
PPDP: A Data Portal of Paris polyphylla for Polyphyllin Biosynthesis and Germplasm Resource Exploration


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and PacBio SMRT Sequencing
2.2. Transcriptome Determination and Evaluation
2.3. Functional Annotation and Classification
2.4. SSR Analysis
2.5. Identification of AS and lncRNAs
2.6. Prediction and Construction of ceRNA Network
2.7. Explore the Biosynthesis Pathway of Secondary Metabolites in P. polyphylla
2.8. Prediction of Transcription Factor and CYP&UGT
2.9. Construction of Co-Expression Network and PPI Network
2.10. Development of the Morphological Identification
2.11. Implementation of PPDP
3. Results
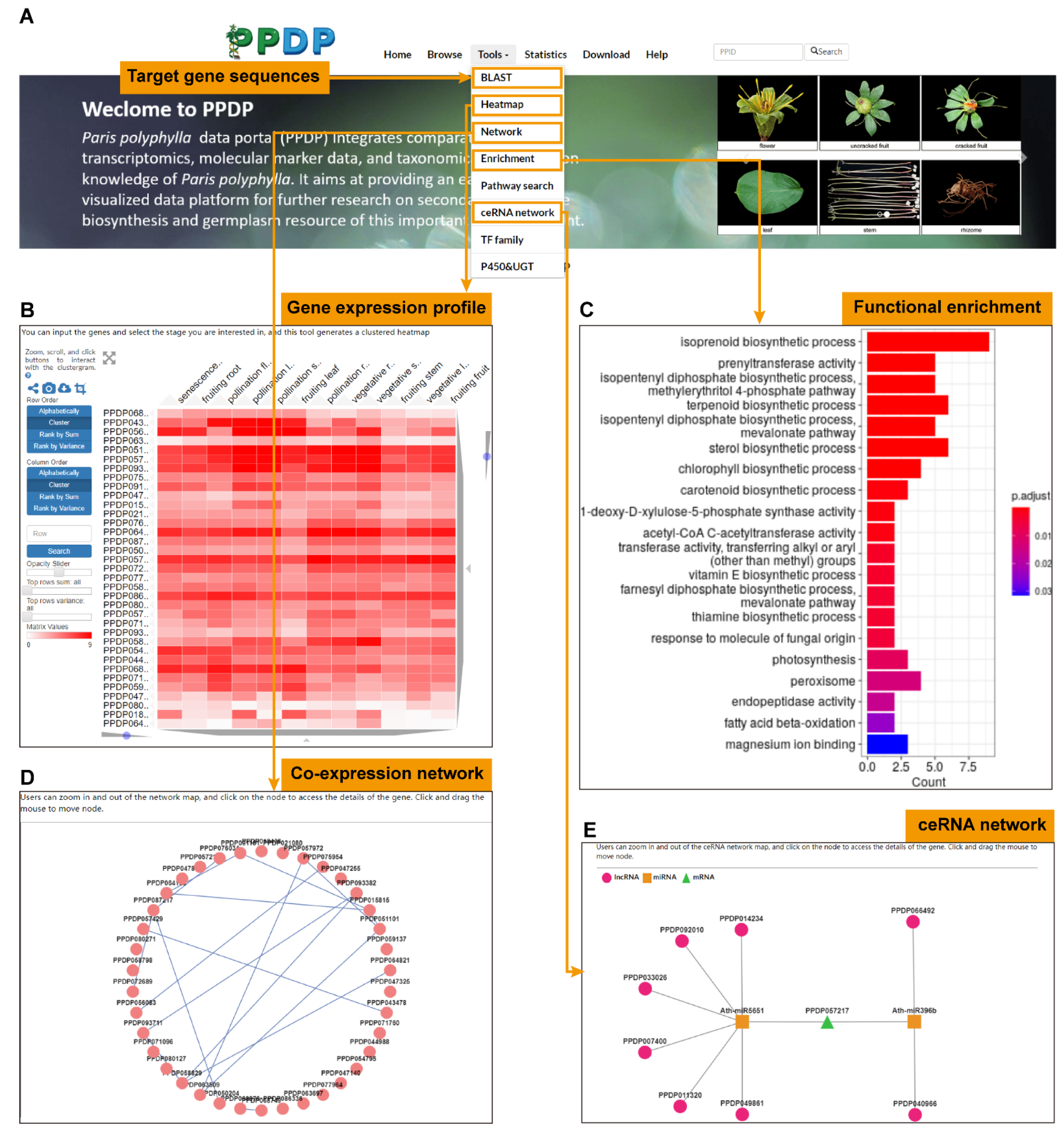
3.1. The Web Interface of PPDP
3.1.1. Browse PPDP
3.1.2. Visualization of Heatmap, Network, and Enrichment
3.1.3. Pathway Search
3.1.4. TF, CYP, and UGT Family
3.1.5. Other Data Resource
3.2. Database Statistics and Use Case
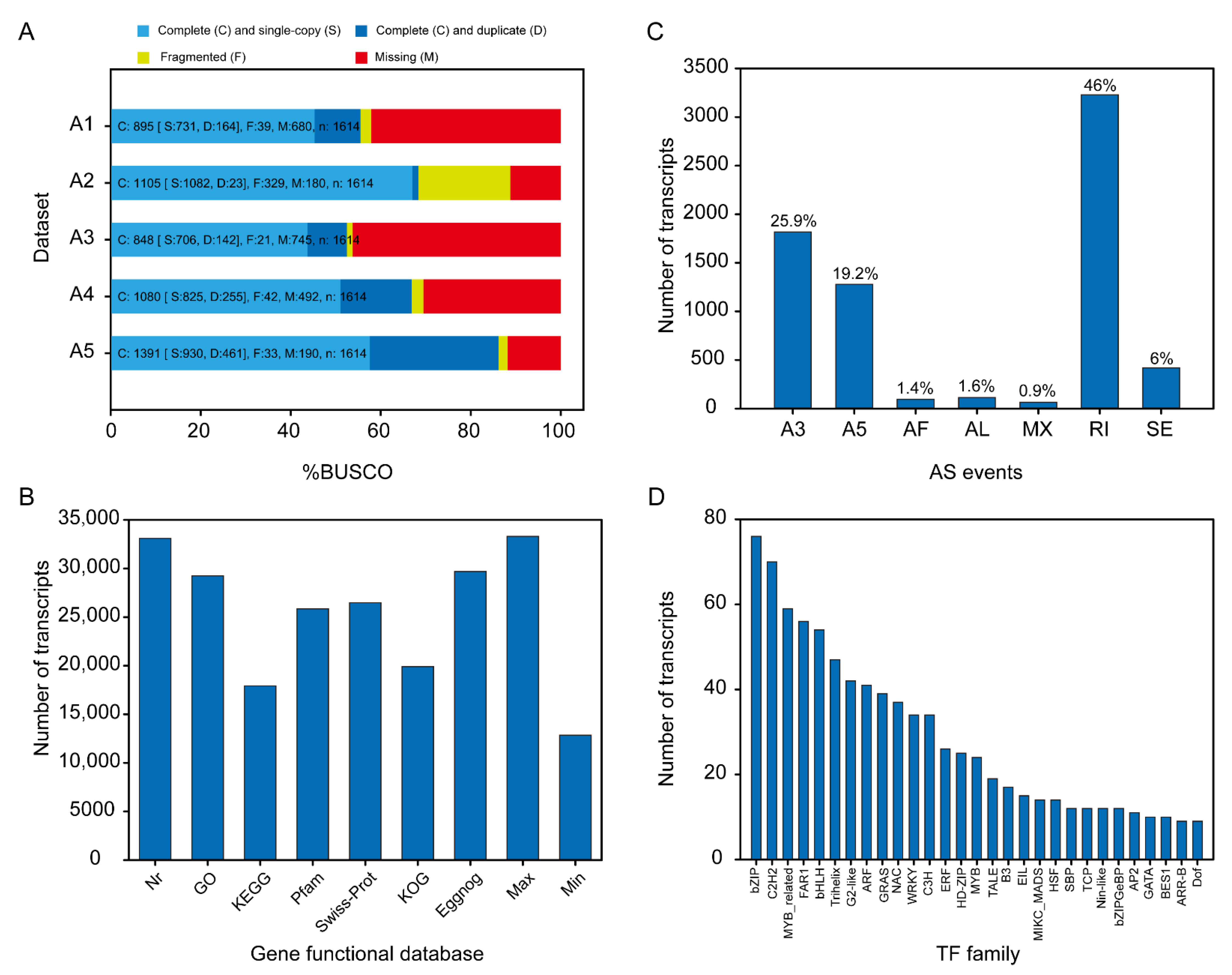
3.2.1. General Properties of PacBio Sequencing Data and Evaluation of Transcriptome
3.2.2. Data Summary
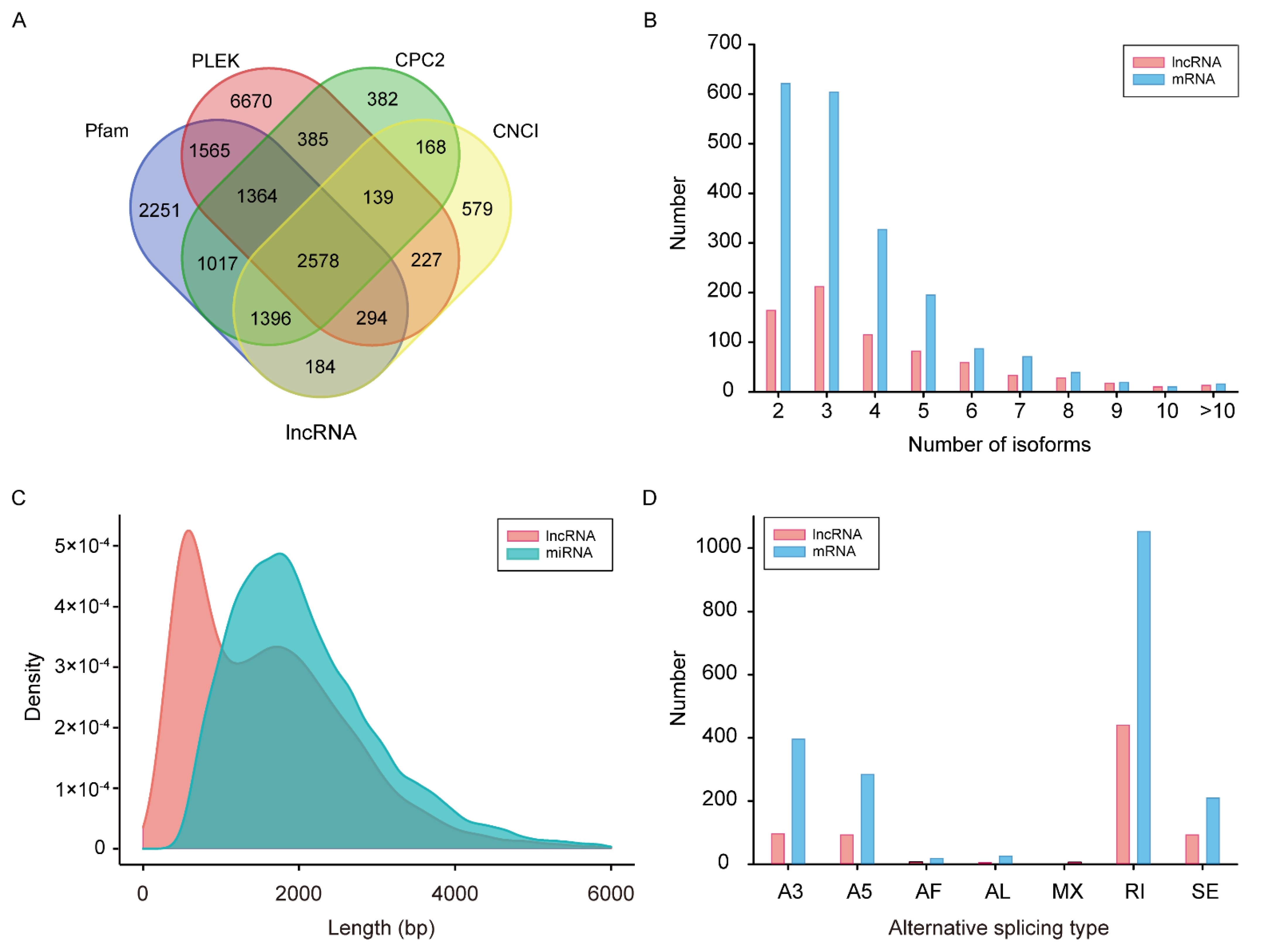
3.2.3. Comparison of lncRNAs and mRNAs
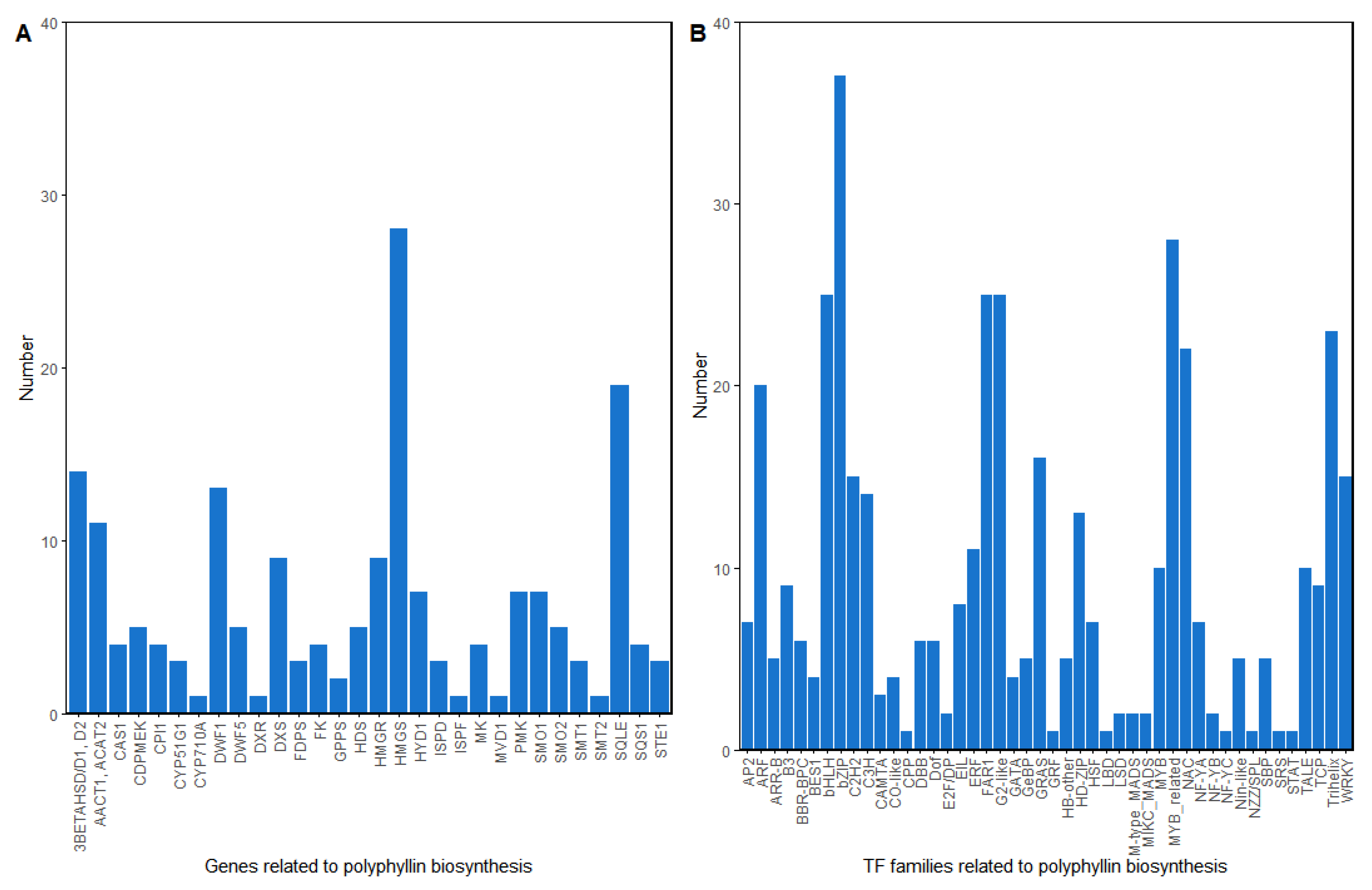
3.2.4. Exploration of the Polyphyllin Biosynthesis Pathway
3.2.5. Data Related to Germplasm Resources
3.2.6. A Case Study of Mining Genes Related to Polyphyllin Synthesis Using PPDP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, H. The Genus Paris Plants; Science Press: Beijing, China, 2008; pp. 1–36. [Google Scholar]
