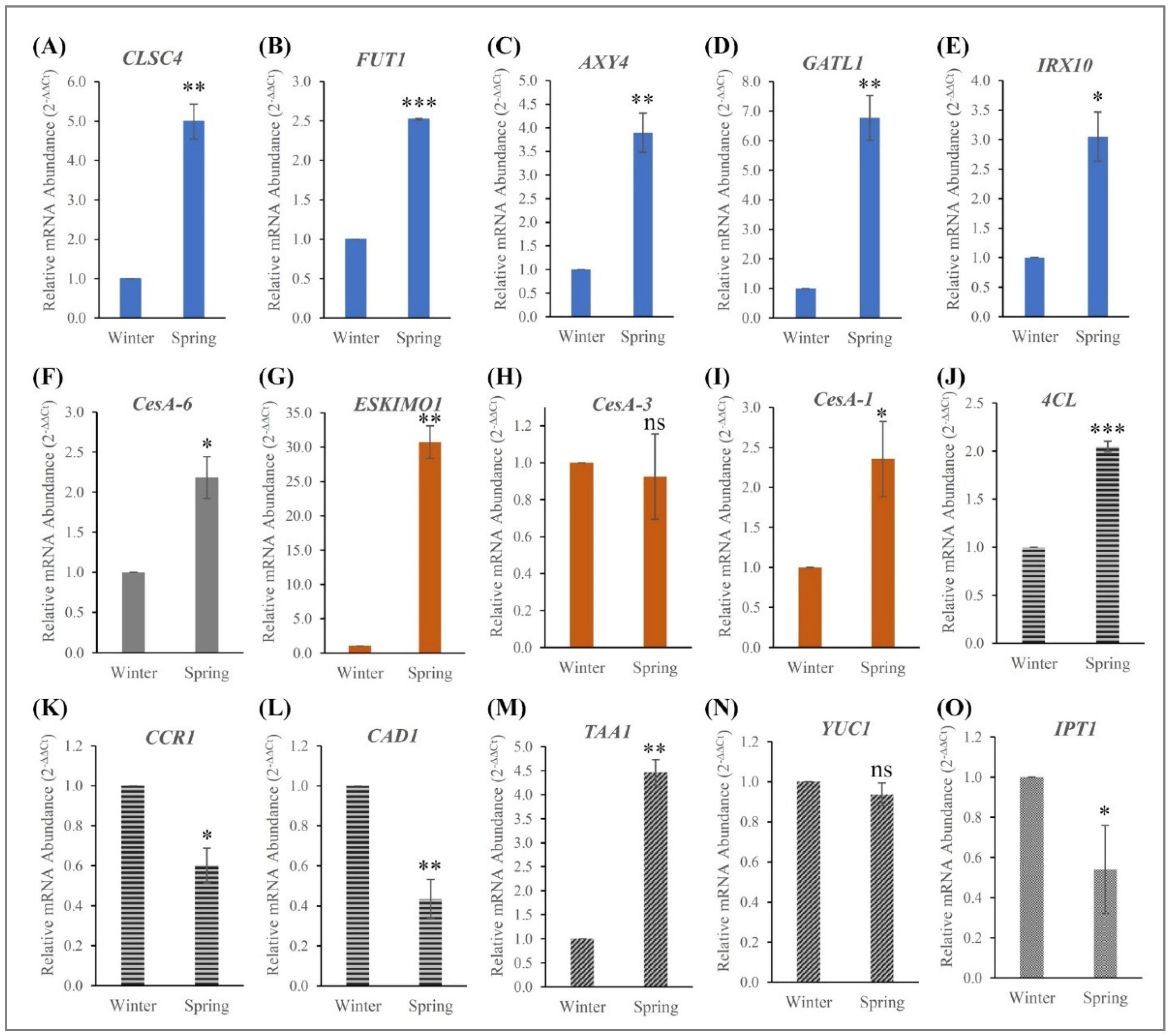
3.4. Expression of Lignocellulosic Pathway Genes and Their Validation
3.5. Analysis of Hormone-Specific Genes and Their Validation
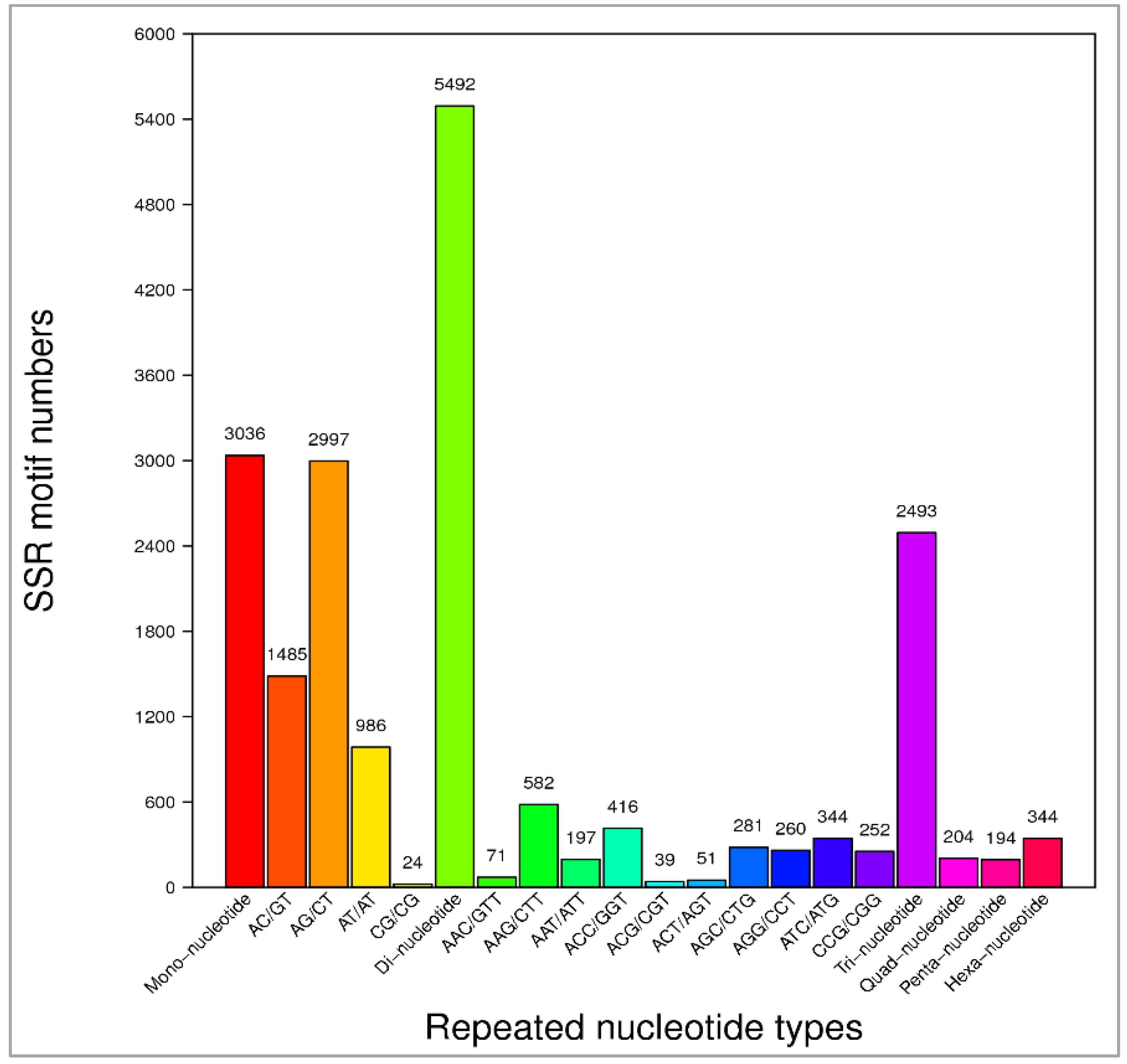
3.6. Analysis of Simple Sequence Repeats (SSRs)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, Z.-H.; Chao, C.-J.; Lu, X.-Y.; Xiong, Y.G. Paulownia in China: Cultivation and Utilization; Asian Network for Biological Sciences and International Development Research Centre and International Development Research Centre: Ottawa, ON, Canada, 1986; pp. 1–65. [Google Scholar]
- Clatterbuck, K.W.; Hodges, D.G. Tree Crops for Marginal Farmland-Paulownia; The University of Tennessee Extension Service: Knoxville, TN, USA, 2004; pp. 1–32. Available online: https://trace.tennessee.edu/cgi/viewcontent.cgi?article=1002&context=utk_agexfores (accessed on 26 May 2021).
- El-Showk, S.; El-Showk, N. The Paulownia Tree. An Alternative for Sustainable Forestry. 2003. Available online: www.cropdevelopment.org (accessed on 30 August 2021).
- Akyildiz, M.H.; Kol Sahin, H. Some technological properties and uses of paulownia (Paulownia tomentosa Steud.) wood. J. Environ. Biol. 2010, 31, 351–355. [Google Scholar]
- Li, P.; Oda, J. Flame retardancy of paulownia wood and its mechanism. J. Mater. Sci. 2007, 42, 8544–8550. [Google Scholar] [CrossRef] [Green Version]
- Yadav, N.K.; Vaidya, B.N.; Henderson, K.; Lee, J.F.; Stewart, W.M.; Dhekney, S.A.; Joshee, N. A Review of Paulownia biotechnology: A short rotation, fast growing multipurpose bioenergy tree. Am. J. Plant. Sci. 2013, 4, 2070–2082. [Google Scholar] [CrossRef] [Green Version]
- Tisserat, B.; Joshee, N.; Mahapatra, A.K.; Selling, G.W.; Finkenstadt, V. Physical and mechanical properties of extruded poly(lactic acid)-based Paulownia elongata biocomposites. Ind. Crop. Prod. 2013, 44, 88–96. [Google Scholar] [CrossRef]
- Tisserat, B.; Reifschneider, L.; Joshee, N.; Finkenstadt, V. Properties of high density polyethylene—Paulownia wood flour composites via injection molding. Bioresources 2013, 8, 4440–4458. [Google Scholar] [CrossRef] [Green Version]
- Tisserat, B.; Reifschneider, L.; Joshee, N.; Finkenstadt, V. Evaluation of Paulownia elongata wood polyethylene composites. J. Thermoplast. Compos. Mater. 2015, 28, 1301–1320. [Google Scholar] [CrossRef]
- Vaughn, S.F.; Dinelli, F.D.; Tisserat, B.; Joshee, N.; Vaughan, M.M.; Peterson, S.C. Creeping bentgrass growth in sand-based root zones with or without biochar. Sci. Hortic. 2015, 197, 592–596. [Google Scholar] [CrossRef]
- Stewart, W.M.; Vaidya, B.N.; Mahapatra, A.K.; Terrill, T.H.; Joshee, N. Potential use of multipurpose paulownia elongata tree as an animal feed resource. Am. J. Plant. Sci. 2018, 9, 1212–1227. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.-H.; Zhong, R. Molecular control of wood formation in trees. J. Exp. Bot. 2015, 66, 4119–4131. [Google Scholar] [CrossRef] [Green Version]
- Samuels, A.L.; Kaneda, M.; Rensing, K. The cell biology of wood formation: From cambial divisions to mature secondary xylem. Can. J. Bot. 2006, 84, 631–639. [Google Scholar] [CrossRef]
- Song, J.; Lu, S.; Chen, Z.-Z.; Lourenco, R.; Chiang, V.L. Genetic transformation of populus trichocarpa genotype nisqually-1: A functional genomic tool for woody plants. Plant. Cell Physiol. 2006, 47, 1582–1589. [Google Scholar] [CrossRef] [Green Version]
