
DNA Barcoding of Fresh and Historical Collections of Lichen-Forming Basidiomycetes in the Genera Cora and Corella (Agaricales: Hygrophoraceae): A Success Story?

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Newly Generated Sequence Data from Fresh and Historical Collections
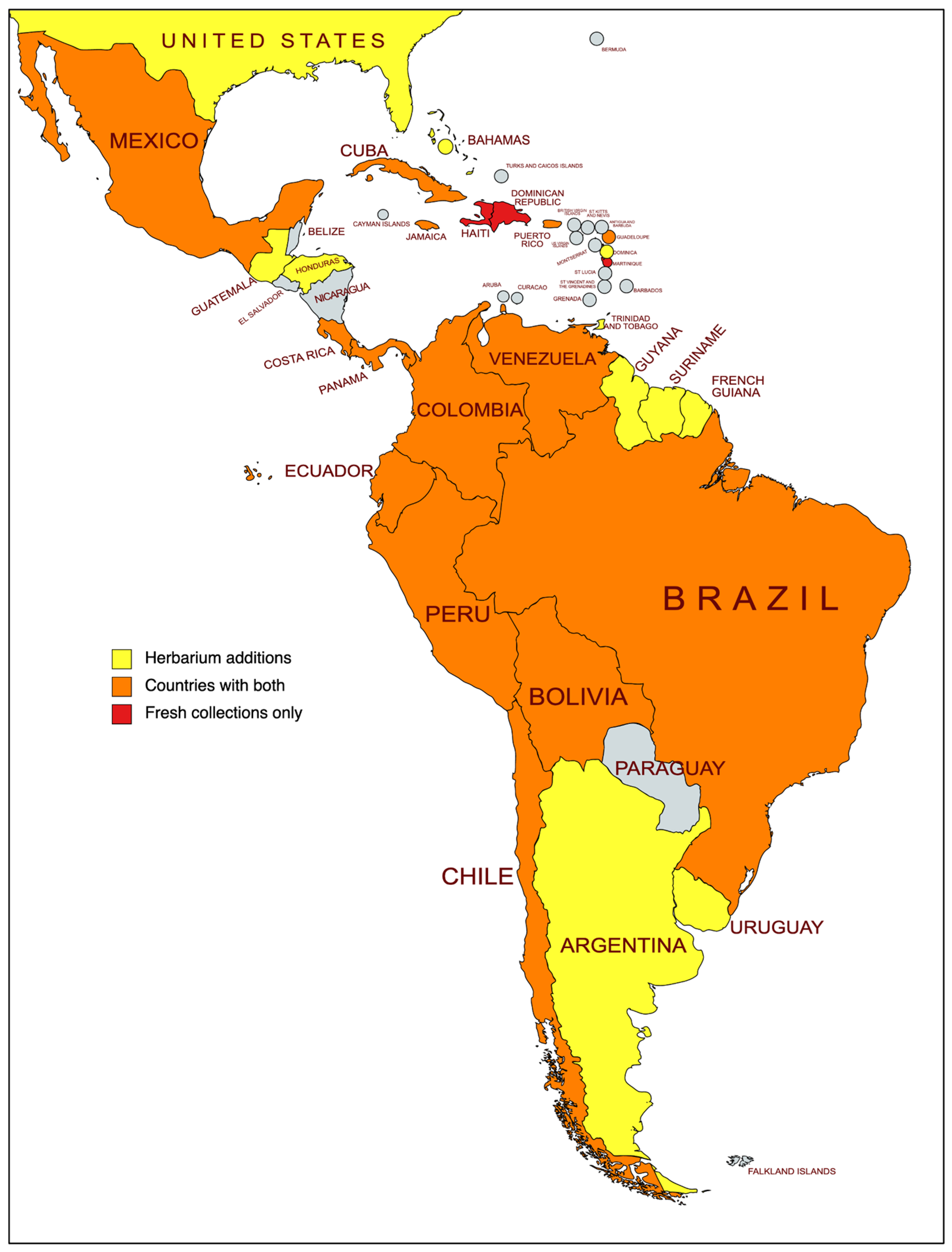
2.1.1. Sampling
2.1.2. DNA Extractions
2.1.3. PCR, ITS Sanger Sequencing and Assembly
2.1.4. PCR, ITS Illumina Sequencing and Bioinformatic Analyses
2.1.5. Microfluidics PCR, Multi-Marker Illumina Sequencing and Bioinformatic Analyses
2.2. Phylogenetic Analyses
2.2.1. ITS-Based Phylogeny
2.2.2. Six-Marker Phylogeny
2.3. Quantitative Species Delimitation
2.3.1. Single-Locus Tree-Based Methods
2.3.2. Distance-Based Methods and Barcoding Gap Analyses
2.3.3. ITS-Based BLAST Performance
2.4. Phenotypic Assessment
3. Results
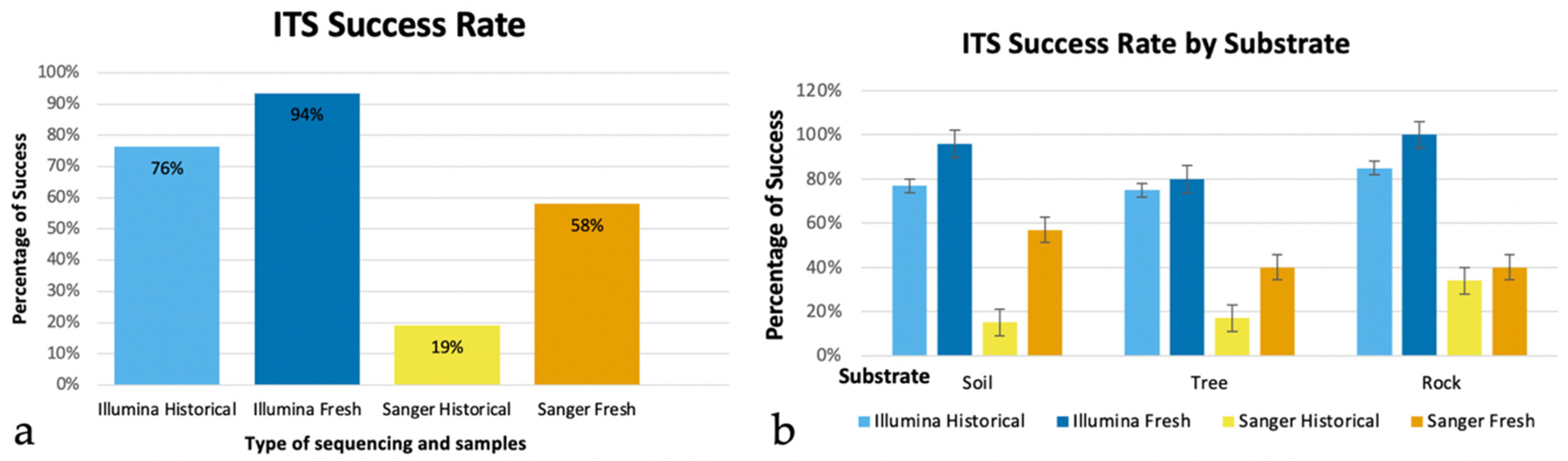
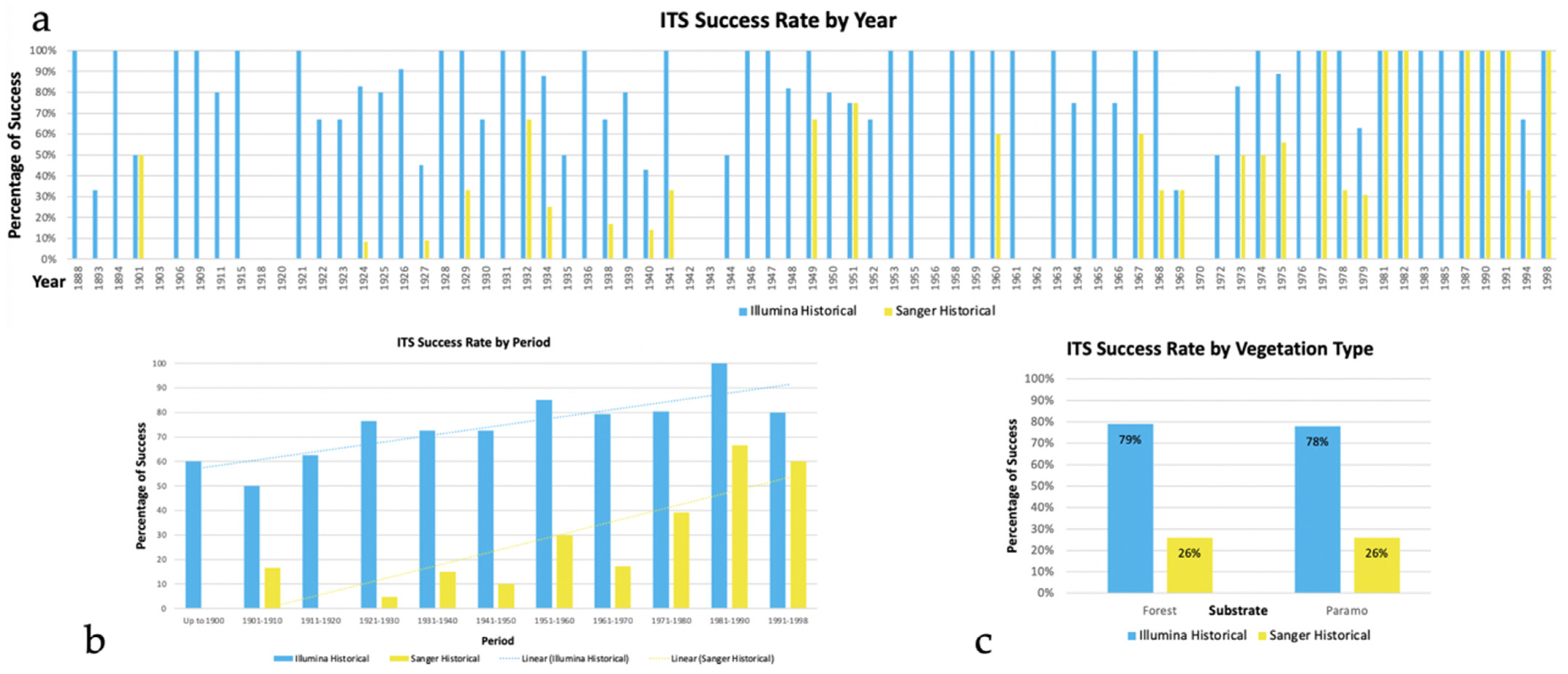
3.1. Comparison of Sequence Performance between Fresh and Historical Collections
- Run 01 (Plates 1 and 2): 331.6 MB (I1 or Indexes), 3.35 GB (R1 or forward reads) and 3.94 GB (R2 or reverse reads) (samples: NSF-001–106 and US-001–086); 8417853 total sequences;
- Run 02 (Plates 3 and 4): 289 MB (I1 or Indexes), 3.06 GB (R1 or forward reads) and 3.55 GB (R2 or reverse reads) (samples: US-087–278); 11250308 total sequences.