- Chase, M.W.; Christenhusz, M.J.M.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; Stevens, P.F. An Update of the Angiosperm Phylogeny Group Classification for the Orders and Families of Flowering Plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Committee of National Pharmacopoeia. Pharmacopoeia of the People’s Republic of China (I); China Medical Science and Technology Press: Beijing, China, 2020. [Google Scholar]
- Ji, Y. A Monograph of Paris (Melanthiaceae): 1–203; Science Press: Beijing, China, 2021; pp. 1–203. [Google Scholar]
- Liu, Z.; Li, N.; Gao, W.; Man, S.; Yin, S.; Liu, C. Comparative study on hemostatic, cytotoxic and hemolytic activities of different species of Paris L. J. Ethnopharmacol. 2012, 142, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-C.; Gao, W.-Y.; Yan, X.-D.; Wang, Y.; Jing, S.-S.; Xiao, P.-G. Chemical Constituents of Plants from the Genus Paris. Chem. Biodivers. 2014, 11, 1277–1297. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, L.; Wang, G.-C.; Wang, H.; Dai, Y.; Yang, X.-X.; Ye, W.-C.; Li, Y.-L. Triterpenoid saponins from rhizomes of Paris polyphylla var. yunnanensis. Carbohydr. Res. 2013, 368, 1–7. [Google Scholar] [CrossRef]
- Xin, G.; Ruoshi, L.; Duan, B.; Ying, W.; Min, F.A.N.; Shuang, W.; Zhang, H.; Xia, C. Advances in research on chemical constituents and pharmacological effects of Paris genus and prediction and analysis of quality markers. Chin. Tradit. Herb. Drugs 2019, 50, 4838–4852. [Google Scholar]
- Cunningham, A.; Brinckmann, J.; Bi, Y.-F.; Pei, S.-J.; Schippmann, U.; Luo, P. Paris in the spring: A review of the trade, conservation and opportunities in the shift from wild harvest to cultivation of Paris polyphylla (Trilliaceae). J. Ethnopharmacol. 2018, 222, 208–216. [Google Scholar] [CrossRef]
