
Identifying Complex DNA Contamination in Pig-Footed Bandicoots Helps to Clarify an Anomalous Ecological Transition



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Authentication of GenBank DNA Accessions
2.2. Phylogenetic Inference
2.3. Molecular Dating
3. Results
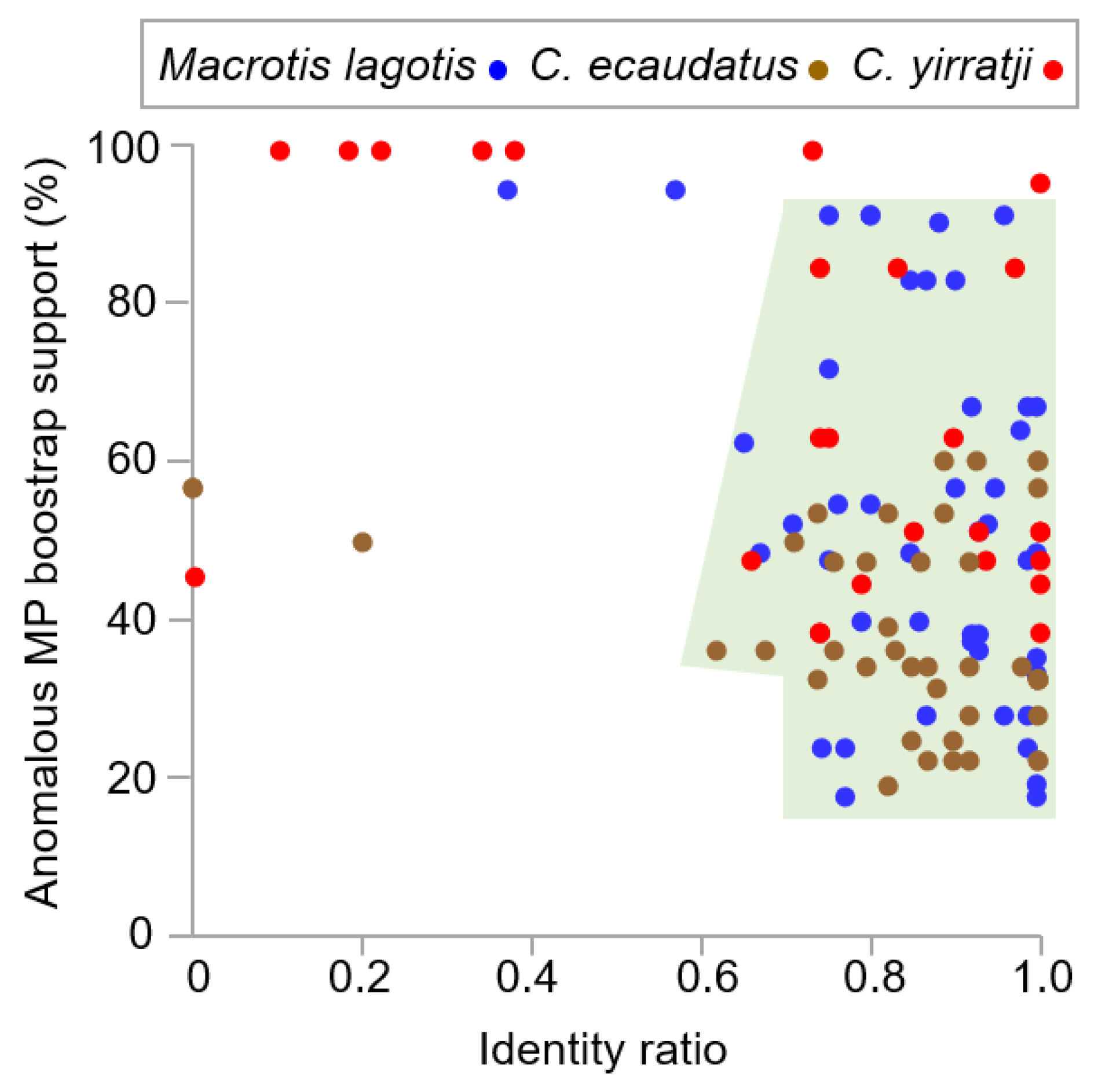
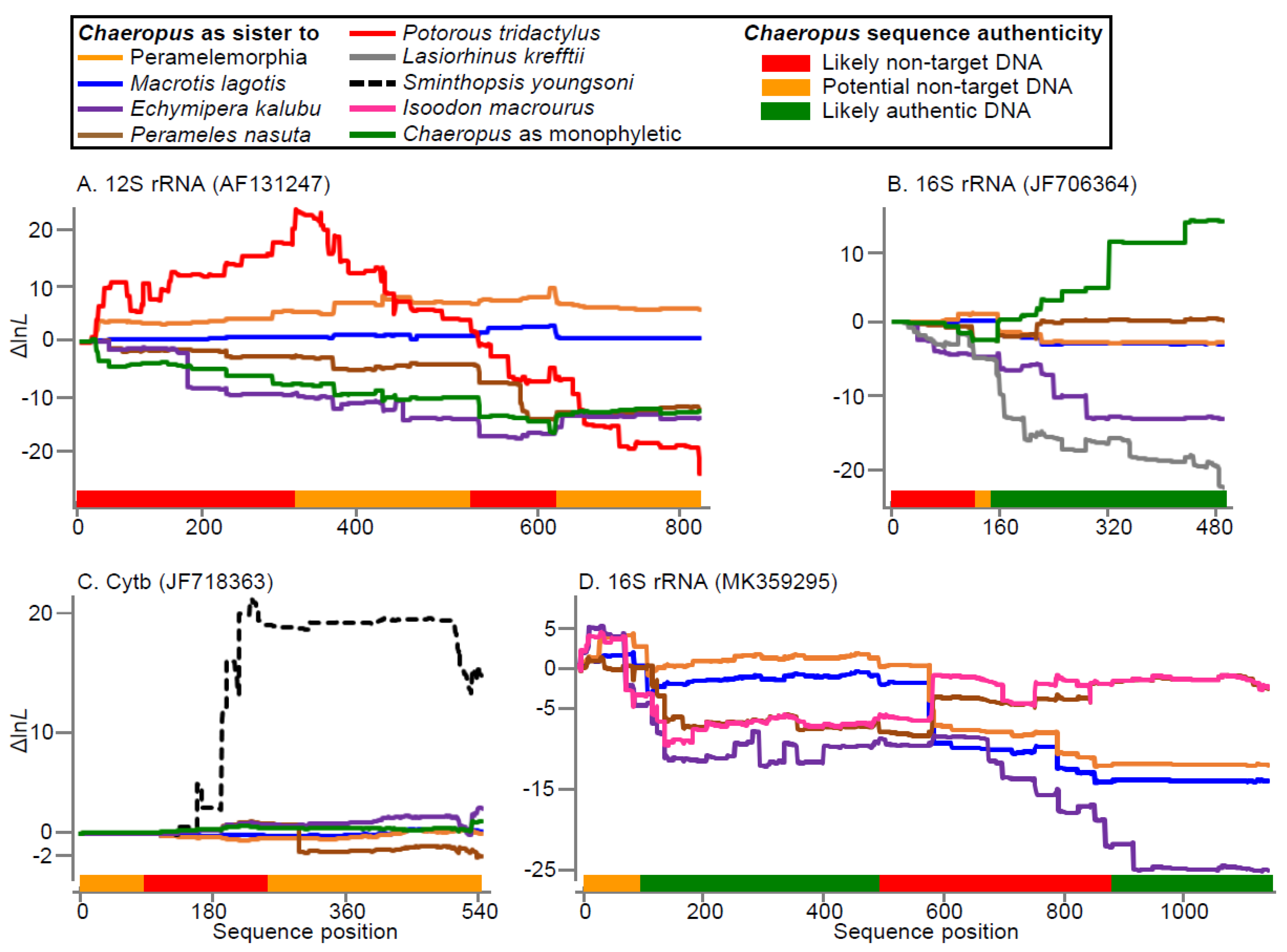
3.1. Authentication of GenBank DNA Sequences
3.1.1. Chaeropus Ecaudatus Sequence Authenticity
3.1.2. Chaeropus yirratji Sequence Authenticity
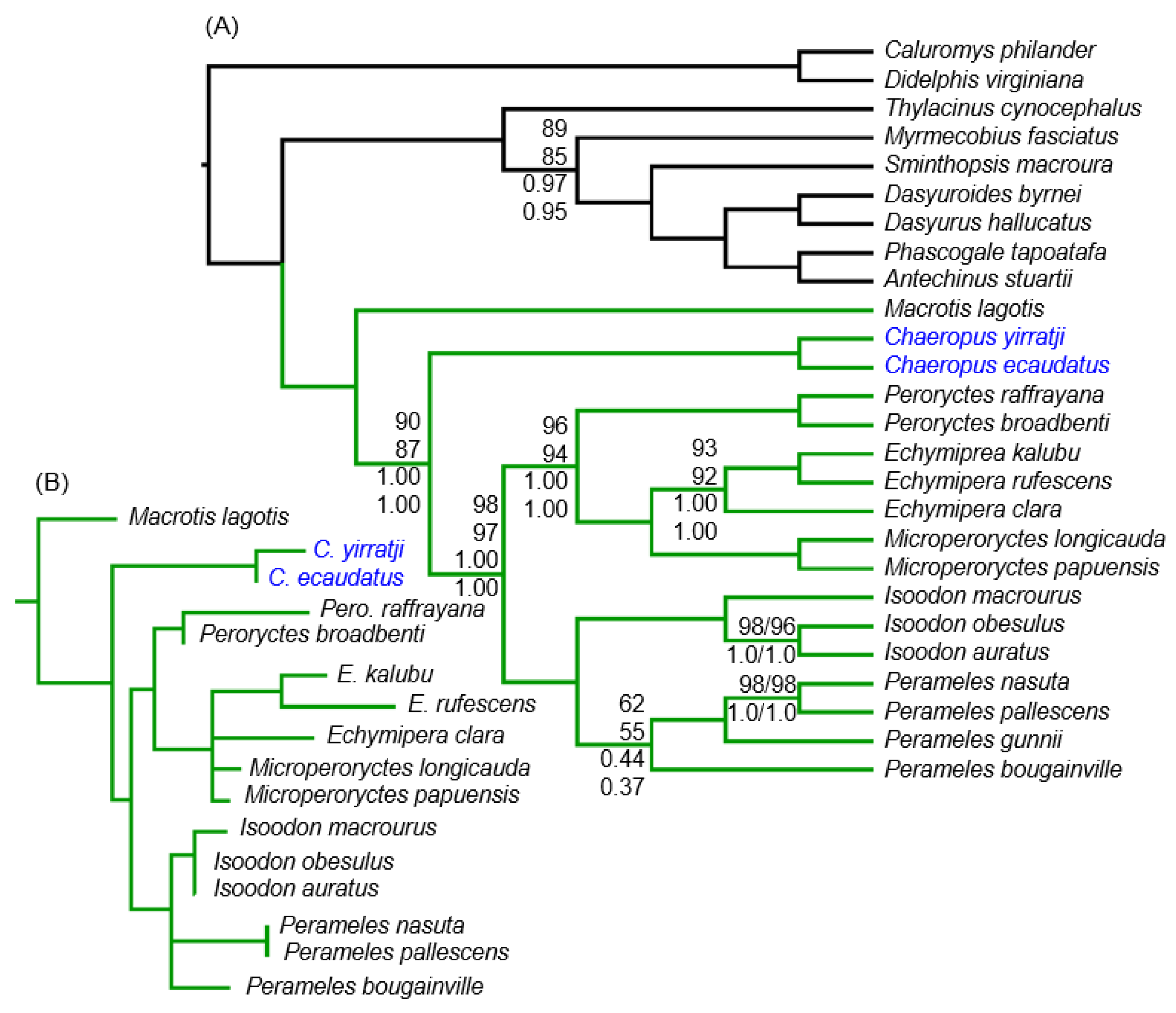
3.2. Phylogenetic Affinities of Chaeropus
3.3. Timescale of Chaeropus Evolution
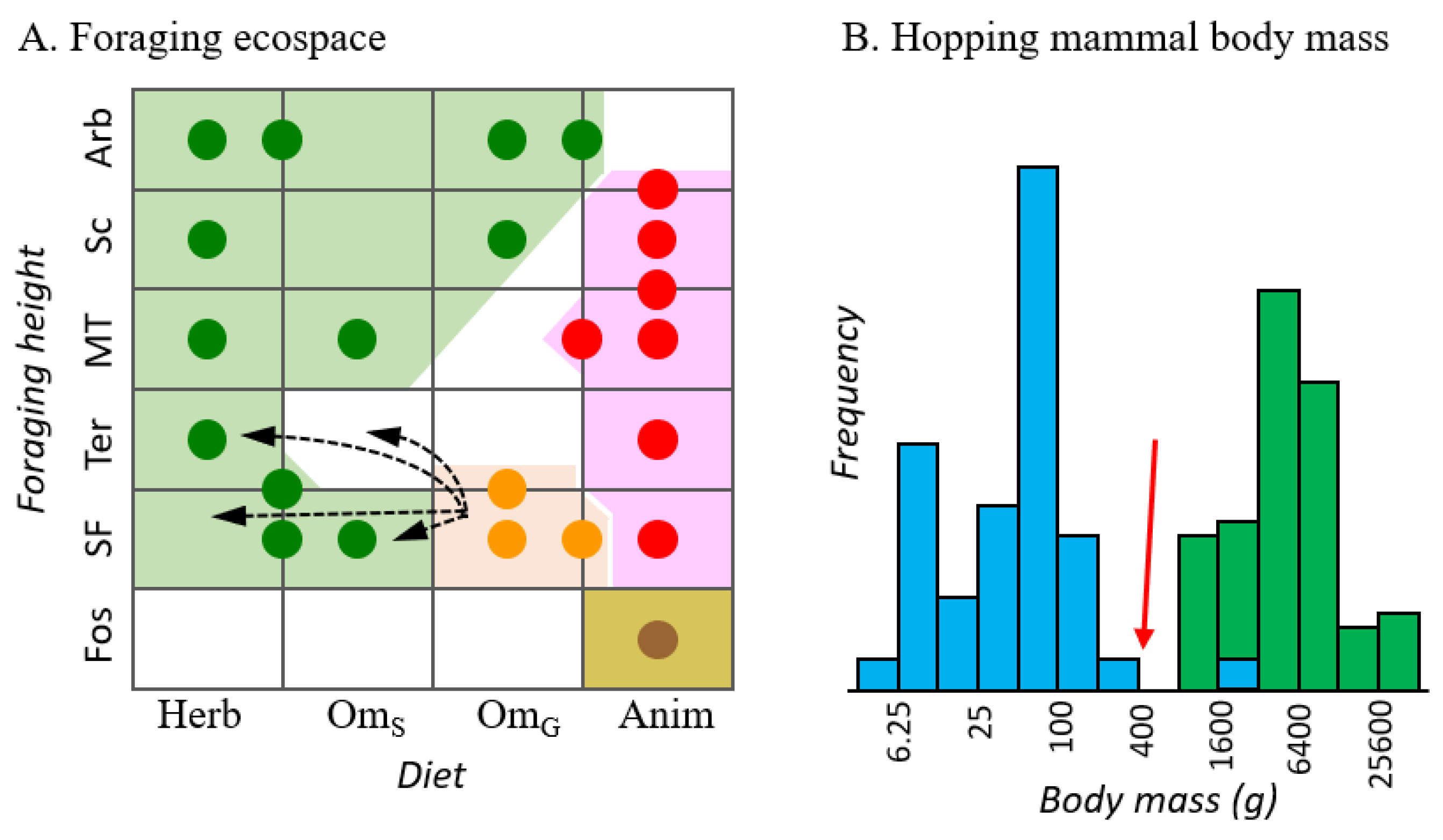
4. Discussion
4.1. MtDNA Authentication
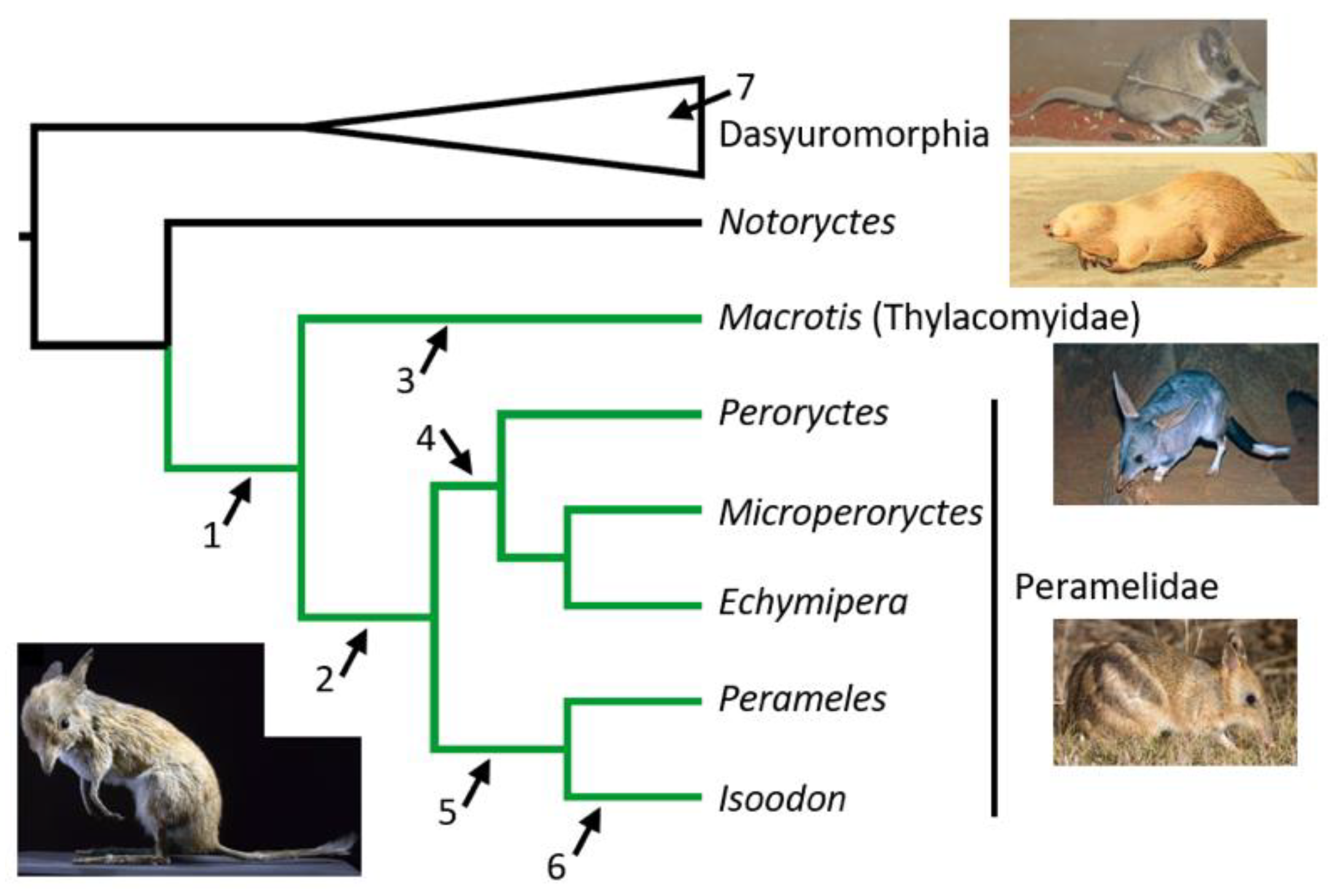
4.2. Peramelemorphian Systematics
4.3. Chaeropus Evolution
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, R.L. Morphological studies on implantation in marsupials. Reproduction 1974, 39, 173–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalay, F.S. Evolutionary History of the Marsupials and an Analysis of Osteological Characters; Cambridge University Press: Cambridge, UK, 1994. [Google Scholar]
- Denyer, A.L.; Regnault, S.; Hutchinson, J.R. Evolution of the patella and patelloid in marsupial mammals. PeerJ 2020, 8, e9760. [Google Scholar] [CrossRef] [PubMed]
- Szalay, F.S.; Sargis, E.J. Model-based analysis of postcranial osteology of marsupials from the Palaeocene of Itaboraí (Brazil) and the phylogenetics and biogeography of Metatheria. Geodiversitas 2001, 23, 139–302. [Google Scholar]
- Wright, W.; Sanson, G.D.; McArthur, C. The diet of the extinct bandicoot Chaeropus ecaudatus. In Vertebrate Palaeontology of Australasia; Vickers-Rich, P., Monaghan, J.M., Baird, R.F., Rich, T.H., Eds.; Pioneer Design Studio: Melbourne, Australia, 1991; pp. 229–245. [Google Scholar]