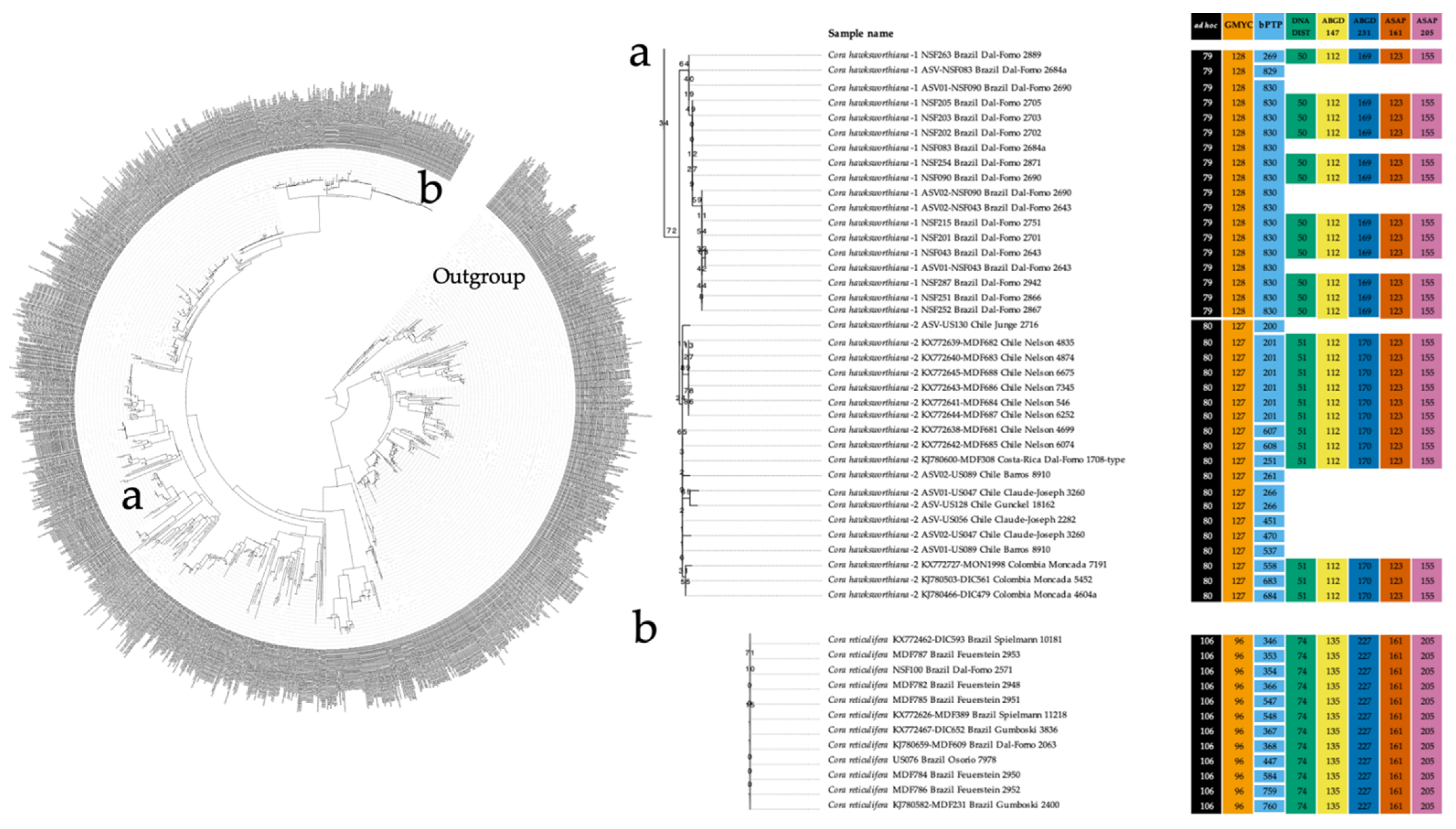
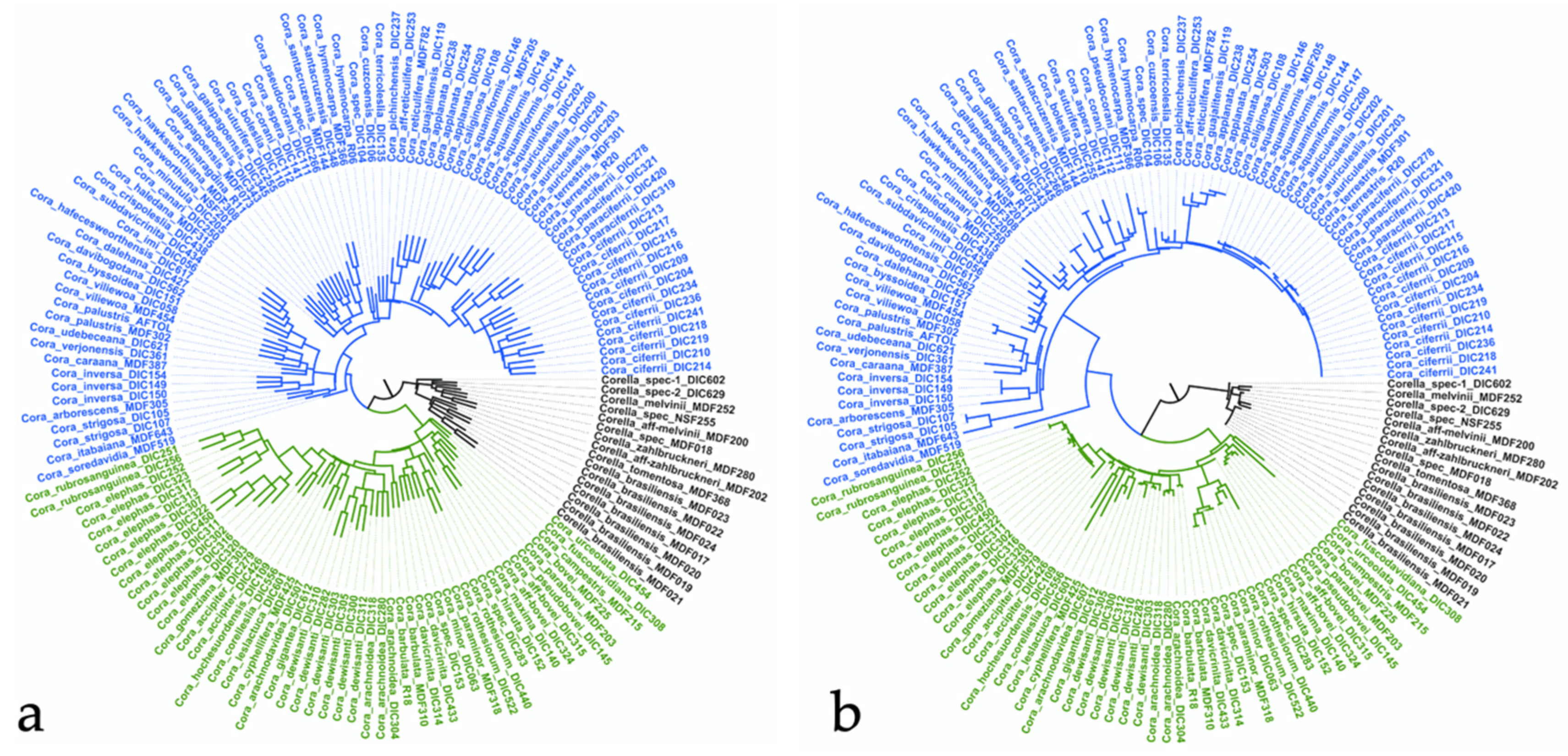
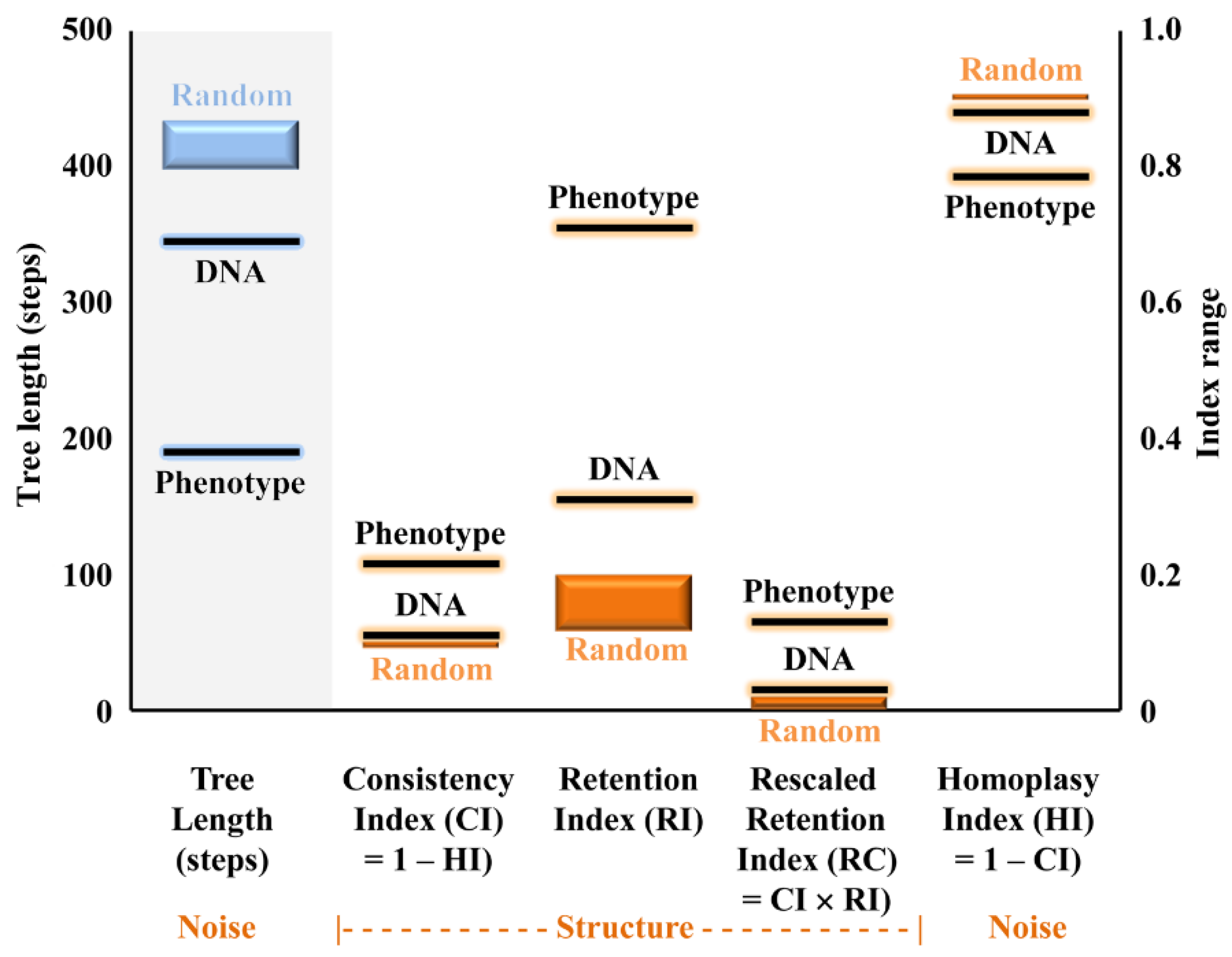
3.2. ITS-Based Phylogeny
3.3. Comparison between ITS and Astral Six-Marker Tree
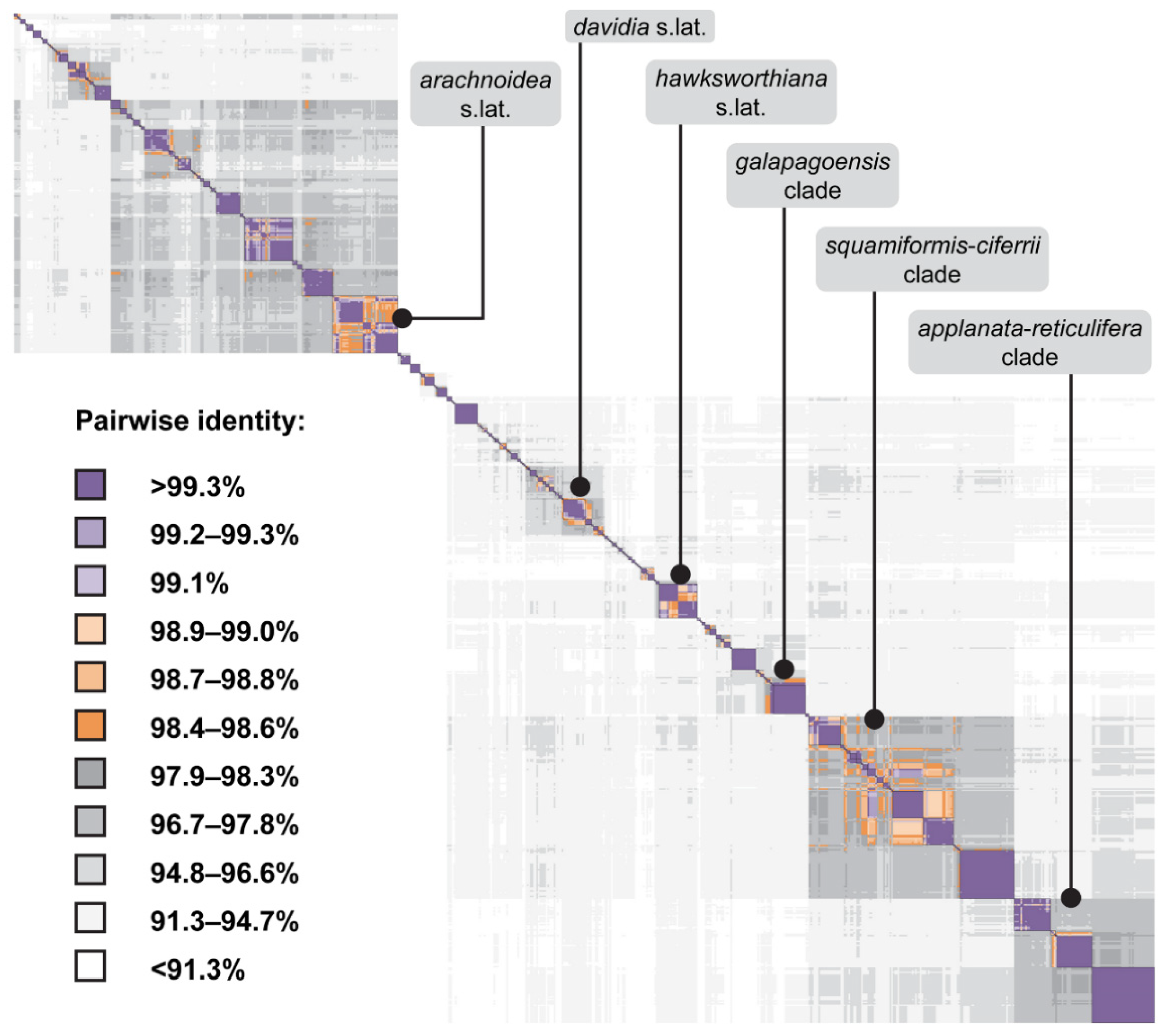
3.4. Quantitative Species Delimitation Methods
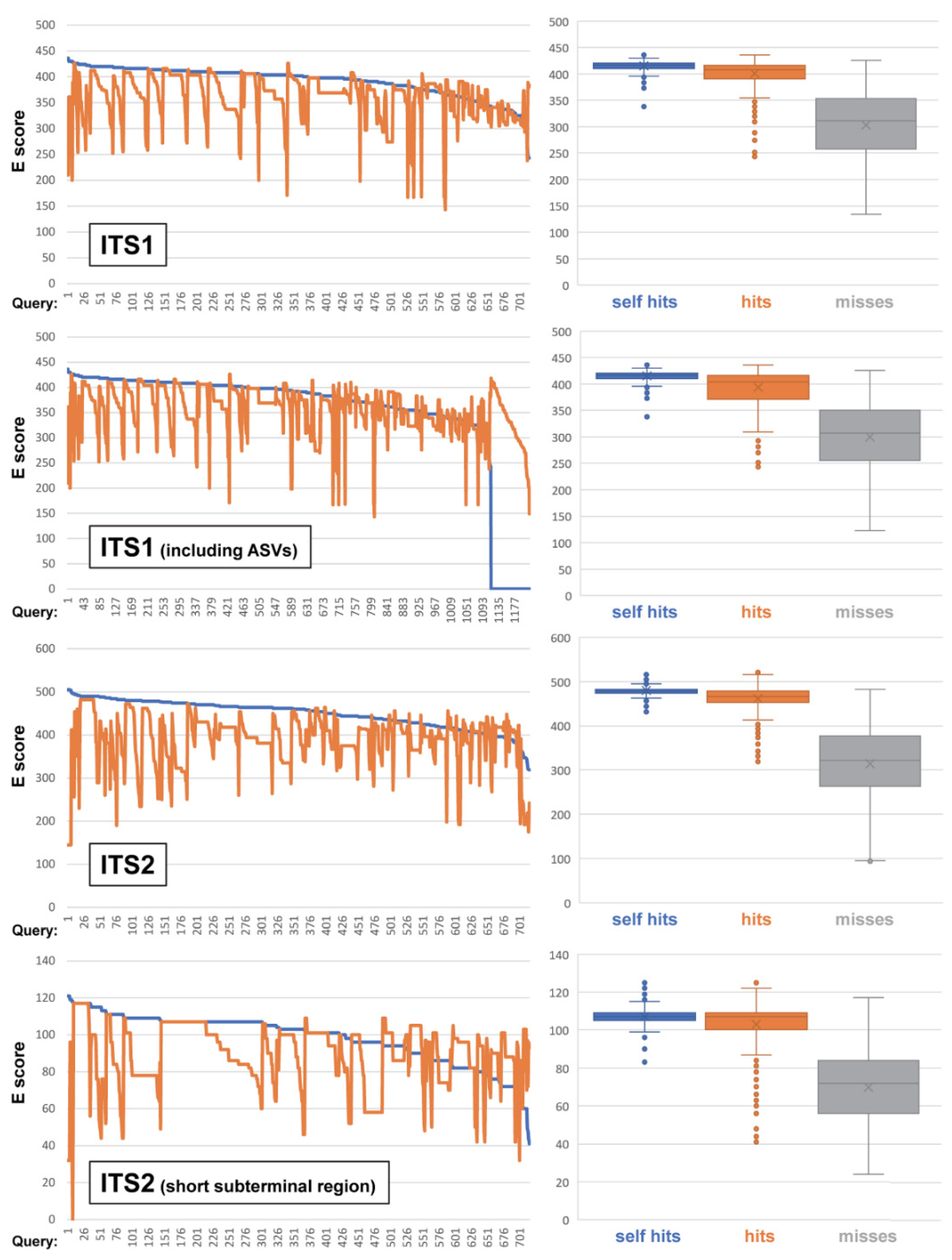
3.5. ITS-Based BLAST Performance
3.6. Phenotype Assessment
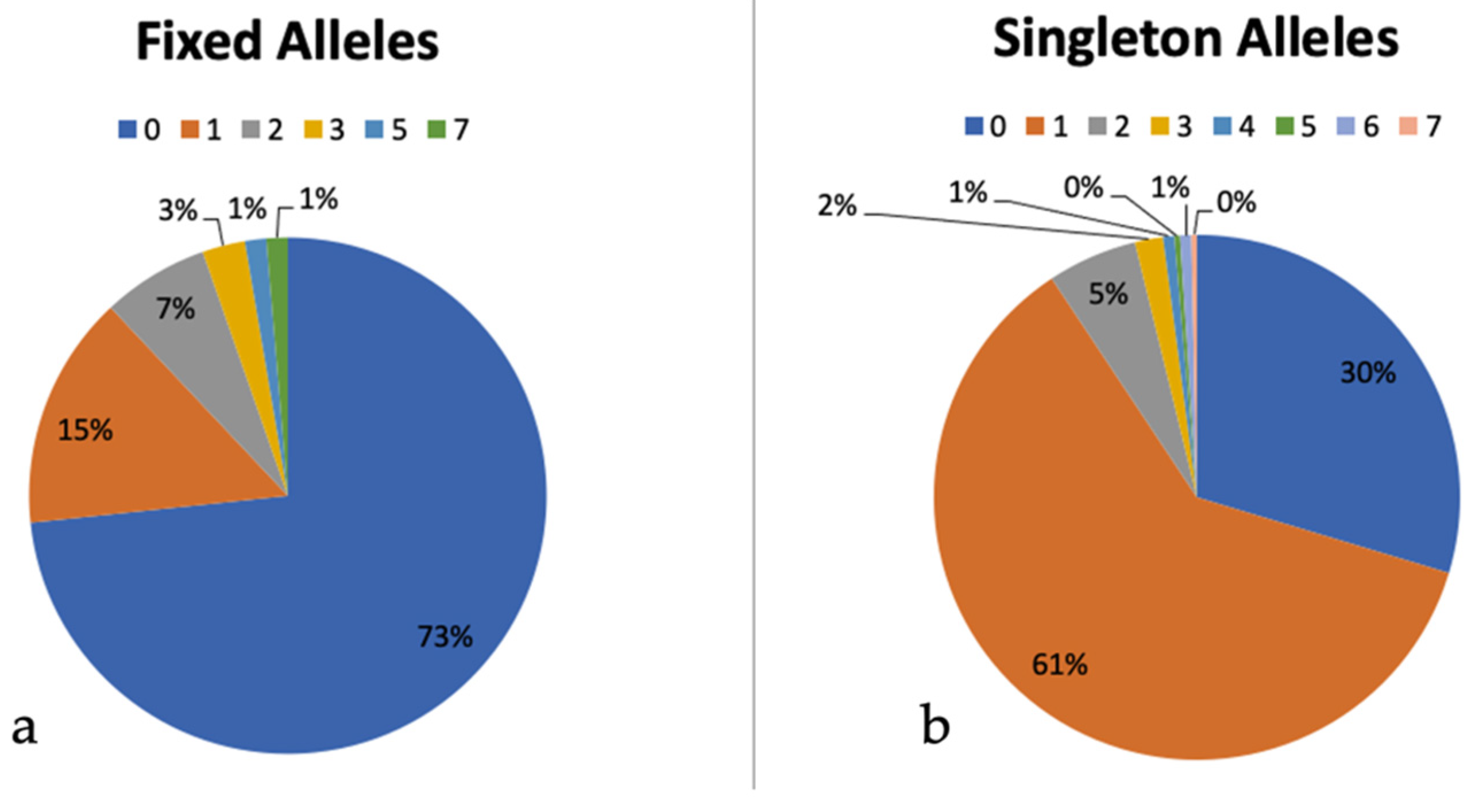
3.7. Intragenomic ITS Variation
4. Discussion
4.1. Use of Archival Specimens
4.2. Assessment of ITS as a Barcoding Marker and Intragenomic Variation within ITS
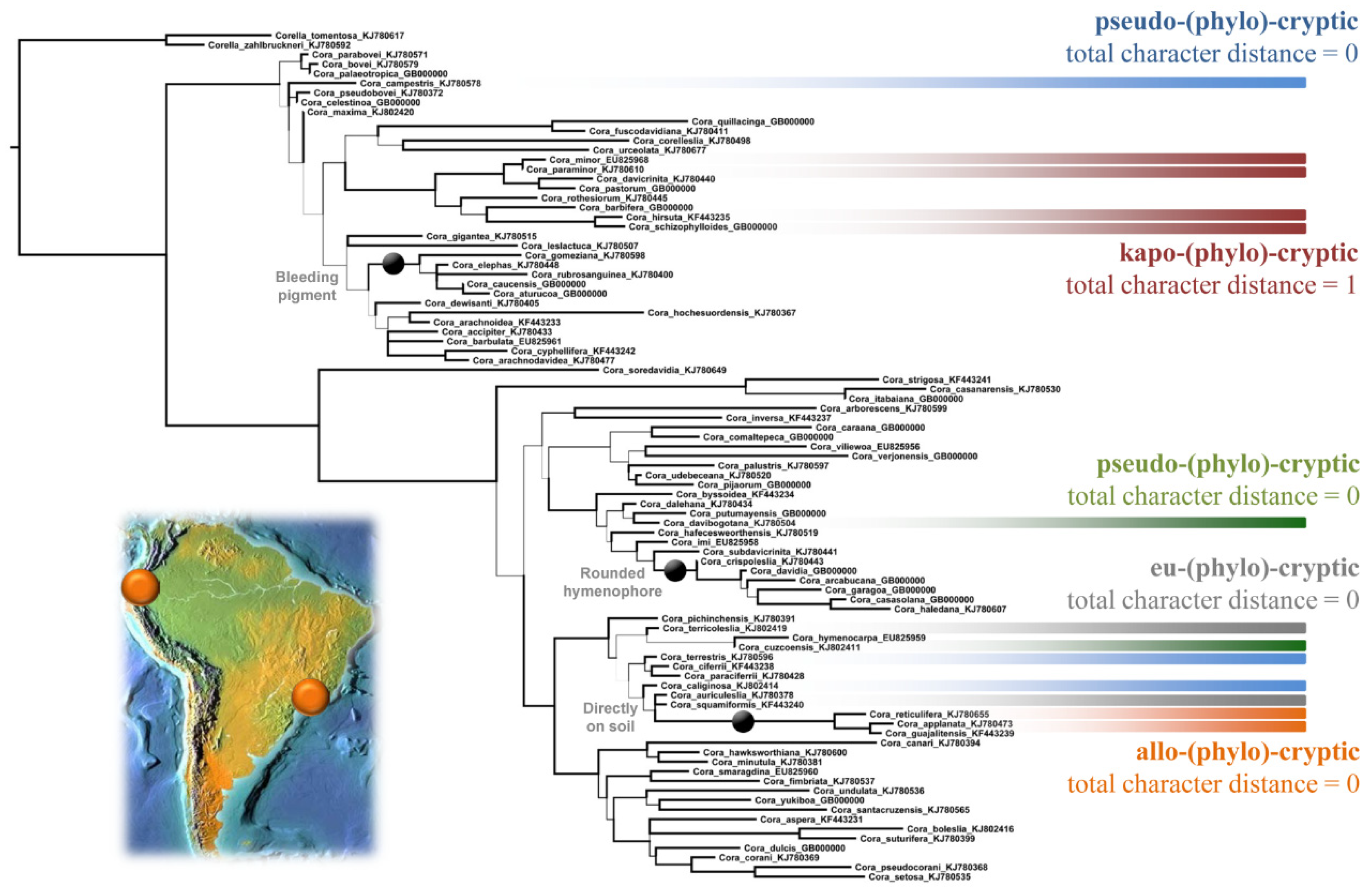
4.3. Accurate Assessment of Phylogenetic Diversity in Cora and Corella
4.4. Level of Cryptic Speciation and Potential Taxonomic Inflation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Funk, V.A. Collections-based science in the 21st Century. J. Syst. Evol. 2018, 56, 175–193. [Google Scholar] [CrossRef] [Green Version]
- Holmes, M.W.; Hammond, T.T.; Wogan, G.; Walsh, R.E.; LaBarbera, K.; Wommack, E.A.; Martins, F.M.; Crawford, J.C.; Mack, K.L.; Bloch, L.M.; et al. Natural history collections as windows on evolutionary processes. Mol. Ecol. 2016, 25, 864–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador, R.B.; Cunha, C.M. Natural history collections and the future legacy of ecological research. Oecologia 2020, 192, 641–646. [Google Scholar] [CrossRef] [PubMed]
- MacLean, H.J.; Nielsen, M.E.; Kingsolver, J.G.; Buckley, L.B. Using museum specimens to track morphological shifts through climate change. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20170404. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, B.P.; Heberling, J.M.; Meineke, E.K.; Turner, K.G.; Grassa, C.J.; Park, D.S.; Kennedy, J.; A Clarke, J.; A Cook, J.; Blackburn, D.C.; et al. Digitization and the Future of Natural History Collections. BioScience 2020, 70, 243–251. [Google Scholar] [CrossRef]
- Schindel, D.E.; Cook, J.A. The next generation of natural history collections. PLOS Biol. 2018, 16, e2006125. [Google Scholar] [CrossRef] [Green Version]
- Kauserud, H.; Stige, L.C.; Vik, J.O.; Økland, R.H.; Høiland, K.; Stenseth, N.C. Mushroom fruiting and climate change. Proc. Natl. Acad. Sci. USA 2008, 105, 3811–3814. [Google Scholar] [CrossRef] [Green Version]
- Ristaino, J.B. The Importance of Mycological and Plant Herbaria in Tracking Plant Killers. Front. Ecol. Evol. 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- Purvis, O.; Dubbin, W.; Chimonides, P.; Jones, G.; Read, H. The multi-element content of the lichen Parmelia sulcata, soil, and oak bark in relation to acidification and climate. Sci. Total Environ. 2008, 390, 558–568. [Google Scholar] [CrossRef]