- Liu, T.; Li, X.; Xie, S.; Wang, L.; Yang, S. RNA-seq analysis of Paris polyphylla var. yunnanensis roots identified candidate genes for saponin synthesis. Plant Divers. 2016, 38, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Peng, L.; Sun, X.; Huang, W.; Wang, N.; He, Y.; Shi, X.; Liu, Y.; Zhang, P.; Yang, X.; et al. Organ-specific transcriptome sequencing and mining of genes involved in polyphyllin biosynthesis in Paris polyphylla. Ind. Crop. Prod. 2020, 156, 112775. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, L.; Liu, C.; Qin, X.; Liu, H.; Chen, J.; Ji, Y. Transcriptome analyses of Paris polyphylla var. chinensis, Ypsilandra thibetica, and Polygonatum kingianum characterize their steroidal saponin biosynthesis pathway. Fitoterapia 2019, 135, 52–63. [Google Scholar] [CrossRef]
- Christ, B.; Xu, C.; Xu, M.; Li, F.-S.; Wada, N.; Mitchell, A.J.; Han, X.-L.; Wen, M.-L.; Fujita, M.; Weng, J.-K. Repeated evolution of cytochrome P450-mediated spiroketal steroid biosynthesis in plants. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Su, Q.; Li, J.; Yang, W.; Yao, B.; Guo, J.; Li, S.; Liu, C. RNA-Seq analysis reveals the important co-expressed genes associated with polyphyllin biosynthesis during the developmental stages of Paris polyphylla. BMC Genom. 2022, 23, 1–17. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, X.; Chen, W.; Li, J.; Yang, W.; Zhang, X.; Li, S.; Liu, C. Transcriptome analysis of Paris polyphylla var. yunnanensis illuminates the biosynthesis and accumulation of steroidal saponins in rhizomes and leaves. Phytochemistry 2020, 178, 112460. [Google Scholar] [CrossRef] [PubMed]
- Jayakodi, M.; Choi, B.-S.; Lee, S.-C.; Kim, N.-H.; Park, J.Y.; Jang, W.; Lakshmanan, M.; Mohan, S.V.G.; Lee, D.-Y.; Yang, T.-J. Ginseng Genome Database: An open-access platform for genomics of Panax ginseng. BMC Plant Biol. 2018, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Li, K.; Yao, F.; Sun, L.; Liu, Y. Comparative transcriptome analyses on terpenoids metabolism in field- and mountain-cultivated ginseng roots. BMC Plant Biol. 2019, 19, 82. [Google Scholar] [CrossRef]
