
Mitochondrial Genomes of two Lycosa spiders (Araneae, Lycosidae): Genome Description and Phylogenetic Implications


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Splicing and Annotation of Mitogenome Sequences
2.3. Sequence Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
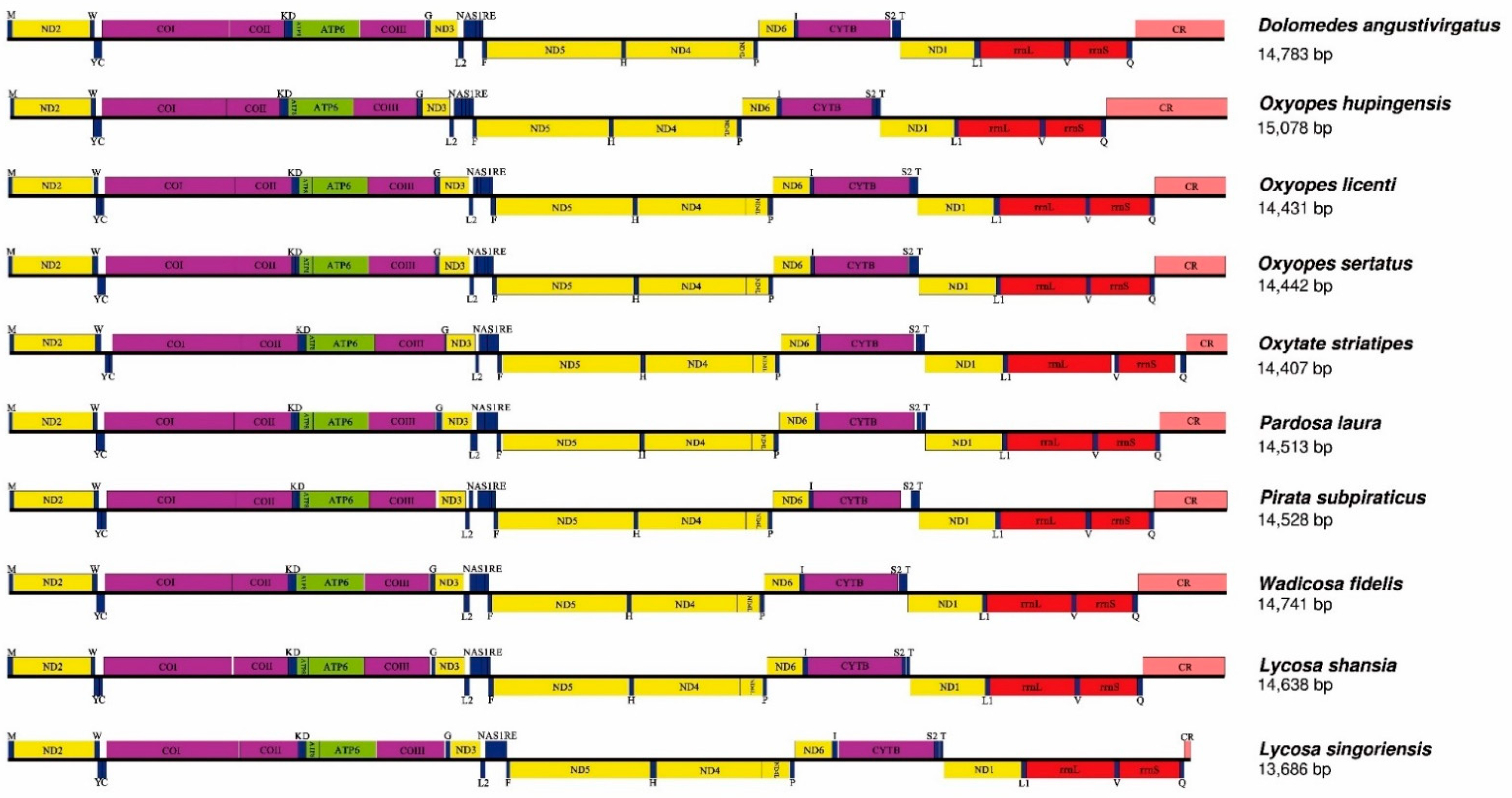
3.1. Mitogenome Composition
3.2. Nucleotide Composition
3.3. Protein-Coding Genes and Codon Usage Patterns
3.4. Ribosomal and Transfer RNA Genes and Control Regions
3.5. Phylogenetic Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hebert, S.L.; Lanza, I.R.; Nair, K.S. Mitochondrial DNA alterations and reduced mitochondrial function in aging. Mech. Ageing Dev. 2010, 131, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Lin, S.; Liu, H. Mitochondrial genomes of five Hyphessobrycon tetras and their phylogenetic implications. Ecol. Evol. 2021, 11, 12754–12764. [Google Scholar] [CrossRef]
- Tyagi, K.; Kumar, V.; Poddar, N.; Prasad, P.; Tyagi, I.; Kundu, S.; Chandra, K. The gene arrangement and phylogeny using mitochondrial genomes in spiders (Arachnida: Araneae). Int. J. Biol. Macromol. 2020, 146, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, W.C.; Coddington, J.A.; Crowle, L.M.; Dimitrov, D.; Goloboff, P.A.; Griswold, C.E.; Hormiga, G.; Prendini, L.; Ramirez, M.J.; Sierwald, P.; et al. The spider tree of life: Phylogeny of Araneae based on target-gene analyses from an extensive taxon sampling. Cladistics 2017, 33, 574–616. [Google Scholar] [CrossRef] [PubMed]
- Oshida, T.; Masuda, R. Phylogeny and Zoogeography of Six Squirrel Species of the Genus Sciurus (Mammalia, Rodentia), Inferred from Cytochrome b Gene Sequences. Zool. Sci. 2000, 17, 405–409. [Google Scholar] [CrossRef] [Green Version]
- Astrin, J.J.; Huber, B.A.; Misof, B.; Klütsch, C.F.C. Molecular taxonomy in pholcid spiders (Pholcidae, Araneae): Evaluation of species identification methods using CO1 and 16S rRNA. Zool. Scr. 2006, 35, 441–457. [Google Scholar] [CrossRef]
- Wang, Z.L.; Yang, X.Q.; Wang, T.Z.; Yu, X. Assessing the effectiveness of mitochondrial COI and 16S rRNA genes for DNA barcoding of farmland spiders in China. Mitochondrial DNA Part A 2017, 29, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Fernández, R.; Hormiga, G.; Giribet, G. Phylogenomic Analysis of Spiders Reveals Nonmonophyly of Orb Weavers. Curr. Biol. 2014, 24, 1772–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-L.; Li, C.; Fang, W.-Y.; Yu, X.