- Travouillon, K.J.; Simões, B.F.; Miguez, R.P.; Brace, S.; Brewer, P.; Stemmer, D.; Price, G.J.; Cramb, J.; Louys, J. Hidden in plain sight: Reassessment of the pig-footed bandicoot, Chaeropus ecaudatus (Peramelemorphia, Chaeropodidae), with a description of a new species from Central Australia, and use of the fossil record to trace its past distribution. Zootaxa 2019, 4566, 1. [Google Scholar] [CrossRef] [PubMed]
- Groves, C.P.; Flannery, T. Revision of the families and genera of bandicoots. In Bandicoots and Bilbies; Seebeck, J.H., Wallis, R.L., Brown, P.R., Kemper, C.M., Eds.; Surrey Beatty: Sydney, Australia, 1990; pp. 1–11. ISBN 978-0-94932-433-7. [Google Scholar]
- Muirhead, J.; Dawson, L.; Archer, M. Perameles bowensis, a new species of Perameles (Peramelemorphia, Marsupialia) from the Pliocene faunas of Bow and Wellington Caves, New South Wales. Proc. Linn. Soc. N. S. W. 1997, 117, 163–173. [Google Scholar]
- Gurovich, Y.; Travouillon, K.J.; Beck, R.M.D.; Muirhead, J.; Archer, M. Biogeographical implications of a new mouse-sized fossil bandicoot (Marsupialia: Peramelemorphia) occupying a dasyurid-like ecological niche across Australia. J. Syst. Palaeontol. 2014, 12, 265–290. [Google Scholar] [CrossRef]
- Travouillon, K.J.; Archer, M.; Hand, S.J.; Muirhead, J. Sexually dimorphic bandicoots (Marsupialia: Peramelemorphia) from the Oligo-Miocene of Australia, first cranial ontogeny for fossil bandicoots and new species descriptions. J. Mamm. Evol. 2015, 22, 141–167. [Google Scholar] [CrossRef]
- Travouillon, K.J.; Gurovich, Y.; Beck, R.M.D.; Muirhead, J. An exceptionally well-preserved short-snouted bandicoot (Marsupialia; Peramelemorphia) from Riversleigh’s Oligo-Miocene deposits, northwestern Queensland, Australia. J. Vert. Paleont. 2010, 30, 1528–1546. [Google Scholar] [CrossRef]
- Westerman, M.; Springer, M.S.; Dixon, J.; Krajewski, C. Molecular relationships of the extinct pig-footed bandicoot Chaeropus ecaudatus (Marsupialia: Perameloidea) using 12S rRNA sequences. J. Mamm. Evol. 1999, 6, 271–288. [Google Scholar] [CrossRef]