- Tien, N.Q.D.; Ma, X.; Man, L.Q.; Chi, D.T.K.; Huy, N.X.; Nhut, D.-T.; Rombauts, S.; Ut, T.; Loc, N.H. De novo whole-genome assembly and discovery of genes involved in triterpenoid saponin biosynthesis of Vietnamese ginseng (Panax vietnamensis Ha et Grushv.). Physiol. Mol. Biol. Plants 2021, 27, 2215–2229. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Gao, L.; Zhang, X.; Gao, W. A cytochrome P450 monooxygenase responsible for the C-22 hydroxylation step in the Paris polyphylla steroidal saponin biosynthesis pathway. Phytochemistry 2018, 156, 116–123. [Google Scholar] [CrossRef]
- Qi, J.J.; Zheng, N.; Zhang, B.; Sun, P.; Hu, S.N.; Xu, W.J.; Ma, Q.; Zhao, T.Z.; Zhou, L.L.; Qin, M.J.; et al. Mining genes involved in the stratification of Paris Polyphylla seeds using high-throughput embryo Transcriptome sequencing. BMC Genom. 2013, 14, 358. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Song, W.; Wang, K.; Yin, X.; Hao, C.; Duan, B.; Xu, Z.; Su, T.; Xue, Z. Effective prediction of biosynthetic pathway genes involved in bioactive polyphyllins in Paris polyphylla. Commun. Biol. 2022, 5, 50. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Kuang, Z.; Wang, Y.; Zhao, Y.; Tao, Y.; Cheng, C.; Yang, J.; Lu, X.; Hao, C.; Wang, T.; et al. PmiREN: A comprehensive encyclopedia of plant miRNAs. Nucleic Acids Res. 2019, 48, D1114–D1121. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2. 0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.L. The TAIR database. Methods Mol. Biol. 2007, 406, 179–212. [Google Scholar] [PubMed]