- Shim, D.; Ko, J.-H.; Kim, W.-C.; Wang, Q.; Keathley, D.E.; Han, K.-H. A molecular framework for seasonal growth-dormancy regulation in perennial plants. Hortic. Res. 2014, 1, 14059. [Google Scholar] [CrossRef] [Green Version]
- Joshee, N. Paulownia: A multipurpose tree for rapid lignocellulosic biomass production. In Handbook of Bioenergy Crop Plants; Kole, C., Joshi, C.P., Shonnard, D., Eds.; Taylor & Francis: Boca Raton, FL, USA, 2012; pp. 671–686. [Google Scholar]
- Aspeborg, H.; Schrader, J.; Coutinho, P.M.; Stam, M.; Kallas, A.; Djerbi, S.; Nilsson, P.; Denman, S.; Amini, B.; Sterky, F.; et al. Carbohydrate-active enzymes involved in the secondary cell wall biogenesis in hybrid aspen. Plant. Physiol. 2005, 137, 983–997. [Google Scholar] [CrossRef] [Green Version]
- Dharmawardhana, P.; Brunner, A.M.; Strauss, S.H. Genome-wide transcriptome analysis of the transition from primary to secondary stem development in Populus trichocarpa. BMC Genom. 2010, 11, 150. [Google Scholar] [CrossRef] [Green Version]
- Pavy, N.; Boyle, B.; Nelson, C.; Paule, C.; Giguère, I.; Caron, S.; Parsons, L.; Dallaire, N.; Bedon, F.; Bérubé, H.; et al. Identification of conserved core xylem gene sets: Conifer cDNA microarray development, transcript profiling and computational analyses. New Phytol. 2008, 180, 766–786. [Google Scholar] [CrossRef]
- Wang, M.; Qi, X.; Zhao, S.; Zhang, S.; Lu, M.-Z. Dynamic changes in transcripts during regeneration of the secondary vascular system in Populus tomentosa Carr. revealed by cDNA microarrays. BMC Genom. 2009, 10, 215. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, O.; Nahal, H.; Foong, J.; Provart, N.J.; Campbell, M.M. Expansion and diversification of the populus R2R3-MYB family of transcription factors. Plant Physiol. 2009, 149, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Fan, G.; Zhao, Z.; Deng, M. Compatible solute, transporter protein, transcription factor, and hormone-related gene expression provides an indicator of drought stress in Paulownia fortunei. Funct. Integr. Genom. 2014, 14, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Fan, G.; Zhao, Z.; Deng, M. Transcriptome expression profiling in response to drought stress in paulownia australis. Int. J. Mol. Sci. 2014, 15, 4583–4607. [Google Scholar] [CrossRef] [Green Version]
- Mou, H.-Q.; Lu, J.; Zhu, S.-F.; Lin, C.-L.; Tian, G.-Z.; Xu, X.; Zhao, W.-J. Transcriptomic analysis of paulownia infected by paulownia witches’-broom phytoplasma. PLoS ONE 2013, 8, e77217. [Google Scholar] [CrossRef]
- Li, B.; Zhai, X.; Cao, Y.; Zhao, H.; Wang, Z.; Liu, H.; Fan, G. Transcriptome and small RNA sequencing analysis revealed roles of PaWB-related miRNAs and genes in Paulownia fortunei. Forests 2018, 9, 397. [Google Scholar] [CrossRef] [Green Version]
- Nitsch, J. Photoperiodism in woody plants. J. Am. Soc. Hort. Sci. 1957, 70, 526–544. [Google Scholar]
- Espinosa-Ruiz, A.; Saxena, S.; Schmidt, J.; Mellerowicz, E.; Miskolczi, P.; Bakó, L.; Bhalerao, R.P. Differential stage-specific regulation of cyclin-dependent kinases during cambial dormancy in hybrid aspen. Plant. J. 2004, 38, 603–615. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Deng, T.; Liu, Z.; Wang, X. Time-coursed transcriptome analysis identifies key expressional regulation in growth cessation and dormancy induced by short days in Paulownia. Sci. Rep. 2019, 9, 16602. [Google Scholar] [CrossRef]