- Meredith, R.W.; Westerman, M.; Springer, M.S. A timescale and phylogeny for “bandicoots” (Peramelemorphia: Marsupialia) based on sequences for five nuclear genes. Mol. Phylogenet. Evol. 2008, 47, 1–20. [Google Scholar] [CrossRef]
- Westerman, M.; Kear, B.P.; Aplin, K.; Meredith, R.W.; Emerling, C.; Springer, M.S. Phylogenetic relationships of living and recently extinct bandicoots based on nuclear and mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2012, 62, 97–108. [Google Scholar] [CrossRef] [PubMed]
- May-Collado, L.J.; Kilpatrick, C.W.; Agnarsson, I. Mammals from ‘down under’: A multi-gene species-level phylogeny of marsupial mammals (Mammalia, Metatheria). PeerJ 2015, 3, e805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travouillon, K.J.; Phillips, M.J. Total evidence analysis of the phylogenetic relationships of bandicoots and bilbies (Marsupialia: Peramelemorphia): Reassessment of two species and description of a new species. Zootaxa 2018, 4378, 224. [Google Scholar] [CrossRef] [PubMed]
- Kear, B.P.; Aplin, K.P.; Westerman, M. Bandicoot fossils and DNA elucidate lineage antiquity amongst xeric-adapted Australasian marsupials. Sci. Rep. 2016, 6, 37537. [Google Scholar] [CrossRef] [Green Version]
- Beck, R.M.D.; Voss, R.; Jansa, S. Craniodental morphology and phylogeny of marsupials. Bull. Am. Mus. Nat. Hist. 2021; accepted. [Google Scholar] [CrossRef]
- Upham, N.S.; Esselstyn, J.A.; Jetz, W. Inferring the mammal tree: Species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLOS Biol. 2019, 17, e3000494. [Google Scholar] [CrossRef]
- Travouillon, K.J.; Beck, R.M.D.; Case, J.A. Upper Oligocene–Lower-Middle Miocene peramelemorphians from the Etadunna, Namba and Wipajiri Formations of South Australia. Alcheringa Australas. J. Palaeontol. 2021, 45, 109–125. [Google Scholar] [CrossRef]