- Guo, A.-Y.; Chen, X.; Gao, G.; Zhang, H.; Zhu, Q.-H.; Liu, X.-C.; Zhong, Y.-F.; Gu, X.; He, K.; Luo, J. PlantTFDB: A comprehensive plant transcription factor database. Nucleic Acids Res. 2007, 36, D966–D969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Li, J.; Huang, S.; Li, X.; Zhang, X.; Hu, X.; Xiang, S.; Liu, C. GCEN: An Easy-to-Use Toolkit for Gene Co-Expression Network Analysis and lncRNAs Annotation. Curr. Issues Mol. Biol. 2022, 44, 1479–1487. [Google Scholar] [CrossRef]
- Li, P.; Zang, W.; Li, Y.; Xu, F.; Wang, J.; Shi, T. AtPID: The overall hierarchical functional protein interaction network interface and analytic platform for Arabidopsis. Nucleic Acids Res. 2010, 39, D1130–D1133. [Google Scholar] [CrossRef] [Green Version]
- Brandão, M.M.; Dantas, L.L.; Silva-Filho, M.C. AtPIN: Arabidopsis thaliana Protein Interaction Network. BMC Bioinform. 2009, 10, 454. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Shen, X.; Chen, X. PAIR: The predicted Arabidopsis interactome resource. Nucleic Acids Res. 2010, 39, D1134–D1140. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.M.; Xuan, Y.; Xu, M.; Wang, R.S.; Ho, C.H.; Lalonde, S.; You, C.H.; Sardi, M.I.; Parsa, S.A.; Smith-Valle, E.; et al. Border control—A membrane-linked interactome of Arabidopsis. Science 2014, 344, 711–716. [Google Scholar] [CrossRef]
- Arabidopsis Interactome Mapping Consortium. Evidence for Network Evolution in an Arabidopsis Interactome Map. Science 2011, 333, 601–607. [Google Scholar] [CrossRef]
