
MolMarker: A Simple Tool for DNA Fingerprinting Studies and Polymorphic Information Content Calculation




Abstract
:1. Introduction
2. Methods
2.1. Programming Language and IDE
2.2. Main Implemented Algorithms
2.2.1. UPGMA Algorithm
2.2.2. Neighbor-Joining Algorithm
2.2.3. The Expectation-Maximization Algorithm
3. Results
Description of the Software and Its Functionalities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References and Note
- Singh, B.D.; Singh, A.K. Marker-Assisted Plant Breeding: Principles and Practices; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Appleby, N.; Edwards, D.; Batley, J. New Technologies for Ultra-High Throughput Genotyping in Plants. Methods Mol. Biol. 2009, 513, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Poczai, P.; Varga, I.; Laos, M.; Cseh, A.; Bell, N.; Valkonen, J.P.T.; Hyvönen, J. Advances in Plant Gene-Targeted and Functional Markers: A Review. Plant Methods 2013, 9, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Lee, C.; Hyung, D.; Jo, Y.J.; Park, J.S.; Cook, D.R.; Choi, H.K. CSGM Designer: A Platform for Designing Cross-Species Intron-Spanning Genic Markers Linked with Genome Information of Legumes. Plant Methods 2015, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a Genetic Linkage Map in Man Using Restriction Fragment Length Polymorphisms. Am. J. Hum. Genet. 1980, 32, 314. [Google Scholar]
- Nagy, S.; Poczai, P.; Cernák, I.; Gorji, A.M.; Hegedűs, G.; Taller, J. PICcalc: An Online Program to Calculate Polymorphic Information Content for Molecular Genetic Studies. Biochem. Genet. 2012, 50, 670–672. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.H. Statistical Genomics: Linkage, Mapping, and QTL Analysis; CRC Press: Boca Raton, FL, USA, 1998; ISBN 9780367400743. [Google Scholar]
- Schwartz, J.J.; Roach, D.J.; Thomas, J.H.; Shendure, J. Primate Evolution of the Recombination Regulator PRDM9. Nat. Commun. 2014, 5, 4370. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-L.; Yang, J.; Boykin, L.M.; Zhao, Q.-Y.; Wang, Y.-J.; Liu, S.-S.; Wang, X.-W. Developing conversed microsatellite markers and their implications in evolutionary analysis of the Bemisia tabaci complex. Sci. Rep. 2014, 4, srep06351. [Google Scholar] [CrossRef] [Green Version]
- Martins, S.; Simões, F.; Matos, J.; Silva, A.P.; Carnide, V. Genetic Relationship among Wild, Landraces and Cultivars of Hazelnut (Corylus Avellana) from Portugal Revealed through ISSR and AFLP Markers. Plant Syst. Evol. 2014, 300, 1035–1046. [Google Scholar] [CrossRef]
- Write Once, Run Anywhere? Computer Weekly. 2002. Available online: http://www.computerweekly.com/Articles/2002/05/02/186793/write-once-run-anywhere.htm (accessed on 25 May 2022).