- Immanen, J.; Nieminen, K.; Smolander, O.-P.; Kojima, M.; Serra, J.A.; Koskinen, P.; Zhang, J.; Elo, A.; Mähönen, A.P.; Street, N. Cytokinin and auxin display distinct but interconnected distribution and signaling profiles to stimulate cambial activity. Curr. Biol. 2016, 26, 1990–1997. [Google Scholar] [CrossRef] [Green Version]
- Nieminen, K.; Blomster, T.; Helariutta, Y.; Mähönen, A.P. Vascular cambium development. Arab. Book 2015, 13, e0177. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; He, Y.; Zhang, Y.; Guo, J.; Zhang, L. Genome-wide identification and profiling of microRNAs in Paulownia to-mentosa cambial tissues in response to seasonal changes. Gene 2018, 677, 32–40. [Google Scholar] [CrossRef]
- Saminathan, T.; Nimmakayala, P.; Manohar, S.; Malkaram, S.; Almeida, A.; Cantrell, R.; Tomason, Y.; Abburi, L.; Rahman, M.A.; Vajja, V.G.; et al. Differential gene expression and alternative splicing between diploid and tetraploid watermelon. J. Exp. Bot. 2015, 66, 1369–1385. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Boil. 1999, 99, 138–148. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [Green Version]
- Doering, A.; Lathe, R.; Persson, S. An Update on xylan synthesis. Mol. Plant. 2012, 5, 769–771. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.-L.; Wang, G.-L.; Xiong, F.; Yu, X.-R.; Xu, Z.-S.; Wang, F.; Xiong, A.-S. De novo assembly, transcriptome characterization, lignin accumulation and anatomic characteristics: Novel insights into lignin biosynthesis during celery leaf development. Sci. Rep. 2015, 5, srep08259. [Google Scholar] [CrossRef] [Green Version]
- Pauly, M.; Gille, S.; Liu, L.; Mansoori, N.; de Souza, A.J.; Schultink, A.; Xiong, G. Hemicellulose biosynthesis. Planta 2013, 238, 627–642. [Google Scholar] [CrossRef]
- Quang, T.H.; Hallingback, H.R.; Gyllenstrand, N.; Von Arnold, S.; Clapham, D. Expression of genes of cellulose and lignin synthesis in Eucalyptus urophylla and its relation to some economic traits. Trees 2011, 26, 893–901. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genom. 2008, 9, 312. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, Y.; Diao, G.; Jiang, J.; Yang, C. Isolation and characterization of expressed sequence tags (ESTs) from cambium tissue of birch (betula platyphylla suk). Plant. Mol. Biol. Rep. 2010, 28, 438–449. [Google Scholar] [CrossRef]
- Xu, E.; Fan, G.; Niu, S.; Zhao, Z.; Deng, M.; Dong, Y. Transcriptome-wide profiling and expression analysis of diploid and autotetraploid paulownia tomentosa × paulownia fortunei under drought stress. PLoS ONE 2014, 9, e113313. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Li, R.; Shang, F. The complete chloroplast genome of Paulownia elongata and phylogenetic implications in Lamiales. Mitochondrial DNA Part B 2019, 4, 2067–2068. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Song, D.; Sun, J.; Wang, X.; Li, L. PtrHB7, a class III HD-zip gene, plays a critical role in regulation of vascular cambium differentiation in populus. Mol. Plant. 2013, 6, 1331–1343. [Google Scholar] [CrossRef] [Green Version]
- Groover, A.T.; Mansfield, S.D.; DiFazio, S.; Dupper, G.; Fontana, J.R.; Millar, R.; Wang, Y. The populus homeobox gene ARBORKNOX1 reveals overlapping mechanisms regulating the shoot apical meristem and the vascular cambium. Plant. Mol. Biol. 2006, 61, 917–932. [Google Scholar] [CrossRef]
- Li, E.; Bhargava, A.; Qiang, W.; Friedmann, M.C.; Forneris, N.; Savidge, R.A.; Johnson, L.A.; Mansfield, S.; Ellis, B.E.; Douglas, C.J. The class II KNOX gene KNAT7 negatively regulates secondary wall formation in Arabidopsis and is functionally conserved in Populus. New Phytol. 2012, 194, 102–115. [Google Scholar] [CrossRef]