- Mukhtar, M.S.; Carvunis, A.-R.; Dreze, M.; Epple, P.; Steinbrenner, J.; Moore, J.; Tasan, M.; Galli, M.; Hao, T.; Nishimura, M.T.; et al. Independently Evolved Virulence Effectors Converge onto Hubs in a Plant Immune System Network. Science 2011, 333, 596–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, K.P.; Remm, M.; Sonnhammer, E.L.L. Inparanoid: A comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005, 33, D476–D480. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Ren, Q.; Sang, S.Y.; Liu, C.N. Design and implementation of Paris plants online classification and identification system. Computer Era 2020, 9, 72–75. [Google Scholar]
- Franz, M.; Lopes, C.T.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape.js: A graph theory library for visualisation and analysis. Bioinformatics 2015, 32, 309–311. [Google Scholar] [CrossRef] [Green Version]
- Izhaki, I. Emodin—A secondary metabolite with multiple ecological functions in higher plants. New Phytol. 2002, 155, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Khare, S.; Singh, N.B.; Singh, A.; Hussain, I.; Niharika, K.; Yadav, V.; Bano, C.; Yadav, R.K.; Amist, N. Plant secondary metabolites synthesis and their regulations under biotic and abiotic constraints. J. Plant Biol. 2020, 63, 203–216. [Google Scholar] [CrossRef]
- Bartwal, A.; Mall, R.; Lohani, P.; Guru, S.K.; Arora, S. Role of Secondary Metabolites and Brassinosteroids in Plant Defense Against Environmental Stresses. J. Plant Growth Regul. 2012, 32, 216–232. [Google Scholar] [CrossRef]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Zhou, Z.; Wang, Z.; Li, W.; Fang, C.; Wu, M.; Ma, Y.; Liu, T.; Kong, L.-A.; Peng, D.-L.; et al. Global Dissection of Alternative Splicing in Paleopolyploid Soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.M.; Ruan, C.J.; Liang, J.H.; Han, P.; Yan, R.; Dong, X.; Liu, L.Y.; Shuai, X.C.; Liu, H.H. Development of SSR Markers of Fanjingshan Paris polyphylla Smith var.chinensis Based on High Throughput RNA-seq. Mol. Plant Breed. 2019, 18, 6059–6065. [Google Scholar]