- Agnan, Y.; Séjalon-Delmas, N.; Probst, A. Comparing early twentieth century and present-day atmospheric pollution in SW France: A story of lichens. Environ. Pollut. 2013, 172, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Minganti, V.; Drava, G.; de Pellegrini, R.; Modenesi, P.; Malaspina, P.; Giordani, P. Temporal trends (1981–2007) of trace and rare earth elements in the lichen Cetraria islandica (L.) Ach. from Italian herbaria. Chemosphere 2014, 99, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Stiller, M.; Knapp, M.; Stenzel, U.; Hofreiter, M.; Meyer, M. Direct multiplex sequencing (DMPS)—A novel method for targeted high-throughput sequencing of ancient and highly degraded DNA. Genome Res. 2009, 19, 1843–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieker, V.; Martin, M.D. Implications and future prospects for evolutionary analyses of DNA in historical herbarium collections. Bot. Lett. 2018, 165, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Staats, M.; Erkens, R.; van de Vossenberg, B.T.; Wieringa, J.; Kraaijeveld, K.; Stielow, B.; Geml, J.; Richardson, J.-E.; Bakker, F.T. Genomic Treasure Troves: Complete Genome Sequencing of Herbarium and Insect Museum Specimens. PLoS ONE 2013, 8, e69189. [Google Scholar] [CrossRef] [Green Version]
- Cong, Q.; Shen, J.; Zhang, J.; Li, W.; Kinch, L.N.; Calhoun, J.V.; Warren, A.D.; Grishin, N.V. Genomics Reveals the Origins of Historical Specimens. Mol. Biol. Evol. 2021, 38, 2166–2176. [Google Scholar] [CrossRef]
- Sohrabi, M.; Myllys, L.; Stenroos, S. Successful DNA sequencing of a 75 year-old herbarium specimen of Aspicilia aschabadensis (J. Steiner) Mereschk. Lichenol. 2010, 42, 626–628. [Google Scholar] [CrossRef]
- Redchenko, O.; Vondrák, J.; KOŠNAR, J. The oldest sequenced fungal herbarium sample. Lichenol. 2012, 44, 715–718. [Google Scholar] [CrossRef]
- Bendiksby, M.; Mazzoni, S.; Jørgensen, M.H.; Halvorsen, R.; Holien, H. Combining genetic analyses of archived specimens with distribution modelling to explain the anomalous distribution of the rare lichen Staurolemma omphalarioides: Long-distance dispersal or vicariance? J. Biogeogr. 2014, 41, 2020–2031. [Google Scholar] [CrossRef]
- Kistenich, S.; Halvorsen, R.; Schrøder-Nielsen, A.; Thorbek, L.; Timdal, E.; Bendiksby, M. DNA Sequencing Historical Lichen Specimens. Front. Ecol. Evol. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Gueidan, C.; Elix, J.A.; McCarthy, P.M.; Roux, C.; Mallen-Cooper, M.; Kantvilas, G. PacBio amplicon sequencing for metabarcoding of mixed DNA samples from lichen herbarium specimens. MycoKeys 2019, 53, 73–91. [Google Scholar] [CrossRef]
- Gueidan, C.; Li, L. A long-read amplicon approach to scaling up the metabarcoding of lichen herbarium specimens. MycoKeys 2022, 86, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Schmull, M.; Dal-Forno, M.; Lücking, R.; Cao, S.; Clardy, J.; Lawrey, J.D. Dictyonema huaorani (Agaricales: Hygrophoraceae), a new lichenized basidiomycete from Amazonian Ecuador with presumed hallucinogenic properties. Bryologist 2014, 117, 386–394. [Google Scholar] [CrossRef]