- Pines, J. Cubism and the cell cycle: The many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 2011, 12, 427–438. [Google Scholar] [CrossRef]
- Hertzberg, M.; Aspeborg, H.; Schrader, J.; Andersson, A.; Erlandsson, R.; Blomqvist, K.; Bhalerao, R.; Uhlen, M.; Teeri, T.T.; Lundeberg, J.; et al. A transcriptional roadmap to wood formation. Proc. Natl. Acad. Sci. USA 2001, 98, 14732–14737. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Liu, Q.; Tan, M.; Yi, H.; Deng, X. Lignin deposition converts juice sacs to “Brown Thorns” in a citrus triploid hybrid. J. Am. Soc. Hortic. Sci. 2008, 133, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Jokipii-Lukkari, S.; Delhomme, N.; Schiffthaler, B.; Mannapperuma, C.; Prestele, J.; Nilsson, O.; Street, N.R.; Tuominen, H. Transcriptional roadmap to seasonal variation in wood formation of norway spruce. Plant. Physiol. 2018, 176, 2851–2870. [Google Scholar] [CrossRef] [Green Version]
- Mauseth, J.D. Plant Anatomy; The Benjamin/Cummings: San Francisco, CA, USA, 1988; p. 560. [Google Scholar]
- Zhong, R.; Ye, Z.-H. Secondary cell walls: Biosynthesis, patterned deposition and transcriptional regulation. Plant. Cell Physiol. 2015, 56, 195–214. [Google Scholar] [CrossRef] [Green Version]
- Villalobos, D.P.; Moreno, R.B.; Said, E.-S.S.; Cañas, R.A.; Osuna, D.; Van Kerckhoven, S.H.E.; Bautista, R.; Claros, M.G.; Canovas, F.M.; Cantón, F.R. Reprogramming of gene expression during compression wood formation in pine: Coordinated modulation of S-adenosylmethionine, lignin and lignan related genes. BMC Plant. Biol. 2012, 12, 100. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, A.; Morin, H.; Krause, C.; Deslauriers, A.; Thibeault-Martel, M. The timing of spring rehydration and its relation with the onset of wood formation in black spruce. Agric. For. Meteorol. 2009, 149, 1403–1409. [Google Scholar] [CrossRef]
- Djerbi, S.; Aspeborg, H.; Nilsson, P.; Sundberg, B.; Mellerowicz, E.; Blomqvist, K.; Teeri, T.T. Identification and expression analysis of genes encoding putative cellulose synthases (CesA) in the hybrid aspen, Populus tremula (L.) P. tremuloides (Michx.). Cellulose 2004, 11, 301–312. [Google Scholar] [CrossRef]
- Davis, J.; Brandizzi, F.; Liepman, A.H.; Keegstra, K. Arabidopsis mannan synthase CSLA9 and glucan synthase CSLC4 have opposite orientations in the Golgi membrane. Plant. J. 2010, 64, 1028–1037. [Google Scholar] [CrossRef]
- Perrin, R.M.; DeRocher, A.E.; Bar-Peled, M.; Zeng, W.; Norambuena, L.; Orellana, A.; Raikhel, N.V.; Keegstra, K. Xyloglucan fucosyltransferase, an enzyme involved in plant cell wall biosynthesis. Science 1999, 284, 1976–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanzin, G.; Madson, M.; Carpita, N.C.; Raikhel, N.V.; Keegstra, K.; Reiter, W.-D. The mur2 mutant of Arabidopsis thaliana lacks fucosylated xyloglucan because of a lesion in fucosyltransferase AtFUT. Proc. Natl. Acad. Sci. USA 2002, 99, 3340–3345. [Google Scholar] [CrossRef] [Green Version]