- Gosling, J.; McGilton, H. The Java Language Environment; Sun Microsystems Computer Company: Hongkong, China, 1996. [Google Scholar]
- Wielenga, G. Java Editor. In Beginning NetBeans IDE; Apress: Berkeley, CA, USA, 2015; pp. 31–83. [Google Scholar] [CrossRef]
- Wielenga, G. Putting the Pieces Together. In Beginning NetBeans IDE; Apress: Berkeley, CA, USA, 2015; pp. 103–123. [Google Scholar] [CrossRef]
- Wielenga, G. Beginning Netbeans Ide: For Java Developers; Apress: Berkeley, CA, USA, 2015. [Google Scholar] [CrossRef]
- Sneath, P.H.A.; Sokal, R.R. Numerical Taxonomy. Nature 1962, 193, 855–860. [Google Scholar] [CrossRef]
- Sokal, R.; Michener, C. A Statistical Method for Evaluating Systematic Relationships. Univ. Kansas Sci. Bull. 1958, 38, 1409–1438. [Google Scholar]
- Vinay, S. Text Book of Bioinformatics; Rakesh Kumar Rastogi for Rastogi Publications: New Delhi, India, 2008. [Google Scholar]
- Gronau, I.; Moran, S. Optimal Implementations of UPGMA and Other Common Clustering Algorithms. Inf. Process. Lett. 2007, 104, 205–210. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Studier, J.A.; Keppler, K.J. A Note on the Neighbor-Joining Algorithm of Saitou and Nei. Mol. Biol. Evol. 1988, 5, 729–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronau, I.; Moran, S. Neighbor Joining Algorithms for Inferring Phylogenies via LCA Distances. J. Comput. Biol. 2007, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rzhetsky, A.; Nei, M. A Simple Method for Estimating and Testing Minimum-Evolution Trees. Mol. Biol. Evol. 1992, 9, 945. [Google Scholar] [CrossRef]
- Gascuel, O.; Bryant, D.; Denis, F. Strengths and Limitations of the Minimum Evolution Principle. Syst. Biol. 2001, 50, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Kuhner, M.K.; Felsenstein, J. A Simulation Comparison of Phylogeny Algorithms under Equal and Unequal Evolutionary Rates. Mol. Biol. Evol. 1994, 11, 459–468. [Google Scholar] [CrossRef]
- Dempster, A.P.; Laird, N.M.; Rubin, D.B. Maximum Likelihood from Incomplete Data Via the EM Algorithm. J. R. Stat. Soc. Ser. B Methodol. 1977, 39, 1–22. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L. Maximum Likelihood Estimation of the Frequency of Null Alleles at Microsatellite Loci. Conserv. Genet. 2006, 7, 991–995. [Google Scholar] [CrossRef]
- Hardy, G.H. Mendelian Proportions in a Mixed Population. Science 1908, 28, 49–50. [Google Scholar] [CrossRef]
- Czekanowski, J. Zarys Metod Statystycnck Anthr. Anz 1913, 9, 227–249. [Google Scholar]
- Czekanowski, J. Zarys Metod Statystycznych W Zastosowaniu Do Antropologii; Towarzystwo Naukowe Warszawskie: Warszawa, Poland, 1913. [Google Scholar]
- Dice, L. Measures of the Amount of Ecological Association between Species. Ecology 1945, 26, 297–302. [Google Scholar] [CrossRef]
- Batzer, D.P.; Rader, R.B.; Wissinger, S.A. Invertebrates in Freshwater Wetlands of North America: Ecology and Management; John Wiley & Sons: Hoboken, NJ, USA, 1999; p. 1100. [Google Scholar]
- MolMarker Download|Source Forge. Available online: https://sourceforge.net/projects/molmarker/ (accessed on 21 May 2022).
- IBM SPSS Statistics. Available online: https://www.ibm.com/products/spss-statistics (accessed on 13 June 2022).
- Wagner, H.; Sefc, K. IDENTITY 1.0 Centre for Applied Genetics. 1999. Available online: https://boku.ac.at/zag/forsch/identity.htm (accessed on 7 January 2019).
- Kalinowski, S. ML-Null Freq-Steven Kalinowski|Montana State University. Available online: https://www.montana.edu/kalinowski/software/null-freq.html (accessed on 12 May 2022).
- Bowers, J.E.; Meredith, C.P. The parentage of a classic wine grape, Cabernet Sauvignon. Nat. Genet. 1997, 16, pp. 84–87.