-P. Characterization of the complete mitogenomes of two Neoscona spiders (Araneae: Araneidae) and its phylogenetic implications. Gene 2016, 590, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Pons, J.; Bover, P.; Bidegaray-Batista, L.; Arnedo, M.A. Arm-less mitochondrial tRNAs conserved for over 30 millions of years in spiders. BMC Genom. 2019, 20, 665. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-L.; Li, C.; Fang, W.-Y.; Yu, X.-P. The Complete Mitochondrial Genome of two Tetragnatha Spiders (Araneae: Tetragnathidae): Severe Truncation of tRNAs and Novel Gene Rearrangements in Araneae. Int. J. Biol. Sci. 2016, 12, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crampton-Platt, A.; Timmermans, M.J.T.N.; Gimmel, M.L.; Kutty, S.N.; Cockerill, T.D.; Khen, C.V.; Vogler, A.P. Soup to Tree: The Phylogeny of Beetles Inferred by Mitochondrial Metagenomics of a Bornean Rainforest Sample. Mol. Biol. Evol. 2015, 32, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, L.N.; Ramírez, M.J. Hunting the wolf: A molecular phylogeny of the wolf spiders (Araneae, Lycosidae). Mol. Phylogenetics Evol. 2019, 136, 227–240. [Google Scholar] [CrossRef]
- Polotow, D.; Carmichael, A.; Griswold, C.E. Total evidence analysis of the phylogenetic relationships of Lycosoidea spiders (Araneae, Entelegynae). Invertebr. Syst. 2015, 29, 124. [Google Scholar] [CrossRef] [Green Version]
- Planas, E.; Fernández-Montraveta, C.; Ribera, C. Molecular systematics of the wolf spider genus Lycosa (Araneae: Lycosidae) in the Western Mediterranean Basin. Mol. Phylogenetics Evol. 2013, 67, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Basso, A.; Babbucci, M.; Pauletto, M.; Riginella, E.; Patarnello, T.; Negrisolo, E. The highly rearranged mitochondrial genomes of the crabs Maja crispata and Maja squinado (Majidae) and gene order evolution in Brachyura. Sci. Rep. 2017, 7, 4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akram, A.M.; Chaudhary, A.; Kausar, H.; Althobaiti, F.; Abbas, A.S.; Hussain, Z.; Fatima, N.; Zafar, E.; Asif, W.; Afzal, U.; et al. Analysis of RAS gene mutations in cytogenetically normal de novo acute myeloid leukemia patients reveals some novel alterations. Saudi J. Biol. Sci. 2021, 28, 3735–3740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Weng, Y.; Ye, D.; You, Y.; Shi, J.; Chen, J. The complete chloroplast genome sequence of Casuarina equisetifolia. Mitochondrial DNA Part B 2021, 6, 3046–3048. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction; Humana: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Li, H.; Winterton, S.L.; Liu, Z. Ancestral Gene Organization in the Mitochondrial Genome of Thyridosmylus langii (McLachlan, 1870) (Neuroptera: Osmylidae) and Implications for Lacewing Evolution. PLoS ONE 2013, 8, e62943. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for Multiple Reversals of Asymmetric Mutational Constraints during the Evolution of the Mitochondrial Genome of Metazoa, and Consequences for Phylogenetic Inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, S.; Masta, S.E. Pseudoscorpion mitochondria show rearranged genes and genome-wide reductions of RNA gene sizes and inferred structures, yet typical nucleotide composition bias. BMC Evol. Biol. 2012, 12, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Chang, H.; Qiu, Z.; Yuan, H.; Wang, X.; Li, X.; Sun, H.; Guo, X.; Lu, Y.; Feng, X.; Majid, M.; et al. Evolutionary rates of and selective constraints on the mitochondrial genomes of Orthoptera insects with different wing types. Mol. Phylogenetics Evol. 2020, 145, 106734. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Kumar, V.; Tyagi, K.; Chakraborty, R.; Prasad, P.; Kundu, S.; Tyagi, I.; Chandra, K. The Complete Mitochondrial Genome of endemic giant tarantula, Lyrognathus crotalus (Araneae: Theraphosidae) and comparative analysis. Sci. Rep. 2020, 10, 74. [Google Scholar] [CrossRef]
- Gong, L.; Shi, W.; Si, L.-Z.; Wang, Z.-M.; Kong, X.-Y. The complete mitochondrial genome of peacock sole Pardachirus pavoninus (Pleuronectiformes: Soleidae) and comparative analysis of the control region among 13 soles. Mol. Biol. 2015, 49, 408–417. [Google Scholar] [CrossRef]
- Padhi, A. Geographic variation within a tandemly repeated mitochondrial DNA D-loop region of a North American freshwater fish, Pylodictis olivaris. Gene 2014, 538, 63–68. [Google Scholar] [CrossRef]
- Xu, W.; Ding, J.; Lin, S.; Xu, R.; Liu, H. Comparative mitogenomes of three species in Moenkhausia: Rare irregular gene rearrangement within Characidae. Int. J. Biol. Macromol. 2021, 183, 1079–1086. [Google Scholar] [CrossRef]
- Mao, M.; Dowton, M. Complete mitochondrial genomes of Ceratobaeus sp. and Idris sp. (Hymenoptera: Scelionidae): Shared gene rearrangements as potential phylogenetic markers at the tribal level. Mol. Biol. Rep. 2014, 41, 6419–6427. [Google Scholar] [CrossRef] [PubMed]