- Gille, S.; de Souza, A.J.; Xiong, G.; Benz, M.; Cheng, K.; Schultink, A.; Reca, I.-B.; Pauly, M. O-Acetylation of arabidopsis hemicellulose xyloglucan requires AXY4 or AXY4L, proteins with a TBL and DUF231 domain. Plant. Cell 2011, 23, 4041–4053. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Zhou, G.; Avci, U.; Gu, X.; Jones, C.; Yin, Y.; Xu, Y.; Hahn, M. Two poplar glycosyltransferase genes, PdGATL1.1 and PdGATL1.2, are functional orthologs to PARVUS/AtGATL1 in arabidopsis. Mol. Plant. 2009, 2, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Hörnblad, E.; Ulfstedt, M.; Ronne, H.; Marchant, A. Partial functional conservation of IRX10 homologs in physcomitrella patens and Arabidopsis thaliana indicates an evolutionary step contributing to vascular formation in land plants. BMC Plant. Biol. 2013, 13, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, V.; Fortabat, M.-N.; Ducamp, A.; North, H.M.; Maia-Grondard, A.; Trouverie, J.; Boursiac, Y.; Mouille, G.; Durand-Tardif, M. ESKIMO1 disruption in arabidopsis alters vascular tissue and impairs water transport. PLoS ONE 2011, 6, e16645. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.-J.; Harding, S.A.; Lung, J.; Popko, J.L.; Ralph, J.; Stokke, D.D.; Tsai, C.-J.; Chiang, V.L. Repression of lignin biosynthesis promotes cellulose accumulation and growth in transgenic trees. Nat. Biotechnol. 1999, 17, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Goujon, T.; Ferret, V.; Mila, I.; Pollet, B.; Ruel, K.; Burlat, V.; Joseleau, J.-P.; Barrière, Y.; Lapierre, C.; Jouanin, L. Down-regulation of the AtCCR1 gene in Arabidopsis thaliana: Effects on phenotype, lignins and cell wall degradability. Planta 2003, 217, 218–228. [Google Scholar] [CrossRef]
- Bouvier d’Yvoire, M.; Bouchabke-Coussa, O.; Voorend, W.; Antelme, S.; Cézard, L.; Legée, F.; Lebris, P.; Legay, S.; Whitehead, C.; McQueen-Mason, S.J. Disrupting the cinnamyl alcohol dehydrogenase 1 gene (Bd CAD 1) leads to altered lignification and improved saccharification in Brachypodium distachyon. Plant J. 2013, 73, 496–508. [Google Scholar] [CrossRef]
- Stepanova, A.; Robertson-Hoyt, J.; Yun, J.; Benavente, L.M.; Xie, D.-Y.; Dolezal, K.; Schlereth, A.; Jürgens, G.; Alonso, J.M. TAA1-mediated auxin biosynthesis is essential for hormone crosstalk and plant development. Cell 2008, 133, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin biosynthesis by the YUCCA flavin monooxygenases controls the formation of floral organs and vascular tissues in Arabidopsis. Genes Dev. 2006, 20, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Won, C.; Shen, X.; Mashiguchi, K.; Zheng, Z.; Dai, X.; Cheng, Y.; Kasahara, H.; Kamiya, Y.; Chory, J.; Zhao, Y. Conversion of tryptophan to indole-3-acetic acid by tryptophan aminotransferases of arabidopsis and yuccas in arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 18518–18523. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Zhang, R.; Feng, S.; Wang, Y.; Wang, Y.; Fan, C.; Li, Y.; Liu, Z.; Schneider, R.; Xia, T. Three AtCesA6-like members enhance biomass production by distinctively promoting cell growth in Arabidopsis. Plant Biotechnol. J. 2018, 16, 976–988. [Google Scholar] [CrossRef] [Green Version]
- Liners, F.; Gaspar, T.; Van Cutsem, P. Acetyl- and methyl-esterification of pectins of friable and compact sugar-beet calli: Consequences for intercellular adhesion. Planta 1994, 192, 545–556. [Google Scholar] [CrossRef]
- Turner, S.R.; Somerville, C.R. Collapsed xylem phenotype of Arabidopsis identifies mutants deficient in cellulose deposition in the secondary cell wall. Plant. Cell 1997, 9, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundberg, B.; Uggla, C.; Tuominen, H.; Savidge, R.; Barnett, J.; Napier, R. Cell and Molecular Biology of Wood Formation; BIOS Scientific Publishers: Oxford, UK; pp. 169–188.