- Travouillon, K.J. Oldest fossil Remains of the enigmatic pig-footed bandicoot show rapid herbivorous evolution. R. Soc. Open Sci. 2016, 3, 160089. [Google Scholar] [CrossRef] [Green Version]
- Whitelaw, M.J. Magnetic polarity stratigraphy of the Fisherman’s Cliff and Bone Gulch vertebrate fossil faunas from the Murray Basin, New South Wales, Australia. Earth Planet. Sci. Lett. 1991, 104, 417–423. [Google Scholar] [CrossRef]
- Poinar, H.N.; Cooper, A. Ancient DNA: Do it right or not at all. Science 2000, 289, 1139. [Google Scholar]
- Key, F.M.; Posth, C.; Krause, J.; Herbig, A.; Bos, K.I. Mining metagenomic data sets for ancient DNA: Recommended protocols for authentication. Trends Genet. 2017, 33, 508–520. [Google Scholar] [CrossRef]
- Hebsgaard, M.B.; Phillips, M.J.; Willerslev, E. Geologically Ancient DNA: Fact or artefact? Trends Microbiol. 2005, 13, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.B.; Jetz, W. Linking global turnover of species and environments. Proc. Natl. Acad. Sci. USA 2008, 105, 17836–17841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedges, S.B.; Schweitzer, M.H. Detecting dinosaur DNA. Science 1995, 268, 1191–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanin, A.; Bonillo, C.; Nguyen, B.X.; Cruaud, C. Comparisons between mitochondrial genomes of domestic goat (Capra hircus) reveal the presence of numts and multiple sequencing errors. Mitochondrial DNA 2010, 21, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Westbury, M.; Baleka, S.; Barlow, A.; Hartmann, S.; Paijmans, J.L.A.; Kramarz, A.; Forasiepi, A.M.; Bond, M.; Gelfo, J.N.; Reguero, M.A.; et al. A mitogenomic timetree for Darwin’s enigmatic South American mammal Macrauchenia patachonica. Nat. Commun. 2017, 8, 15951. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Tromp, J.; Li, M. PatternHunter: Faster and more sensitive homology search. Bioinformatics 2002, 18, 440–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods); Version 4.0b10; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Duchêne, D.A.; Bragg, J.G.; Duchêne, S.; Neaves, L.E.; Potter, S.; Moritz, C.; Johnson, R.N.; Ho, S.Y.W.; Eldridge, M.D.B. Analysis of phylogenomic tree space resolves relationships among marsupial families. Syst. Biol. 2018, 67, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, K.J.; Pratt, R.C.; Watson, L.N.; Gibb, G.C.; Llamas, B.; Kasper, M.; Edson, J.; Hopwood, B.; Male, D.; Armstrong, K.N.; et al. Molecular phylogeny, biogeography, and habitat preference evolution of marsupials. Mol. Biol. Evol. 2014, 31, 2322–2330. [Google Scholar] [CrossRef] [Green Version]
- Westerman, M.; Krajewski, C.; Kear, B.P.; Meehan, L.; Meredith, R.W.; Emerling, C.A.; Springer, M.S. Phylogenetic relationships of dasyuromorphian marsupials revisited. Zool. J. Linn. Soc. 2016, 176, 686–701. [Google Scholar] [CrossRef] [Green Version]
- Celik, M.; Cascini, M.; Haouchar, D.; Van Der Burg, C.; Dodt, W.; Evans, A.R.; Prentis, P.; Bunce, M.; Fruciano, C.; Phillips, M.J. A molecular and morphometric assessment of the systematics of the Macropus complex clarifies the tempo and mode of kangaroo evolution. Zool. J. Linn. Soc. 2019, 186, 793–812. [Google Scholar] [CrossRef]
- Dodt, W.G.; McComish, B.J.; Nilsson, M.A.; Gibb, G.C.; Penny, D.; Phillips, M.J. The complete mitochondrial genome of the eastern grey kangaroo (Macropus giganteus). Mitochondrial DNA 2016, 27, 1366–1367. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.A.; Zheng, Y.; Kumar, V.; Phillips, M.J.; Janke, A. Speciation generates mosaic genomes in kangaroos. Genome Biol. Evol. 2018, 10, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. Sequence Alignment Editor. 1996. Available online: http://tree.bio.ed.ac.uk/software/seal (accessed on 23 March 2014).