- Speed, T. Neighbour Joining Method (Saitou and Nei, 1987). 2006. [Google Scholar]








{kind=link}
{kind=link}
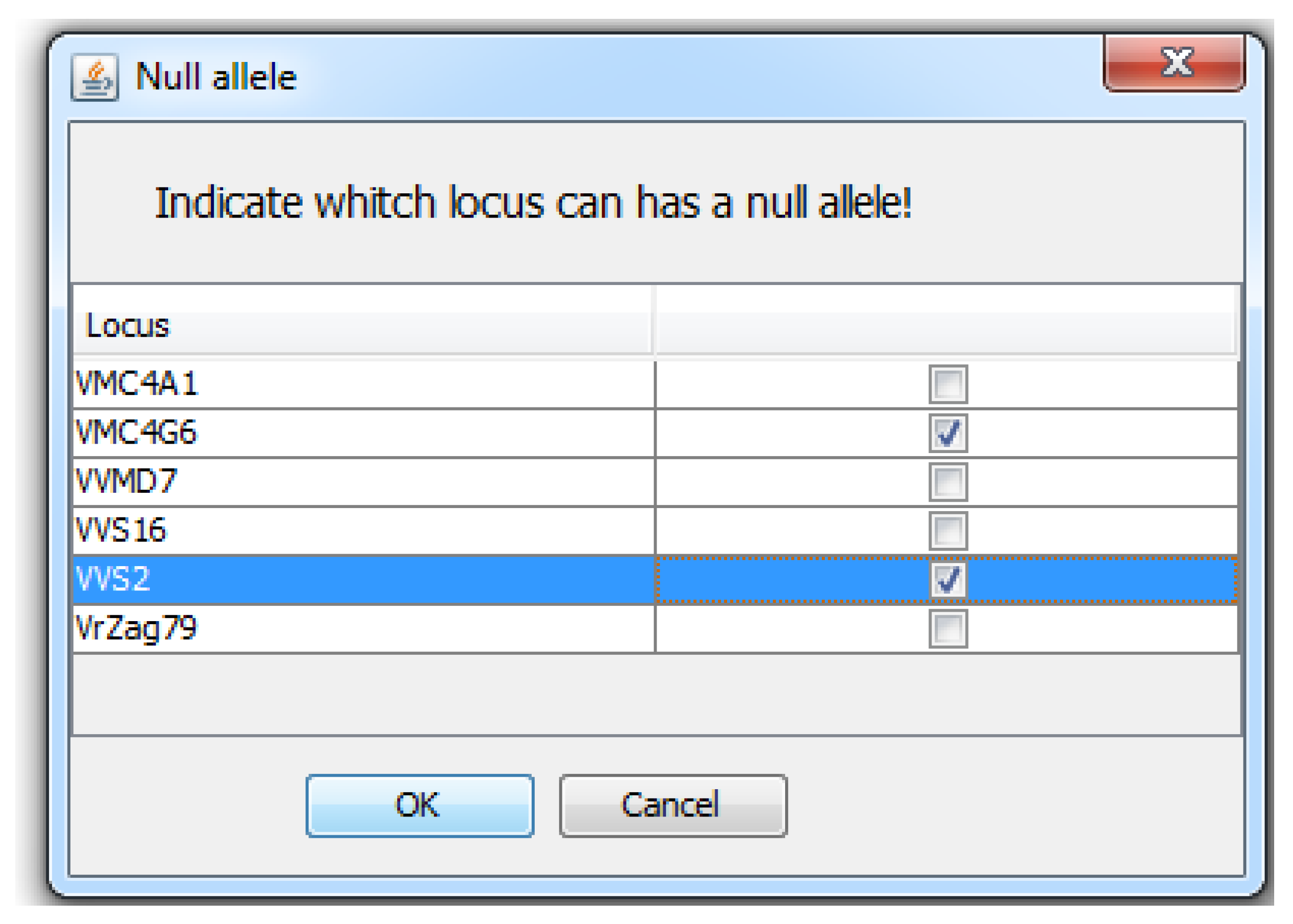{kind=link}
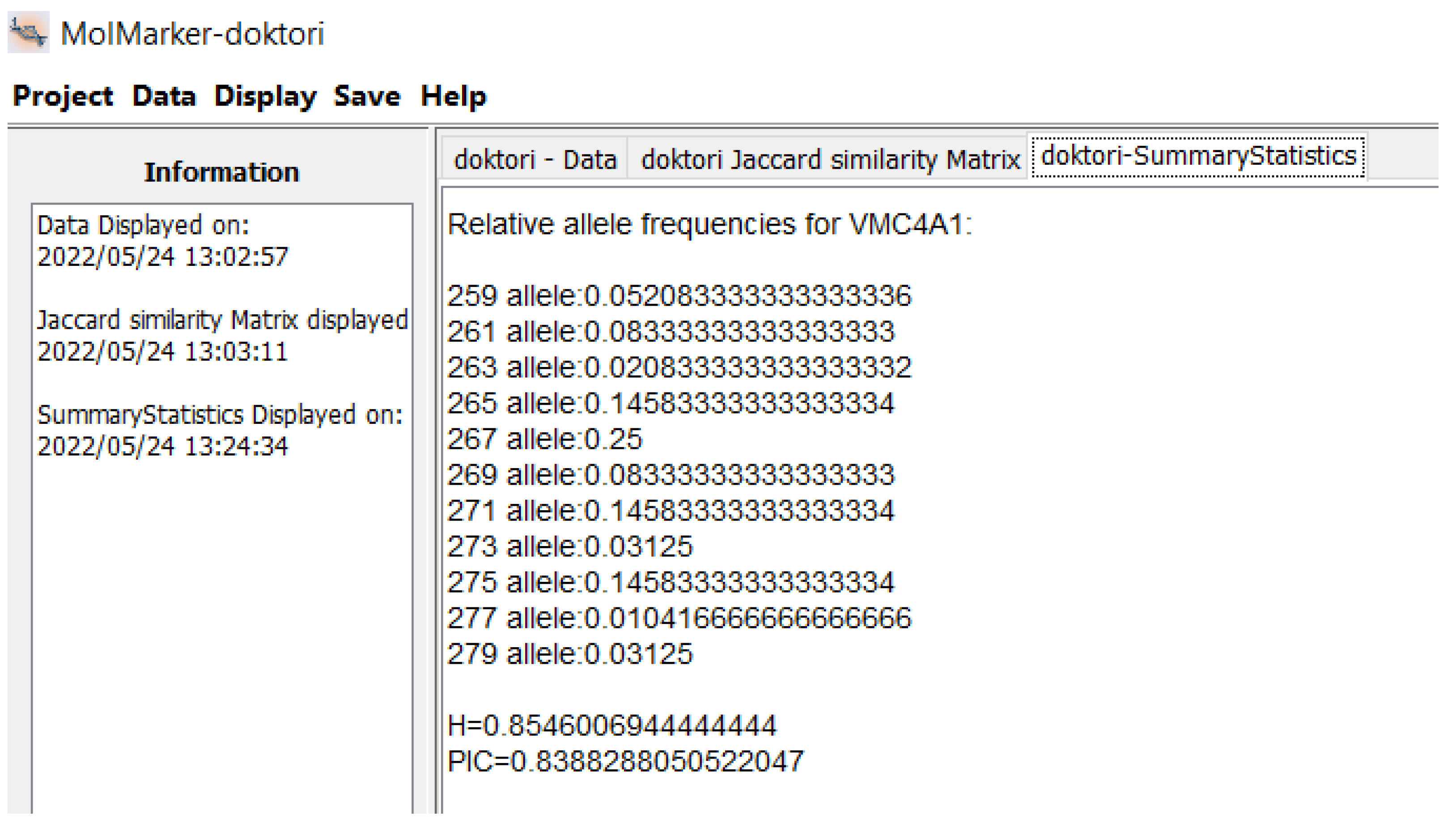{kind=link}
{kind=link}
{kind=link}
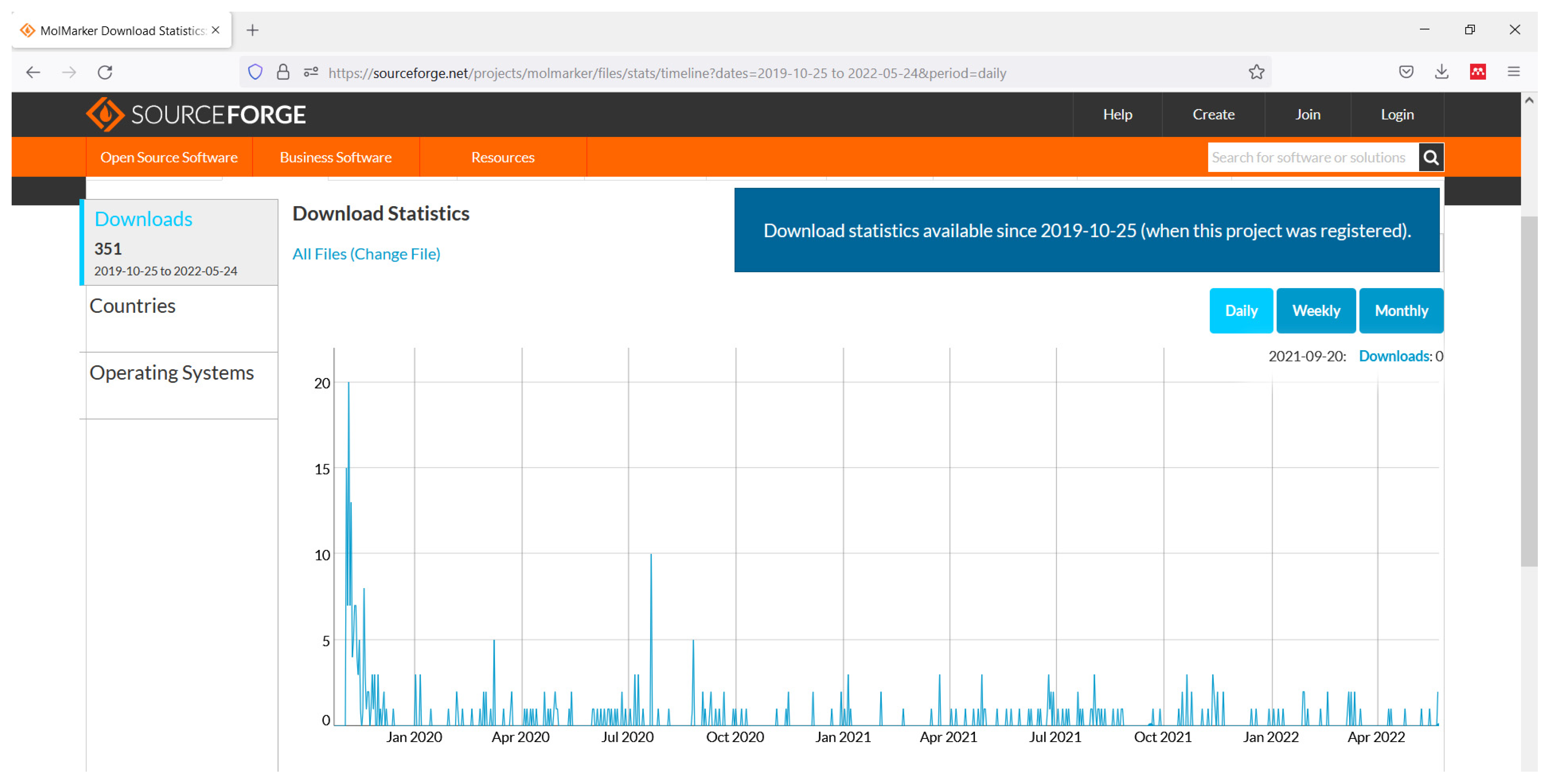{kind=link}
| Project | Data | Display | Save | Help | ||
|---|---|---|---|---|---|---|
| New Project | Input Data | Data | Save Project | Open Manual | ||
| Rename Project | Read Data From File | Similarity Matrix | Save Project As… | Support | ||
| Change Active Project | Read Data From Database | Summary Statistics > | Genetic | Similarity Matrix | About | |