- Lücking, R.; Kaminsky, L.; Perlmutter, G.B.; Lawrey, J.D.; Forno, M.D. Cora timucua (Hygrophoraceae), a new and potentially extinct, previously misidentified basidiolichen of Florida inland scrub documented from historical collections. Bryologist 2020, 123, 657–673. [Google Scholar] [CrossRef]
- Osmundson, T.W.; Robert, V.A.; Schoch, C.L.; Baker, L.J.; Smith, A.; Robich, G.; Mizzan, L.; Garbelotto, M.M. Filling Gaps in Biodiversity Knowledge for Macrofungi: Contributions and Assessment of an Herbarium Collection DNA Barcode Sequencing Project. PLoS ONE 2013, 8, e62419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voitk, A.; Saar, I.; Lücking, R.; Moreau, P.-A.; Corriol, G.; Krisai-Greilhuber, I.; Thorn, R.G.; Hay, C.R.; Moncada, B.; Gulden, G. Surprising Morphological, Ecological and ITS Sequence Diversity in the Arrhenia acerosa Complex (Basidiomycota: Agaricales: Hygrophoraceae). Sydowia 2021, 73, 133. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium; Fungal Barcoding Consortium Author List; Bolchacova, E.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Lawrey, J.D.; Lücking, R.; Sipman, H.J.; Chaves, J.L.; Redhead, S.A.; Bungartz, F.; Sikaroodi, M.; Gillevet, P.M. High concentration of basidiolichens in a single family of agaricoid mushrooms (Basidiomycota: Agaricales: Hygrophoraceae). Mycol. Res. 2009, 113, 1154–1171. [Google Scholar] [CrossRef]
- Dal-Forno, M.; Lawrey, J.D.; Sikaroodi, M.; Bhattarai, S.; Gillevet, P.M.; Sulzbacher, M.; Lücking, R. Starting from scratch: Evolution of the lichen thallus in the basidiolichen Dictyonema (Agaricales: Hygrophoraceae). Fungal Biol. 2013, 117, 584–598. [Google Scholar] [CrossRef]
- Lodge, D.J.; Padamsee, M.; Matheny, P.B.; Aime, M.C.; Cantrell, S.A.; Boertmann, D.; Kovalenko, A.; Vizzini, A.; Dentinger, B.T.M.; Kirk, P.M.; et al. Molecular phylogeny, morphology, pigment chemistry and ecology in Hygrophoraceae (Agaricales). Fungal Divers. 2014, 64, 1–99. [Google Scholar] [CrossRef] [Green Version]
- Parmasto, E. The Genus Dictyonema (‘Thelephorolichenes’). Nova Hedwig. 1978, 29, 99–144. [Google Scholar]
- Hawksworth, D.L. A New Name for Dictyonema pavonium (Swartz) Parmasto. Lichenologist 1988, 20, 101. [Google Scholar] [CrossRef]
- Lücking, R.; Dal-Forno, M.; Sikaroodi, M.; Gillevet, P.M.; Bungartz, F.; Moncada, B.; Yánez-Ayabaca, A.; Chaves, J.L.; Coca, L.F.; Lawrey, J.D. A single macrolichen constitutes hundreds of unrecognized species. Proc. Natl. Acad. Sci. USA 2014, 111, 11091–11096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücking, R.; Forno, M.D.; Moncada, B.; Coca, L.F.; Vargas-Mendoza, L.Y.; Aptroot, A.; Arias, L.J.; Besal, B.; Bungartz, F.; Cabrera-Amaya, D.M.; et al. Turbo-taxonomy to assemble a megadiverse lichen genus: Seventy new species of Cora (Basidiomycota: Agaricales: Hygrophoraceae), honouring David Leslie Hawksworth’s seventieth birthday. Fungal Divers. 2016, 84, 139–207. [Google Scholar] [CrossRef]