- Masta, S.E. Mitochondrial Sequence Evolution in Spiders: Intraspecific Variation in tRNAs Lacking the TΨC Arm. Mol. Biol. Evol. 2000, 17, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Masta, S.E.; Boore, J.L. Parallel Evolution of Truncated Transfer RNA Genes in Arachnid Mitochondrial Genomes. Mol. Biol. Evol. 2008, 25, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimov, P.B.; Oconnor, B.M. Improved tRNA prediction in the American house dust mite reveals widespread occurrence of extremely short minimal tRNAs in acariform mites. BMC Genom. 2009, 10, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidergar, N.; Toplak, N.; Kuntner, M. Streamlining DNA Barcoding Protocols: Automated DNA Extraction and a New cox1 Primer in Arachnid Systematics. PLoS ONE 2014, 9, e113030. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P. A phylogenetic classification of jumping spiders (Araneae: Salticidae). J. Arachnol. 2015, 43, 231–292. [Google Scholar] [CrossRef]
- Azevedo, G.H.F.; Griswold, C.E.; Santos, A.J. Systematics and evolution of ground spiders revisited (Araneae, Dionycha, Gnaphosidae). Cladistics 2017, 34, 579–626. [Google Scholar] [CrossRef]
- Ban, X.C.; Shao, Z.K.; Wu, L.J.; Sun, J.T.; Xue, X.F. Highly diversified mitochondrial genomes provide new evidence for interordinal relationships in the Arachnida. Cladistics 2022. [Google Scholar] [CrossRef]
- Heath, T.A.; Hedtke, S.M.; Hillis, D.M. Taxon sampling and the accuracy of phylogenetic analyses. J. Syst. Evol. 2008, 46, 239–257. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Region | Forward Primer Sequence (5′→3′) | Reverse Primer Sequence (5′→3′) |
|---|---|---|---|
| Lysi-1 | trnM-COI | AGGTCAGCTAATAAAGCTAA | AACCAATTACAAACCCACC |
| Lysi-2 | trnK-ATP6 | AGGTGTTAGTCTCTTAAATT | GCYATTATATTAGCAGCYAA |
| Lysi-3 | COIII-ND5 | GGATTTGAAGCAGCAGCTTG | GGATTACCATTCACATCAGG |
| Lysi-4 | ND5 | CCTGATGTGAATGGTAATCC | ATTATAGACTGAATCTCATC |
| Lysi-5 | ND5-ND4 | ATGAGATTCAGTCTATAATG | CCTTAATCGCTTATTCATCA |
| Lysi-6 | ND4-CYTB | GGTAGGTGATATTAAGATTA | ATWCTAGCWCCATTACATG |
| Lysi-7 | CYTB-ND1 | TRTTCATATTCAACCTGAAT | ATYGGATGATCWACTAATTC |
| Lysi-8 | rrnL-rrnS | AGTTCGATAGGGTCTTATCG | CCTATTTATAATGGCGGCAT |
| Lysi-9 | rrnS-ND2 | AGGTTCCTCTAATAAGATGA | CATGAACCAATCATCTCTAC |
| Order | Family | Species | Accession Number | Total Length (bp) | Total A + T (%) |
|---|---|---|---|---|---|
| Araneomorphae | Araneidae | Araneus angulatus | KU365988.1 | 14,205 | 75.1 |
| Araneidae | Araneus ventricosus | KM588668.1 | 14,617 | 73.4 | |