- Gao, X.; Su, Q.; Yao, B.; Yang, W.; Ma, W.; Yang, B.; Liu, C. Development of EST-SSR Markers Related to Polyphyllin Biosynthesis Reveals Genetic Diversity and Population Structure in Paris polyphylla. Diversity 2022, 14, 589. [Google Scholar] [CrossRef]
- Chen, Z.S.Z.; Tian, B.; Cai, C.T. Genetic diversity of Paris polyphylla var. yunnanensis by SSR marker. Chin. Tradit. Herb. Drugs 2017, 9, 1834–1838. [Google Scholar]
- Yang, W.Z.; Xu, Z.L.; Yang, S.B.; Yang, M.Q.; Zuo, Y.M.; Yang, T.M.; Zhang, J.Y. Transferability Analysis of EST-SSR Marker of Three Plants to Paris polyphylla Smith var. yunnanensis (Franch.) Hand Mazz. Southwest China J. Agric. Sci. 2014, 4, 1686–1690. [Google Scholar]
- Fayyad, U.; Piatetsky-Shapiro, G.; Smyth, P. From data mining to knowledge discovery in databases. AI Mag. 1996, 17, 37. [Google Scholar]
- Xiao, Q.; Li, Z.; Qu, M.; Xu, W.; Su, Z.; Yang, J. LjaFGD: Lonicera japonica functional genomics database. J. Integr. Plant Biol. 2021, 63, 1422–1436. [Google Scholar] [CrossRef]
- Gu, K.J.; Lin, C.F.; Wu, J.J.; Zhao, Y.P. GinkgoDB: An ecological genome database for the living fossil, Ginkgo biloba. Database J. Biol. Databases Curation 2022, 2022, baac046. [Google Scholar] [CrossRef]
- Liu, H.; Wang, X.; Liu, S.; Huang, Y.; Guo, Y.-X.; Xie, W.-Z.; Liu, H.; Qamar, M.T.U.; Xu, Q.; Chen, L.-L. Citrus Pan-genome to Breeding Database (CPBD): A comprehensive genome database for citrus breeding. Mol. Plant 2022, 15, 1503–1505. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Value |
|---|---|
| High-quality isoforms | 110,666 |
| Low-quality isoforms | 824 |
| Non-reductant transcripts | 36,812 |