- Lücking, R.; Leavitt, S.D.; Hawksworth, D.L. Species in lichen-forming fungi: Balancing between conceptual and practical considerations, and between phenotype and phylogenomics. Fungal Divers. 2021, 109, 99–154. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 1–32. [Google Scholar] [CrossRef]
- Stielow, J.; Lévesque, C.; Seifert, K.; Meyer, W.; Irinyi, L.; Smits, D.; Renfurm, R.; Verkley, G.; Groenewald, M.; Chaduli, D.; et al. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia-Mol. Phylogeny Evol. Fungi 2015, 35, 242–263. [Google Scholar] [CrossRef] [Green Version]
- Al-Hatmi, A.M.; Ende, A.G.V.D.; Stielow, J.B.; van Diepeningen, A.D.; Seifert, K.A.; McCormick, W.; Assabgui, R.; Gräfenhan, T.; De Hoog, G.S.; Levesque, C.A. Evaluation of two novel barcodes for species recognition of opportunistic pathogens in Fusarium. Fungal Biol. 2016, 120, 231–245. [Google Scholar] [CrossRef]
- Kelly, L.J.; Hollingsworth, P.M.; Coppins, B.J.; Ellis, C.J.; Harrold, P.; Tosh, J.; Yahr, R. DNA barcoding of lichenized fungi demonstrates high identification success in a floristic context. N. Phytol. 2011, 191, 288–300. [Google Scholar] [CrossRef]
- Pino-Bodas, R.; Martín, M.P.; Burgaz, A.R.; Lumbsch, H.T. Species delimitation in Cladonia (Ascomycota): A challenge to the DNA barcoding philosophy. Mol. Ecol. Resour. 2013, 13, 1058–1068. [Google Scholar] [CrossRef]
- Dal Forno, M.; Bungartz, F.; Yánez-Ayabaca, A.; Lücking, R.; Lawrey, J.D. High levels of endemism among Galapagos basidiolichens. Fungal Divers. 2017, 85, 45–73. [Google Scholar] [CrossRef]
- Lücking, R.; Lawrey, J.D.; Gillevet, P.M.; Sikaroodi, M.; Dal-Forno, M.; Berger, S.A. Multiple ITS Haplotypes in the Genome of the Lichenized Basidiomycete Cora inversa (Hygrophoraceae): Fact or Artifact? J. Mol. Evol. 2014, 78, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, M.; Grewe, F.; Thomas, A.; Harrison, C.H.; Lindgren, H.; Muggia, L.; Clair, L.L.S.; Lumbsch, H.T.; Leavitt, S.D. Characterizing the ribosomal tandem repeat and its utility as a DNA barcode in lichen-forming fungi. BMC Evol. Biol. 2020, 20, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Forno, M.; Lawrey, J.D.; Sikaroodi, M.; Gillevet, P.M.; Schuettpelz, E.; Lücking, R. Extensive photobiont sharing in a rapidly radiating cyanolichen clade. Mol. Ecol. 2021, 30, 1755–1776. [Google Scholar] [CrossRef] [PubMed]
- Dal Forno, M.; Kaminsky, L.; Lücking, R. IUCN Red List of Threatened Species: Cora timucua. 2020. Available online: https://www.iucnredlist.org/ (accessed on 23 January 2022).