| Open Project | Write Data To Database | Molecular | Summary Statistics > | Genetic | ||
| Close Project | Phylogeny > | Paretage | Molecular | |||
| Merge Projects | Dendogram | Phylogeny > | Paretage | |||
| Exit | Dendogram | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahnke, G.; Smidla, J.; Poczai, P. MolMarker: A Simple Tool for DNA Fingerprinting Studies and Polymorphic Information Content Calculation. Diversity 2022, 14, 497. https://doi.org/10.3390/d14060497
Jahnke G, Smidla J, Poczai P. MolMarker: A Simple Tool for DNA Fingerprinting Studies and Polymorphic Information Content Calculation. Diversity. 2022; 14(6):497. https://doi.org/10.3390/d14060497
Chicago/Turabian StyleJahnke, Gizella, József Smidla, and Peter Poczai. 2022. "MolMarker: A Simple Tool for DNA Fingerprinting Studies and Polymorphic Information Content Calculation" Diversity 14, no. 6: 497. https://doi.org/10.3390/d14060497
APA StyleJahnke, G., Smidla, J., & Poczai, P. (2022). MolMarker: A Simple Tool for DNA Fingerprinting Studies and Polymorphic Information Content Calculation. Diversity, 14(6), 497. https://doi.org/10.3390/d14060497