| High-quality non-redundant transcripts | 36,504 |
| Total bases (bp) | 73,313,381 |
| N50 (bp) | 2330 |
| Average length (bp) | 2008 |
| Reads mapping (%) | 86.80 |
| Percent GC (%) | 45.88 |
| Item | Value |
|---|---|
| Total number of sequences examined | 36,504 |
| Total size of examined sequences (bp) | 73,313,381 |
| Total number of identified SSRs | 25,767 |
| SSR-containing sequences | 16,120 |
| sequences containing more than 1 SSR | 6169 |
| SSRs present in compound formation | 4027 |
| Mononucleotide repeats | 10,164 |
| Dinucleotide repeats | 12,045 |
| Trinucleotide repeats | 3129 |
| Tetranucleotide repeats | 88 |
| Pentanucleotide repeats | 71 |
| Hexanucleotide repeats | 270 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Q.; Zhang, X.; Li, J.; Yang, W.; Ren, Q.; Gao, X.; Liu, C. PPDP: A Data Portal of Paris polyphylla for Polyphyllin Biosynthesis and Germplasm Resource Exploration. Diversity 2022, 14, 1057. https://doi.org/10.3390/d14121057
Su Q, Zhang X, Li J, Yang W, Ren Q, Gao X, Liu C. PPDP: A Data Portal of Paris polyphylla for Polyphyllin Biosynthesis and Germplasm Resource Exploration. Diversity. 2022; 14(12):1057. https://doi.org/10.3390/d14121057
Chicago/Turabian StyleSu, Qixuan, Xuan Zhang, Jing Li, Wenjing Yang, Qiang Ren, Xiaoyang Gao, and Changning Liu. 2022. "PPDP: A Data Portal of Paris polyphylla for Polyphyllin Biosynthesis and Germplasm Resource Exploration" Diversity 14, no. 12: 1057. https://doi.org/10.3390/d14121057
APA StyleSu, Q., Zhang, X., Li, J., Yang, W., Ren, Q., Gao, X., & Liu, C. (2022). PPDP: A Data Portal of Paris polyphylla for Polyphyllin Biosynthesis and Germplasm Resource Exploration. Diversity, 14(12), 1057. https://doi.org/10.3390/d14121057