- Marotz, C.; Amir, A.; Humphrey, G.; Gaffney, J.; Gogul, G.; Knight, R. DNA extraction for streamlined metagenomics of diverse environmental samples. BioTechniques 2017, 62, 290–293. [Google Scholar] [CrossRef] [Green Version]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes—Application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- ITS Illumina Amplicon Protocol. Earth Microbiome Project. Available online: https://earthmicrobiome.org/protocols-and-standards/its/ (accessed on 5 July 2017).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. QIIME 2: Reproducible, Interactive, Scalable, and Extensible Microbiome Data Science. Peer J. Prepr. 2018. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Abarenkov, K.; Zirk, A.; Piirmann, T.; Pöhönen, R.; Ivanov, F.; Nilsson, R.H.; Kõljalg, U. UNITE QIIME Release Fungi 2; UNITE Community: Tuckingmill, UK, 2020. [Google Scholar] [CrossRef]
- Hydra Smithsonian Institution. High Performance Computing Cluster; Smithsonian Institution: Washington, DC, USA, 2022. [Google Scholar]
- Gostel, M.R.; Coy, K.A.; Weeks, A. Microfluidic PCR-based target enrichment: A case study in two rapid radiations of Commiphora (Burseraceae) from Madagascar. J. Syst. Evol. 2015, 53, 411–431. [Google Scholar] [CrossRef]
- Gostel, M.R.; Zúñiga, J.D.; Kress, W.J.; Funk, V.A.; Puente-Lelievre, C. Microfluidic Enrichment Barcoding (MEBarcoding): A new method for high throughput plant DNA barcoding. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sela, I.; Ashkenazy, H.; Katoh, K.; Pupko, T. GUIDANCE2: Accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015, 43, W7–W14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: Enabling high-impact science for phylogenetics researchers with limited resources. In Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the Extreme to the Campus and Beyond, New York, NY, USA, 16–19 July 2012; pp. 1–8. [Google Scholar]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.R. TreeDist: Distances between Phylogenetic Trees. Comprehensive R Archive Network. Bioinformatics 2020, 36, 5007–5013. [Google Scholar] [CrossRef]
- Smith, M.R. Information theoretic generalized Robinson–Foulds metrics for comparing phylogenetic trees. Bioinformatics 2021, 37, 2077–2078. [Google Scholar] [CrossRef]
- Fujisawa, T.; Barraclough, T.G. Delimiting Species Using Single-Locus Data and the Generalized Mixed Yule Coalescent Approach: A Revised Method and Evaluation on Simulated Data Sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. PHYLIP—Phylogeny Inference Package (Version 3.57c); University of Washington: Seattle, WA, USA, 1995. [Google Scholar]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.; Biosciences, I.; Carlsbad, C. BioEdit: An Important Software for Molecular Biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Madden, T. The BLAST Sequence Analysis Tool. In The NCBI Handbook; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2003. [Google Scholar]
- Swofford, D. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods); Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar] [CrossRef]
- Swofford, D.L.; Sullivan, J. Phylogeny Inference Based on Parsimony and Other Methods Using PAUP*. The Phylogenetic Handbook: A Practical Approach to DNA and Protein Phylogeny; Sinauer Associates: Sunderland, MA, USA, 2003; Volume 7, pp. 160–206. [Google Scholar]
- Wong, B. Points of view: Color blindness. Nat. Methods 2011, 8, 441. [Google Scholar] [CrossRef]
- Bakker, F.T.; Bieker, V.C.; Martin, M.D. Editorial: Herbarium Collection-Based Plant Evolutionary Genetics and Genomics. Front. Ecol. Evol. 2020, 8, 603948. [Google Scholar] [CrossRef]
- Ohmura, Y.; Uno, K.; Hosaka, K.; Hosoya, T. DNA Fragmentation of herbarium specimens of lichens, and significance of epitypification for threatened species of Japan. In Proceedings of the 10th International Mycological Congress, Bangkok, Thailand, 3–9 August 2014; p. 151. [Google Scholar]
- Gueidan, C.; Aptroot, A.; Cáceres, M.E.D.S.; Binh, N.Q. Molecular phylogeny of the tropical lichen family Pyrenulaceae: Contribution from dried herbarium specimens and FTA card samples. Mycol. Prog. 2016, 15. [Google Scholar] [CrossRef]
- Kranner, I.; Zorn, M.; Turk, B.; Wornik, S.; Beckett, R.P.; Batič, F. Biochemical traits of lichens differing in relative desiccation tolerance. N. Phytol. 2003, 160, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranner, I.; Beckett, R.; Hochman, A.; Nash, T.H. Desiccation-Tolerance in Lichens: A Review. Bryologist 2008, 111, 576–593. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Grube, M. Lichens redefined as complex ecosystems. N. Phytol. 2020, 227, 1281–1283. [Google Scholar] [CrossRef]
- Sierra, M.A.; Danko, D.C.; Sandoval, T.A.; Pishchany, G.; Moncada, B.; Kolter, R.; Mason, C.E.; Zambrano, M.M. The Microbiomes of Seven Lichen Genera Reveal Host Specificity, a Reduced Core Community and Potential as Source of Antimicrobials. Front. Microbiol. 2020, 11, 398. [Google Scholar] [CrossRef] [Green Version]
- Hodkinson, B.P.; Gottel, N.R.; Schadt, C.W.; Lutzoni, F. Photoautotrophic symbiont and geography are major factors affecting highly structured and diverse bacterial communities in the lichen microbiome. Environ. Microbiol. 2012, 14, 147–161. [Google Scholar] [CrossRef]
- Prillinger, H.; Kraepelin, G.; Lopandic, K.; Schweigkofler, W.; Molnár, O.; Weigang, F.; Dreyfuss, M.M. New species of Fellomyces Isolated from Epiphytic Lichen Species. Syst. Appl. Microbiol. 1997, 20, 572–584. [Google Scholar] [CrossRef]
- Mark, K.; Laanisto, L.; Bueno, C.G.; Niinemets, Ü.; Keller, C.; Scheidegger, C. Contrasting co-occurrence patterns of photobiont and cystobasidiomycete yeast associated with common epiphytic lichen species. N. Phytol. 2020, 227, 1362–1375. [Google Scholar] [CrossRef]
- Arnold, A.E.; Miadlikowska, J.; Higgins, K.L.; Sarvate, S.D.; Gugger, P.; Way, A.; Hofstetter, V.; Kauff, F.; Lutzoni, F. A Phylogenetic Estimation of Trophic Transition Networks for Ascomycetous Fungi: Are Lichens Cradles of Symbiotrophic Fungal Diversification? Syst. Biol. 2009, 58, 283–297. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.; Passarini, M.R.Z.; Delforno, T.; Pellizzari, F.; Cipro, C.V.Z.; Montone, R.C.; Petry, M.; Putzke, J.; Rosa, L.H.; Sette, L.D. Yeasts from macroalgae and lichens that inhabit the South Shetland Islands, Antarctica. Environ. Microbiol. Rep. 2016, 8, 874–885. [Google Scholar] [CrossRef]
- Lindgren, H.; Diederich, P.; Goward, T.; Myllys, L. The phylogenetic analysis of fungi associated with lichenized ascomycete genus Bryoria reveals new lineages in the Tremellales including a new species Tremella huuskonenii hyperparasitic on Phacopsis huuskonenii. Fungal Biol. 2015, 119, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Millanes, A.M.; Diederich, P.; Wedin, M. Cyphobasidium gen. nov., a new lichen-inhabiting lineage in the Cystobasidiomycetes (Pucciniomycotina, Basidiomycota, Fungi). Fungal Biol. 2015, 120, 1468–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePriest, P.T.; Ivanova, N.V.; Fahselt, D.; Alstrup, V.; Gargas, A. Sequences of Psychrophilic Fungi Amplified from Glacier-Preserved Ascolichens. Can. J. Bot. 2000, 78, 1450–1459. [Google Scholar] [CrossRef] [Green Version]
- Spribille, T.; Tuovinen, V.; Resl, P.; Vanderpool, D.; Wolinski, H.; Aime, M.C.; Schneider, K.; Stabentheiner, E.; Toome-Heller, M.; Thor, G.; et al. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 2016, 353, 488–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrey, J.D.; Diederich, P. Lichenicolous Fungi: Interactions, Evolution, and Biodiversity. Bryologist 2003, 106, 80–120. [Google Scholar] [CrossRef]
- Diederich, P.; Lawrey, J.D.; Ertz, D. The 2018 classification and checklist of lichenicolous fungi, with 2000 non-lichenized, obligately lichenicolous taxa. Bryologist 2018, 121. [Google Scholar] [CrossRef]