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.J.; Pratt, R.C. Family-level relationships among the Australasian marsupial “herbivores” (Diprotodontia: Koala, wombats, kangaroos and possums). Mol. Phylogenet. Evol. 2008, 46, 594–605. [Google Scholar] [CrossRef]
- Cascini, M.; Mitchell, K.J.; Cooper, A.; Phillips, M.J. Reconstructing the evolution of giant extinct kangaroos: Comparing the utility of DNA, morphology, and total evidence. Syst. Biol. 2019, 68, 520–537. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Grant, J.R.; Katz, L.A. Building a phylogenomic pipeline for the eukaryotic tree of life—Addressing deep phylogenies with genome-scale data. PLoS Curr. 2014, 6. [Google Scholar] [CrossRef]
- Phillips, M.J.; McLenachan, P.A.; Down, C.; Gibb, G.C.; Penny, D. Combined mitochondrial and nuclear DNA sequences resolve the interrelations of the major Australasian marsupial radiations. Syst. Biol. 2006, 55, 122–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajewski, C.; Anderson, F.E.; Woolley, P.A.; Westerman, M. Molecular evidence for a deep clade of dunnarts (Marsupialia: Dasyuridae: Sminthopsis). J. Mamm. Evol. 2012, 19, 265–276. [Google Scholar] [CrossRef]
- Phillips, M.J.; Westerman, M.; Cascini, M. The value of updating GenBank accessions for supermatrix phylogeny: The case of the New Guinean carnivore genus Myoictis. Mol. Phylogenet. Evol. 2022, 160, 107328. [Google Scholar] [CrossRef] [PubMed]
- Meredith, R.W.; Westerman, M.; Springer, M.S. A phylogeny of Diprotodontia (Marsupialia) based on sequences for five nuclear genes. Mol. Phylogenet. Evol. 2009, 51, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Groves, C. Order Peramelemorphia. In Mammal Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; Wilson, D.E., Reader, D.M., Eds.; Johns Hopkins University Press: Baltimore, MA, USA, 2005; pp. 38–42. [Google Scholar]
- Travouillon, K.J.; Hand, S.J.; Archer, M.; Black, K.H. Earliest modern bandicoot and bilby (Marsupialia, Peramelidae and Thylacomyidae) from the Miocene of the Riversleigh World Heritage Area, Northwestern Queensland, Australia. J. Vertebr. Paleontol. 2014, 34, 375–382. [Google Scholar] [CrossRef]