- Savidge, R.A. The role of plant hormones in higher plant cellular differentiation. II. Experiments with the vascular cambium, and sclereid and tracheid differentiation in the pine, Pinus contorta. J. Mol. Histol. 1983, 15, 447–466. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto-Kitano, M.; Kusumoto, T.; Tarkowski, P.; Kinoshita-Tsujimura, K.; Václavíková, K.; Miyawaki, K.; Kakimoto, T. Cytokinins are central regulators of cambial activity. Proc. Natl. Acad. Sci. USA 2008, 105, 20027–20031. [Google Scholar] [CrossRef] [Green Version]
- Bevan, M.; Northcote, D.H. The interaction of auxin and cytokinin in the induction of phenylalanine ammonia-lyase in suspension cultures of Phaseolus vulgaris. Planta 1979, 147, 77–81. [Google Scholar] [CrossRef]
- Stepanova, A.; Yun, J.; Robles, L.M.; Novak, O.; He, W.; Guo, H.; Ljung, K.; Alonso, J.M. The arabidopsis YUCCA1 flavin monooxygenase functions in the indole-3-pyruvic acid branch of auxin biosynthesis. Plant. Cell 2011, 23, 3961–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbará, T.; Palma-Silva, C.; Paggi, G.; Bered, F.; Fay, M.F.; Lexer, C. Cross-species transfer of nuclear microsatellite markers: Potential and limitations. Mol. Ecol. 2007, 16, 3759–3767. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Varshney, R.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; Zhu, L.; Zhang, K.; Li, L.; Zhao, Z.; Zeng, W.; Lin, X. Development and characterization of EST-SSR markers from RNA-seq data in phyllostachys violascens. Front. Plant. Sci. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Dong, W.; Lyu, T.; Lyu, Y. An RNA sequencing transcriptome analysis and development of EST-SSR markers in chinese hawthorn through illumina sequencing. Forest 2019, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- Hiremath, P.J.; Kumar, A.; Penmetsa, R.V.; Farmer, A.; Schlueter, J.; Chamarthi, S.K.; Whaley, A.M.; Carrasquilla-Garcia, N.; Gaur, P.; Upadhyaya, H.D.; et al. Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant. Biotechnol. J. 2012, 10, 716–732. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.-K.; Tripathi, S.; Cho, Y.-I.; Kim, J.-H.; Lee, H.-E.; Kim, D.-S.; Woo, J.-G.; Cho, M.-C. De novo transcriptome assembly and novel microsatellite marker information in Capsicum annuum varieties Saengryeg 211 and Saengryeg 213. Bot. Stud. 2013, 54, 58. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yu, G.; Shi, B.; Wang, X.; Qiang, H.; Gao, H. Development and characterization of simple sequence repeat (SSR) markers based on RNA-sequencing of medicago sativa and in silico mapping onto the m. truncatula genome. PLoS ONE 2014, 9, e92029. [Google Scholar] [CrossRef] [PubMed]
- Shahinnia, F.; Sayed-Tabatabaei, B.E. Conversion of barley SNPs into PCR-based markers using dCAPS method. Genet. Mol. Biol. 2009, 32, 564–567. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Winter Wood | Spring Wood | |
|---|---|---|
| Total raw reads | 51,005,253 ± 2,639,904 | 52,359,187 ± 737,515 |
| Total clean reads | 49,766,422 ± 2,349,991 | 50,689,696 ± 1,090,856 |
| Percentage of reads | 97.60 ± 0.52 | 96.64 ± 0.63 |
| Q20 Percentage | 97.96 ± 0.36 | 97.51 ± 0.35 |
| Contigs | ||
| Total Number | 129,428 ± 610 | 104,388 ± 1779 |
| Total Length (nt) | 43,730,308 ± 513,387 | 35,501,692 ± 721,304 |
| Mean Length (nt) | 338 ± 3 | 340 ± 1 |
| N50 | 605 ± 7 | 642 ± 4 |
| Unigenes | ||
| Total Number | 64,142 ± 1229 | 45,671 ± 1225 |
| Total Length (nt) | 61,610,800 ± 2,101,797 | 38,465,680 ± 1,457,846 |
| Mean Length (nt) | 960 ± 14 | 842 ± 9 |
| N50 | 1551 ± 25 | 1354 ± 22 |
| Unigene/Contig | Length | WW Expression | SW Expression | log2Fold Change (SW/WW) | Probability | Gene |
|---|---|---|---|---|---|---|
| Unigene8688 | 995 | 0.01 | 42.67 | 12.06 | 0.9997 | Anaphase-promoting complex, cyclosome, subunit 4 |
| Unigene11861 | 477 | 0.01 | 27.14 | 11.41 | 0.9994 | Heat shock protein |
| Unigene6740 | 2339 | 0.01 | 17.72 | 10.79 | 0.9989 | Phosphoenolpyruvate carboxykinase |
| Unigene1612 | 1191 | 0.01 | 11.95 | 10.22 | 0.9978 | Cysteine-type peptidase activity |
| Unigene3797 | 247 | 0.01 | 8.11 | 9.66 | 0.9953 | Calcium-binding domain |