- Tuovinen, V.; Ekman, S.; Thor, G.; Vanderpool, D.; Spribille, T.; Johannesson, H. Two Basidiomycete Fungi in the Cortex of Wolf Lichens. Curr. Biol. 2019, 29, 476–483.e5. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, L.; Feng, H.; Tu, W.; Bao, Z.; Xiong, C.; Wang, X.; Qing, Y.; Huang, W. Characterization of the Complete Mitochondrial Genome of Basidiomycete Yeast Hannaella oryzae: Intron Evolution, Gene Rearrangement, and Its Phylogeny. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Fernández-Mendoza, F.; Fleischhacker, A.; Kopun, T.; Grube, M.; Muggia, L. ITS1 metabarcoding highlights low specificity of lichen mycobiomes at a local scale. Mol. Ecol. 2017, 26, 4811–4830. [Google Scholar] [CrossRef]
- Banchi, E.; Ametrano, C.G.; Stanković, D.; Verardo, P.; Moretti, O.; Gabrielli, F.; Lazzarin, S.; Borney, M.; Tassan, F.; Tretiach, M.; et al. DNA metabarcoding uncovers fungal diversity of mixed airborne samples in Italy. PLoS ONE 2018, 13, e0194489. [Google Scholar] [CrossRef]
- Wright, B.; Clair, L.L.S.; Leavitt, S.D. Is targeted community DNA metabarcoding suitable for biodiversity inventories of lichen-forming fungi? Ecol. Indic. 2019, 98, 812–820. [Google Scholar] [CrossRef]
- Olds, C.G.; Berta-Thompson, J.W.; Loucks, J.J.; Levy, R.A.; Wilson, A.W. Applying a Modified Metabarcoding Approach for the Sequencing of Macrofungal Specimens from Fungarium Collections. bioRxiv 2021. [Google Scholar] [CrossRef]
- Leavitt, S.D.; Johnson, L.; St. Clair, L.L. Species Delimitation and Evolution in Morphologically and Chemically Diverse Communities of the Lichen-forming Genus Xanthoparmelia (Parmeliaceae, Ascomycota) in Western North America. Am. J. Bot. 2011, 98, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-H.; Lücking, R.; Liu, H.-J.; Wei, X.-L.; Xavier-Leite, A.B.; Portilla, C.V.; Ren, Q.; Wei, J.-C. Twelve New Species Reveal Cryptic Diversification in Foliicolous Lichens of Strigula s.lat. (Strigulales, Ascomycota). J. Fungi 2022, 8, 2. [Google Scholar] [CrossRef]
- Leavitt, S.D.; Grewe, F.; Widhelm, T.; Muggia, L.; Wray, B.; Lumbsch, H.T. Resolving evolutionary relationships in lichen-forming fungi using diverse phylogenomic datasets and analytical approaches. Sci. Rep. 2016, 6, 22262. [Google Scholar] [CrossRef]
- Pizarro, D.; Divakar, P.K.; Grewe, F.; Leavitt, S.D.; Huang, J.-P.; Grande, F.D.; Schmitt, I.; Wedin, M.; Crespo, A.; Lumbsch, T. Phylogenomic analysis of 2556 single-copy protein-coding genes resolves most evolutionary relationships for the major clades in the most diverse group of lichen-forming fungi. Fungal Divers. 2018, 92, 31–41. [Google Scholar] [CrossRef]
- Lücking, R.; Moncada, B.; Widhelm, T.J.; Lumbsch, H.T.; Blanchon, D.J.; de Lange, P.J. The Sticta Filix—Sticta Lacera Conundrum (Lichenized Ascomycota: Peltigeraceae Subfamily Lobarioideae): Unresolved Lineage Sorting or Developmental Switch? Bot. J. Linn. Soc. 2021, boab083. [Google Scholar] [CrossRef]
- Grewe, F.; Lagostina, E.; Wu, H.; Printzen, C.; Lumbsch, T. Population genomic analyses of RAD sequences resolves the phylogenetic relationship of the lichen-forming fungal species Usnea antarctica and Usnea aurantiacoatra. MycoKeys 2018, 43, 91–113. [Google Scholar] [CrossRef]
- Lagostina, E.; Grande, F.D.; Andreev, M.; Printzen, C. The use of microsatellite markers for species delimitation in Antarctic Usnea subgenus Neuropogon. Mycologia 2018, 110, 1047–1057. [Google Scholar] [CrossRef]
- Widhelm, T.J.; Grewe, F.; Huang, J.; Ramanauskas, K.; Mason-Gamer, R.; Lumbsch, H.T. Using RADseq to understand the circum-Antarctic distribution of a lichenized fungus, Pseudocyphellaria glabra. J. Biogeogr. 2021, 48, 78–90. [Google Scholar] [CrossRef]
- Boluda, C.; Rico, V.; Divakar, P.; Nadyeina, O.; Myllys, L.; McMullin, T.; Zamora, J.C.; Scheidegger, C.; Hawksworth, D. Evaluating methodologies for species delimitation: The mismatch between phenotypes and genotypes in lichenized fungi (Bryoria sect. Implexae, Parmeliaceae). Persoonia-Mol. Phylogeny Evol. Fungi 2019, 42, 75–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücking, R.; Hodkinson, B.P.; Leavitt, S.D. The 2016 classification of lichenized fungi in the Ascomycota and Basidiomycota—Approaching one thousand genera. Bryologist 2017, 119, 361. [Google Scholar] [CrossRef]
- Larcher, W.; Vareschi, V. Variation in Morphology and Functional Traits of Dictyonema glabratum from Contrasting Habitats in the Venezuelan Andes. Lichenologist 1988, 20, 269–277. [Google Scholar] [CrossRef]
- Hawksworth, D.L. Some advances in the study of lichens since the time of E. M. Holmes. Bot. J. Linn. Soc. 1973, 67, 3–31. [Google Scholar] [CrossRef]
- Geml, J.; Laursen, G.A.; O’Neill, K.; Nusbaum, H.C.; Taylor, D.L. Beringian origins and cryptic speciation events in the fly agaric (Amanita muscaria). Mol. Ecol. 2006, 15, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, W.; Endo, N.; Fukuda, M.; Yamada, A. Phylogenetic analyses of Japanese golden chanterelles and a new species description, Cantharellus anzutake sp. nov. Mycoscience 2018, 59, 153–165. [Google Scholar] [CrossRef]
- Leacock, P.R.; Riddell, J.; Wilson, A.W.; Zhang, R.; Ning, C.; Mueller, G.M. Cantharellus chicagoensis sp. nov. is supported by molecular and morphological analysis as a new yellow chanterelle in midwestern United States. Mycologia 2016, 108, 765–772. [Google Scholar] [CrossRef]
- Buyck, B.; Hofstetter, V. The contribution of tef-1 sequences to species delimitation in the Cantharellus cibarius complex in the southeastern USA. Fungal Divers. 2011, 49, 35–46. [Google Scholar] [CrossRef]
- Hapuarachchi, K.K.; Wen, T.C.; Deng, C.Y.; Kang, J.C.; Hyde, K.D. Mycosphere Essays 1: Taxonomic Confusion in the Ganoderma lucidum Species Complex. Mycosphere 2015, 6, 542–559. [Google Scholar] [CrossRef]
- Du, X.-H.; Zhao, Q.; O’Donnell, K.; Rooney, A.P.; Yang, Z.L. Multigene molecular phylogenetics reveals true morels (Morchella) are especially species-rich in China. Fungal Genet. Biol. 2012, 49, 455–469. [Google Scholar] [CrossRef]
- Moncada, B.; Reidy, B.; Lücking, R. A phylogenetic revision of Hawaiian Pseudocyphellaria sensu lato (lichenized Ascomycota: Lobariaceae) reveals eight new species and a high degree of inferred endemism. Bryologist 2014, 117, 119–160. [Google Scholar] [CrossRef]
- Lumbsch, H.T.; Elix, J. Lecanora. In Flora of Australia Volume 56A, Lichens 4; CSIRO Publishing: Clayton, VIC, Australia, 2004; ISBN 0-643-09056-8. [Google Scholar]
- Guderley, R. Die Lecanora subfusca-Gruppe in Süd-Und Mittelamerika. J. Hattori Bot. Lab. 1999, 87, 131–257. [Google Scholar] [CrossRef]
- Guderley, R.; Lumbsch, H.T. Notes on Multispored Species of Lecanora Sensu Stricto. Lichenologist 1999, 31, 197–203. [Google Scholar] [CrossRef]
- Lumbsch, H.T.; Plümper, M.; Guderley, R.; Feige, G.B. The Corticolous Species of Lecanora Sensu Stricto with Pruinose Apothecial Discs. Symb. Bot. Ups. 1997, 32, 131–161. [Google Scholar]
- Lumbsch, H.T.; Feige, G.B.; Elix, J.A. A Revision of the Usnic Acid Containing Taxa Belonging to Lecanora sensu stricto (Lecanorales: Lichenized Ascomycotina). Bryologist 1995, 98, 561–577. [Google Scholar] [CrossRef]
- Lumbsch, H.T.; Guderley, R.; Elix, J.A. A Revision of Some Species in Lecanora sensu stricto with a Dark Hypothecium (Lecanorales, Ascomycotina). Bryologist 1996, 99, 269–291. [Google Scholar] [CrossRef]
- Zhao, X.; Leavitt, S.D.; Zhao, Z.T.; Zhang, L.L.; Arup, U.; Grube, M.; Pérez-Ortega, S.; Printzen, C.; Śliwa, L.; Kraichak, E.; et al. Towards a revised generic classification of lecanoroid lichens (Lecanoraceae, Ascomycota) based on molecular, morphological and chemical evidence. Fungal Divers. 2016, 78, 293–304. [Google Scholar] [CrossRef]
- Bungartz, F.; Elix, J.A.; Printzen, C. Lecanoroid lichens in the Galapagos Islands: The genera Lecanora, Protoparmeliopsis, and Vainionora (Lecanoraceae, Lecanoromycetes). Phytotaxa 2020, 431, 1–85. [Google Scholar] [CrossRef]
- Guzow-Krzemińska, B.; Łubek, A.; Malíček, J.; Tønsberg, T.; Oset, M.; Kukwa, M. Lecanora stanislai, a new, sterile, usnic acid containing lichen species from Eurasia and North America. Phytotaxa 2017, 329, 201–211. [Google Scholar] [CrossRef]
- Tripp, E.A.; Morse, C.A.; Keepers, K.G.; Stewart, C.A.; Pogoda, C.S.; White, K.H.; Hoffman, J.R.; Kane, N.C.; McCain, C.M. Evidence of substrate endemism of lichens on Fox Hills Sandstone: Discovery and description of Lecanora lendemeri as new to science. Bryologist 2019, 122, 246–259. [Google Scholar] [CrossRef]