- Martin, H. Cenozoic climatic change and the development of the arid vegetation in Australia. J. Arid. Environ. 2006, 66, 533–563. [Google Scholar] [CrossRef]
- Couzens, A.M.C.; Prideaux, G.J. Rapid Pliocene adaptive radiation of modern kangaroos. Science 2018, 362, 72–75. [Google Scholar] [CrossRef] [Green Version]
- Warburton, N.M.; Travouillon, K.J. The biology and palaeontology of the Peramelemorphia: A review of current knowledge and future research directions. Aust. J. Zool. 2016, 64, 151. [Google Scholar] [CrossRef]
- Dixon, J. Notes on the diet of three mammals presumed to be extinct: The Pig-footed bandicoot, the lesser bilby and the desert rat kangaroo. Vic. Nat. 1988, 105, 208–211. [Google Scholar]
- Burbidge, A.A.; Johnson, K.A.; Fuller, P.J.; Southgate, R.I. Aboriginal knowledge of the mammals of the central deserts of Australia. Austr. Wildlife Res. 1988, 15, 9–39. [Google Scholar] [CrossRef]
- Lee, A.K.; Cockburn, A. Evolutionary Ecology of Marsupials (Monographs on Marsupial Biology); Cambridge University Press: Cambridge, UK, 1985. [Google Scholar]
- Cork, S.J. Digestive constraints on dietary scope in small and moderately-small mammals: How much do we really understand? In The Digestive System in Mammals: Food, Form and Function; Chivers, D.J., Langer, P., Eds.; Cambridge University Press: Cambridge, UK, 1994; pp. 337–369. [Google Scholar]
- Poole, A.M.; Phillips, M.J.; Penny, D. Prokaryote and eukaryote evolvability. Biosystems 2003, 69, 163–185. [Google Scholar] [CrossRef]
- Gillespie, A.K.; Archer, M.; Hand, S.J. A tiny new marsupial lion (Marsupialia, Thylacoleonidae) from the early Miocene of Australia. Palaeontol. Electron. 2016, 19, 1–26. [Google Scholar] [CrossRef]