| Unigene10820 | 977 | 0.03 | 24.79 | 9.54 | 0.9992 | Class IV heat shock protein |
| Unigene17843 | 560 | 0.01 | 6.50 | 9.34 | 0.9926 | Pericarp peroxidase |
| Unigene1476 | 971 | 0.01 | 6.17 | 9.27 | 0.9918 | Sulfated surface glycoprotein |
| Unigene11860 | 940 | 0.10 | 58.64 | 9.24 | 0.9996 | Class I heat shock protein |
| Unigene34010 | 359 | 0.01 | 5.87 | 9.20 | 0.9910 | Photosystem II oxygen-evolving complex protein 2 precursor |
| Unigene15576 | 532 | 0.01 | 5.42 | 9.08 | 0.9893 | Gibberellin-regulated protein |
| Unigene35083 | 779 | 0.82 | 359.96 | 8.78 | 0.9995 | Heat shock protein |
| Unigene1551 | 276 | 0.01 | 4.14 | 8.69 | 0.9814 | Large subunit ribosomal protein |
| Unigene8720 | 500 | 0.01 | 4.08 | 8.67 | 0.9808 | Actin-depolymerizing factor |
| Unigene19860 | 1028 | 0.01 | 4.01 | 8.65 | 0.9801 | Leucine-rich repeat extensin |
| Unigene4065 | 510 | 0.01 | 4.00 | 8.64 | 0.9801 | Actin depolymerization factor |
| Unigene4063 | 527 | 0.06 | 23.35 | 8.60 | 0.9986 | Aquaporin PIP2 |
| Unigene3966 | 521 | 0.01 | 3.62 | 8.50 | 0.9754 | Tubulin/FtsZ family |
| Unigene8766 | 606 | 0.01 | 3.59 | 8.49 | 0.9750 | Class II heat shock protein |
| Unigene11017 | 503 | 0.01 | 3.54 | 8.47 | 0.9742 | Cyclophilin peptidyl-prolyl cis-trans isomerase |
| Unigene/Contig | Length | WW Expression | SW Expression | log2Fold Change (SW/WW) | Probability | Gene |
|---|---|---|---|---|---|---|
| Unigene22837 | 205 | 73.99 | 0.01 | −12.85 | 0.9998 | Galactinol synthase |
| Unigene6926 | 252 | 21.15 | 0.01 | −11.05 | 0.9991 | MATE efflux family protein |
| CL7319.Contig2 | 890 | 16.20 | 0.01 | −10.66 | 0.9987 | Coproporphyrinogen-III oxidase |
| Unigene13375 | 584 | 14.56 | 0.01 | −10.51 | 0.9985 | Rosmarinate synthase |
| Unigene10966 | 1645 | 11.55 | 0.01 | −10.17 | 0.9977 | Receptor-like protein kinase |
| Unigene10726 | 1857 | 8.84 | 0.01 | −9.79 | 0.9961 | Valencene synthase |
| CL698.Contig3 | 3901 | 6.65 | 0.01 | −9.38 | 0.9929 | Pectin methyltransferase |
| CL8528.Contig1 | 1476 | 6.63 | 0.01 | −9.37 | 0.9929 | Root phototropism protein |
| CL7708.Contig2 | 888 | 6.54 | 0.01 | −9.35 | 0.9927 | Splicing factor U2af large subunit |
| CL889.Contig1 | 1018 | 6.46 | 0.01 | −9.33 | 0.9925 | Tropinone reductase homolog |
| CL7009.Contig1 | 1844 | 6.39 | 0.01 | −9.32 | 0.9924 | Auxin efflux carrier family protein |
| Unigene13264 | 1617 | 6.04 | 0.01 | −9.24 | 0.9915 | Ethylene-responsive transcription factor |
| Unigene15599 | 228 | 5.86 | 0.01 | −9.20 | 0.9909 | Nitrate transporter |
| CL8637.Contig1 | 863 | 4.75 | 0.01 | −8.89 | 0.9859 | DNA repair protein RadA |
| CL799.Contig5 | 3446 | 4.72 | 0.01 | −8.88 | 0.9857 | Serine/threonine-protein kinase |
| Unigene1474 | 1021 | 4.55 | 0.01 | −8.83 | 0.9845 | Xanthoxin dehydrogenase |
| Unigene25603 | 518 | 4.52 | 0.01 | −8.82 | 0.9844 | SPX domain-containing membrane protein |
| Unigene6422 | 1304 | 45.72 | 0.10 | −8.79 | 0.9993 | Ethylene-responsive transcription factor |
| CL822.Contig5 | 1916 | 4.24 | 0.01 | −8.73 | 0.9823 | Putative dual-specificity protein phosphatase |
| Unigene13350 | 451 | 4.13 | 0.01 | −8.69 | 0.9813 | CBL-interacting protein kinase |
| Pathway | Number of DEGs Genes | p-Value | Pathway ID |
|---|---|---|---|
| Metabolic pathways | 1387 | 1.32E−12 | ko01100 |
| Biosynthesis of secondary metabolites | 827 | 7.55E−10 | ko01110 |
| Plant hormone signal transduction | 320 | 1.58E−05 | ko04075 |
| Plant-pathogen interaction | 266 | 0.2430222 | ko04626 |
| Ribosome | 204 | 0.200625 | ko03010 |
| Spliceosome | 178 | 0.9468082 | ko03040 |
| Starch and sucrose metabolism | 172 | 1.22E−06 | ko00500 |
| Protein processing in E.R. | 162 | 0.3255727 | ko04141 |
| Carbon metabolism | 161 | 0.04321163 | ko01200 |
| RNA transport | 151 | 0.986695 | ko03013 |
| Glycerophospholipid metabolism | 138 | 0.1244536 | ko00564 |
| Endocytosis | 134 | 0.6458048 | ko04144 |
| Biosynthesis of amino acids | 123 | 0.7258124 | ko01230 |
| Glycolysis/Gluconeogenesis | 115 | 5.48E−05 | ko00010 |
| Phenylpropanoid biosynthesis | 106 | 1.50E−06 | ko00940 |
| Circadian rhythm—plant | 105 | 5.10E−09 | ko04712 |