- Gerlach, A.D.C.L.; Clerc, P.; da Silveira, R.M.B. Taxonomy of the corticolous, shrubby, esorediate, neotropical species of Usnea Adans. (Parmeliaceae) with an emphasis on southern Brazil. Lichenologist 2017, 49, 199–238. [Google Scholar] [CrossRef]
- Gerlach, A.D.C.L.; Toprak, Z.; Naciri, Y.; Caviró, E.A.; da Silveira, R.M.B.; Clerc, P. New insights into the Usnea cornuta aggregate (Parmeliaceae, lichenized Ascomycota): Molecular analysis reveals high genetic diversity correlated with chemistry. Mol. Phylogenet. Evol. 2019, 131, 125–137. [Google Scholar] [CrossRef]
- Truong, C.; Clerc, P. The lichen genus Usnea (Parmeliaceae) in tropical South America: Species with a pigmented medulla, reacting C+ yellow. Lichenologist 2012, 44, 625–637. [Google Scholar] [CrossRef] [Green Version]
- Truong, C.; Bungartz, F.; Clerc, P. The lichen genus Usnea (Parmeliaceae) in the tropical Andes and the Galapagos: Species with a red-orange cortical or subcortical pigmentation. Bryologist 2011, 114, 477–503. [Google Scholar] [CrossRef]
- Truong, C.; Rodriguez, J.M.; Clerc, P. Pendulous Usnea species (Parmeliaceae, lichenized Ascomycota) in tropical South America and the Galapagos. Lichenologist 2013, 45, 505–543. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Campos, M.A.; Clerc, P.; Nash, T.H. Pendulous Species of Usnea from the Temperate Forests in Mexico. Bryologist 1998, 101, 303–329. [Google Scholar] [CrossRef]
- Thell, A.; Crespo, A.; Divakar, P.K.; Kärnefelt, I.; Leavitt, S.D.; Lumbsch, H.T.; Seaward, M.R.D. A review of the lichen family Parmeliaceae—History, phylogeny and current taxonomy. Nord. J. Bot. 2012, 30, 641–664. [Google Scholar] [CrossRef]
- Kirika, P.M.; Divakar, P.K.; Buaruang, K.; Leavitt, S.D.; Crespo, A.; Gatheri, G.W.; Mugambi, G.; Benatti, M.N.; Lumbsch, H.T. Molecular phylogenetic studies unmask overlooked diversity in the tropical lichenized fungal genus Bulbothrix s.l. (Parmeliaceae, Ascomycota). Bot. J. Linn. Soc. 2017, 184, 387–399. [Google Scholar] [CrossRef]
- Vondrák, J.; Frolov, I.; Arup, U.; Khodosovtsev, A. Methods for phenotypic evaluation of crustose lichens with emphasis on Teloschistaceae. Chornomorski Bot. J. 2013, 9, 382–405. [Google Scholar] [CrossRef]
- Frolov, I.; Vondrák, J.; Fernández-Mendoza, F.; Wilk, K.; Khodosovtsev, A.; Halıcı, M.G. Three New, Seemingly-Cryptic Species in the Lichen Genus Caloplaca (Teloschistaceae) Distinguished in Two-Phase Phenotype Evaluation. Ann. Bot. Fenn. 2016, 53, 243–262. [Google Scholar] [CrossRef] [Green Version]
- Lücking, R.; Archer, A.W.; Aptroot, A. A world-wide key to the genus Graphis (Ostropales: Graphidaceae). Lichenologist 2009, 41, 363–452. [Google Scholar] [CrossRef]
- Lücking, R.; Kalb, K. Formal Instatement of Allographa (Graphidaceae): How to Deal with a Hyperdiverse Genus Complex with Cryptic Differentiation and Paucity of Molecular Data. Herzogia 2018, 31, 535–561. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Character\State | 0 | 1 | 2 | 3 | 4 | 5 | ? | |
|---|---|---|---|---|---|---|---|---|
| 1 | Substrate a | — | rock | soil | ground | epiphytic | — | — |
| 2 | Size | — | small (lobes > 1 cm) | 10édium (lobes 1–3 cm) | large (lobes > 3cm) | — | — | — |
| 3 | Sutures b | absent | short | long | — | — | — | — |
| 4 | Color c | — | grey | (grey-) brown | olive (-green) | green | aeruginous | — |
| 5 | Surface | even | granular- rugose | pitted | broadly undulate | narrowly undulate | — | — |
| 6 | Trichomes d | absent | present (felty) | present (setose) | present (strigose) | — | — | — |
| 7 | Margin | glabrous | pilose | granular | granular-pilose | — | — | — |
| 8 | Soredia | absent | present | — | — | — | — | — |
| 9 | Cortex | absent | viaduct- shaped | collapsed | compacted | prosoplecten- chymatous | paraplecten- chymatous | — |
| 10 | Papillae | absent | present | — | — | — | — | — |
| 11 | Hymenophore | — | rounded- confluent | concentric | resupinate- cyphelloid | cyphelloid | — | not observed |
| 12 | Bleeding pigment | absent | present | — | — | — | — | — |
| 13 | Central America | absent | present | — | — | — | — | — |
| 14 | Caribbean | absent | present | — | — | — | — | — |
| 15 | Galapagos | absent | present | — | — | — | — | — |
| 16 | Northern Andes | absent | present | — | — | — | — | — |
| 17 | Central Andes | absent | present | — | — | — | — | — |
| 18 | Southern Andes | absent | present | — | — | — | — | — |
| 19 | Eastern Brazil | absent | present | — | — | — | — | — |
| 20 | Asia | absent | present | — | — | — | — | — |
| Proposed Term | Etymology | Phenotype Distance | Phylogenetic Relationship | Distribution |
|---|---|---|---|---|
| Eu-(phylo-)cryptic (“cryptic”) | Greek: eús, eû = good | zero (same phenotype) | closely related or sister species | sympatric |
| Kapo-(phylo-)cryptic (“near-cryptic”) | Greek: kápos = somewhat | very low (similar phenotype) | closely related or sister species | sympatric |
| Allo-(phylo-)cryptic (“semi-cryptic”) | Greek: állos = other | zero or very low (same or similar phenotype) | closely related or sister species | allopatric |
| Pseudo-(phylo-)cryptic (“homoplasic”) | Greek: pseudḗs = false | zero or very low (same or similar phenotype) | unrelated or distantly related | sympatric or allopatric |
| Dataset | Method | Settings | Estimate | Other Metrics: | |||
|---|---|---|---|---|---|---|---|
| Cora 1325 Terminals | bPTP | min | 791 (mean) | 708 (min) | 889 (max) | ||
| Cora 1325 Terminals | GMYC | single | 189 | 145–237 (confidence interval) | 36.02878 (likelihood ratio) | 1.50E-08 (LR Test) | |
| Cora 1325 Terminals | ad hoc | 265 | |||||
| Extrapolated | Barcode gap/threshold distance | ||||||
| Cora 716 Terminals | ad hoc | 175 | 265 | N/A | |||
| Cora 716 Terminals | DNADIST | 171 | 259 | 0.0060 | |||
| Prior maximal distance | Barcode gap/threshold distance | ||||||
| Cora 716 Terminals | ABGD | Partition 2 | 231 | 350 | 0.001668 | 0.0040 | |
| Cora 716 Terminals | ABGD | Partition 3 | 231 | 350 | 0.002783 | 0.0040 | |
| Cora 716 Terminals | ABGD | Partition 4 | 147 | 223 | 0.004642 | 0.0110 | |
| ASAP score | p-value | Barcode gap/threshold distance | |||||
| Cora 716 Terminals | ASAP | K80-2 | 161 | 244 | 11.0 | 0.1620 | 0.0086 |
| Cora 716 Terminals | ASAP | K80-2 | 128 | 194 | 13.5 | 0.0025 | 0.0130 |
| Cora 716 Terminals | ASAP | K80-2 | 205 | 310 | 17.5 | 0.2020 | 0.0046 |
| Cora 716 Terminals | ASAP | JC69 | 161 | 244 | 9.5 | 0.1300 | 0.0086 |
| Cora 716 Terminals | ASAP | JC69 | 143 | 217 | 18.5 | 0.0796 | 0.0102 |
| Cora 716 Terminals | ASAP | JC69 | 205 | 310 | 18.5 | 0.246 | 0.0046 |
| Cora 716 Terminals | ASAP | Simple | 161 | 244 | 8.5 | 0.1400 | 0.0086 |
| Cora 716 Terminals | ASAP | Simple | 205 | 310 | 17.0 | 0.2260 | 0.0046 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Forno, M.; Lawrey, J.D.; Moncada, B.; Bungartz, F.; Grube, M.; Schuettpelz, E.; Lücking, R. DNA Barcoding of Fresh and Historical Collections of Lichen-Forming Basidiomycetes in the Genera Cora and Corella (Agaricales: Hygrophoraceae): A Success Story? Diversity 2022, 14, 284. https://doi.org/10.3390/d14040284
Dal Forno M, Lawrey JD, Moncada B, Bungartz F, Grube M, Schuettpelz E, Lücking R. DNA Barcoding of Fresh and Historical Collections of Lichen-Forming Basidiomycetes in the Genera Cora and Corella (Agaricales: Hygrophoraceae): A Success Story? Diversity. 2022; 14(4):284. https://doi.org/10.3390/d14040284
Chicago/Turabian StyleDal Forno, Manuela, James D. Lawrey, Bibiana Moncada, Frank Bungartz, Martin Grube, Eric Schuettpelz, and Robert Lücking. 2022. "DNA Barcoding of Fresh and Historical Collections of Lichen-Forming Basidiomycetes in the Genera Cora and Corella (Agaricales: Hygrophoraceae): A Success Story?" Diversity 14, no. 4: 284. https://doi.org/10.3390/d14040284
APA StyleDal Forno, M., Lawrey, J. D., Moncada, B., Bungartz, F., Grube, M., Schuettpelz, E., & Lücking, R. (2022). DNA Barcoding of Fresh and Historical Collections of Lichen-Forming Basidiomycetes in the Genera Cora and Corella (Agaricales: Hygrophoraceae): A Success Story? Diversity, 14(4), 284. https://doi.org/10.3390/d14040284