- Arman, S.D.; Prideaux, G.J. Dietary classification of extant kangaroos and their relatives (Marsupialia: Macropodoidea): Diets of extant macropodoids. Austral Ecol. 2015, 40, 909–922. [Google Scholar] [CrossRef]
- Moore, T.Y.; Cooper, K.L.; Biewener, A.A.; Vasudevan, R. Unpredictability of escape trajectory explains predator evasion ability and microhabitat preference of desert rodents. Nat. Commun. 2017, 8, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGowan, C.P.; Collins, C.E. Why do mammals hop? Understanding the ecology, biomechanics and evolution of bipedal hopping. J. Exp. Biol. 2018, 221, jeb161661. [Google Scholar] [CrossRef] [Green Version]
- Webster, K.; Dawson, T. Is the energetics of mammalian hopping locomotion advantageous in arid environments? Aust. Mammal. 2004, 26, 153. [Google Scholar] [CrossRef]
- Emerling, C.A.; Delsuc, F.; Nachman, M.W. Chitinase genes (CHIAs) provide genomic footprints of a post-Cretaceous dietary radiation in placental mammals. Sci. Adv. 2018, 4, eaar6478. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phillips, M.J.; Cascini, M.; Celik, M. Identifying Complex DNA Contamination in Pig-Footed Bandicoots Helps to Clarify an Anomalous Ecological Transition. Diversity 2022, 14, 352. https://doi.org/10.3390/d14050352
Phillips MJ, Cascini M, Celik M. Identifying Complex DNA Contamination in Pig-Footed Bandicoots Helps to Clarify an Anomalous Ecological Transition. Diversity. 2022; 14(5):352. https://doi.org/10.3390/d14050352
Chicago/Turabian StylePhillips, Matthew J., Manuela Cascini, and Mélina Celik. 2022. "Identifying Complex DNA Contamination in Pig-Footed Bandicoots Helps to Clarify an Anomalous Ecological Transition" Diversity 14, no. 5: 352. https://doi.org/10.3390/d14050352
APA StylePhillips, M. J., Cascini, M., & Celik, M. (2022). Identifying Complex DNA Contamination in Pig-Footed Bandicoots Helps to Clarify an Anomalous Ecological Transition. Diversity, 14(5), 352. https://doi.org/10.3390/d14050352