| Ether lipid metabolism | 104 | 0.02664109 | ko00565 |
| Ubiquitin mediated proteolysis | 99 | 0.7266876 | ko04120 |
| Pentose and glucuronate interconversions | 93 | 1.57E−05 | ko00040 |
| Purine metabolism | 92 | 0.9911015 | ko00230 |
| Amino sugar and nucleotide sugar metabolism | 86 | 0.006516035 | ko00520 |
| Pyrimidine metabolism | 78 | 0.992583 | ko00240 |
| mRNA surveillance pathway | 77 | 0.9938776 | ko03015 |
| Flavonoid biosynthesis | 70 | 8.18E−07 | ko00941 |
| RNA degradation | 68 | 0.940714 | ko03018 |
| Gene Name | Unigene/Contig | Macromolecule | Enzyme/Protein Name | Activity | Function |
|---|---|---|---|---|---|
| CESA3 | Unigene9908 | Cellulose | Cellulose synthase A catalytic subunit 3 [UDP-forming] | Cellulose synthase | Catalytic subunit of cellulose synthase terminal complexes required for cell wall formation. |
| CESA1 | Unigene21132 | ” | ” | ” | ” |
| CESA6 | Unigene13924 | ” | ” | ” | ” |
| CSLC4 | CL7362.Contig2 | Hemicellulose | Xyloglucan glycosyltransferase 4 | Glucan synthesis | Involved in the synthesis of the xyloglucan backbone. |
| FUT1 | Unigene2841 | Hemicellulose | Galactoside 2-alpha-L-fucosyltransferase | Fucosyl transferase | Addition of the terminal fucosyl residue on xyloglucan side chains. |
| AXY4 | Unigene14391 | Hemicellulose | Protein ALTERED XYLOGLUCAN 4 | Acetyl transferase | Involved in xyloglucan-specific O-acetylation in roots and rosette leaves. |
| GATL1 | Unigene14440 | Hemicellulose | Galacuronosyltransferase-like 1 | Xylan synthase | Family 8 glycosyl transferase that contributes to xylan biosynthesis. |
| IRX10 | Unigene2644 | Hemicellulose | Beta-1,4-xylosyltransferase | Xylan synthase | Synthesis of the hemicellulose glucuronoxylan, a major component of secondary cell walls. |
| ESKIMO1 | CL7514.Contig1 | Hemicellulose | Protein ESKIMO 1 | Acetyl transferase | Xylan acetyltransferase required for 2-O- and 3-O-monoacetylation of xylosyl residues in xylan. |
| 4CL | CL764.Contig3 | Lignin | 4-coumarate--CoA ligase 1 | Monolignol synthesis | Produces CoA thioesters of a variety of hydroxy- and methoxy-substituted cinnamic acids. |
| CCR1 | CL6693.Contig1 | Lignin | Cinnamoyl-CoA reductase 1 | Monolignol synthesis | Involved in monolignol biosynthesis, the conversion of cinnamoyl-CoAs into cinnamaldehydes. |
| CAD1 | Unigene17183 | Lignin | Cinnamyl alcohol dehydrogenase 1 | Monolignol synthesis | Involved in lignin biosynthesis. Catalyzes the final step specific for the production of lignin monomers. |
| TAA1 | CL8952.Contig1 | Auxin | L-tryptophan--pyruvate aminotransferase 1 | Auxin synthesis | Performs first two reactions in auxin pathway. |
| YUC1 | CL1596.Contig1 | Auxin | Indole-3-pyruvate monooxygenase YUCCA1 | Auxin synthesis | Catalyzes the N-oxidation of tryptamine to form N-hydroxyl tryptamine. |
| IPT1 | Unigene8131 | Cytokinin | Adenylate isopentenyltransferase 1, chloroplastic | Cytokinin synthesis | Catalyzes the transfer of an isopentenyl group from dimethylallyl diphosphate (DMAPP) to ATP, ADP, and AMP. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perry, Z.D.; Saminathan, T.; Arun, A.; Vaidya, B.N.; Basu, C.; Reddy, U.K.; Joshee, N. Transcriptome Analysis of Cambium Tissue of Paulownia Collected during Winter and Spring. Diversity 2021, 13, 423. https://doi.org/10.3390/d13090423
Perry ZD, Saminathan T, Arun A, Vaidya BN, Basu C, Reddy UK, Joshee N. Transcriptome Analysis of Cambium Tissue of Paulownia Collected during Winter and Spring. Diversity. 2021; 13(9):423. https://doi.org/10.3390/d13090423
Chicago/Turabian StylePerry, Zachary D., Thangasamy Saminathan, Alok Arun, Brajesh N. Vaidya, Chhandak Basu, Umesh K. Reddy, and Nirmal Joshee. 2021. "Transcriptome Analysis of Cambium Tissue of Paulownia Collected during Winter and Spring" Diversity 13, no. 9: 423. https://doi.org/10.3390/d13090423
APA StylePerry, Z. D., Saminathan, T., Arun, A., Vaidya, B. N., Basu, C., Reddy, U. K., & Joshee, N. (2021). Transcriptome Analysis of Cambium Tissue of Paulownia Collected during Winter and Spring. Diversity, 13(9), 423. https://doi.org/10.3390/d13090423