| Araneidae | Argiope amoena | KJ607907.1 | 14121 | 72.1 | |
| Araneidae | Argiope bruennichi | KJ594561.1 | 14,063 | 73.4 | |
| Araneidae | Argiope perforata | MK512574.1 | 14,032 | 74.2 | |
| Araneidae | Cyclosa argenteoalba | KP862583.1 | 14,575 | 73.7 | |
| Araneidae | Cyclosa japonica | MK512575.1 | 14,687 | 73.0 | |
| Araneidae | Cyrtarachne nagasakiensis | KR259802.1 | 14,402 | 75.7 | |
| Araneidae | Hypsosinga pygmaea | KR259803.1 | 14,193 | 76.1 | |
| Araneidae | Neoscona adianta | KR259805.1 | 14,161 | 74.6 | |
| Araneidae | Neoscona multiplicans | MK052682.1 | 14,074 | 74.8 | |
| Araneidae | Neoscona nautica | KR259804.1 | 14,049 | 78.8 | |
| Araneidae | Neoscona scylla | MK086023.1 | 14,092 | 74.6 | |
| Araneidae | Neoscona theisi | KP100667.1 | 14,156 | 75.2 | |
| Agelenidae | Agelena silvatica | KX290739.1 | 14,776 | 74.5 | |
| Tetragnathidae | Tetragnatha maxillosa | KP306789.1 | 14,578 | 74.5 | |
| Tetragnathidae | Tetragnatha nitens | KP306790.1 | 14,639 | 74.3 | |
| Nephilidae | Trichonephila clavata | AY452691.1 | 14,436 | 76.0 | |
| Nephilidae | Trichonephila clavipes | LC619787.1 | 14,902 | 77.2 | |
| Dictynidae | Argyroneta aquatica | KJ907736.1 | 16,000 | 72.2 | |
| Thomisidae | Ebrechtella tricuspidata | KU852748.1 | 14,532 | 76.2 | |
| Thomisidae | Oxytate striatipes | KM507783.1 | 14,407 | 78.2 | |
| Salticidae | Carrhotus xanthogramma | KP402247.1 | 14,563 | 75.1 | |
| Salticidae | Epeus alboguttatus | MH922026.1 | 14,625 | 77.6 | |
| Salticidae | Cheliceroides longipalpis | MH891570.1 | 14,334 | 79.0 | |
| Salticidae | Habronattus oregonensis | AY571145.1 | 14,381 | 74.4 | |
| Salticidae | Phanuelus gladstone | MT773150.1 | 14,458 | 75.1 | |
| Salticidae | Phintella cavaleriei | MW540530.1 | 14,325 | 78.1 | |
| Salticidae | Plexippus paykulli | KM114572.1 | 14,316 | 73.5 | |
| Salticidae | Telamonia vlijmi | KJ598073.1 | 14,601 | 77.3 | |
| Desidae | Desis jiaxiangi | MW178198.1 | 14,610 | 77.0 | |
| Selenopidae | Selenops bursarius | KM114573.1 | 14,272 | 74.4 | |
| Pisauridae | Dolomedes angustivirgatus | KU354434.1 | 14,783 | 76.8 | |
| Oxyopidae | Oxyopes hupingensis | MK518391.1 | 15,078 | 77.9 | |
| Oxyopidae | Oxyopes licenti | MT741489.1 | 14,431 | 78.1 | |
| Oxyopidae | Oxyopes sertatus | KM272950.1 | 14,442 | 75.9 | |
| Lycosidae | Pardosa laura | KM272948.1 | 14,513 | 77.4 | |
| Lycosidae | Pirata subpiraticus | KM486623.1 | 14,528 | 75.6 | |
| Lycosidae | Wadicosa fidelis | KP100666.1 | 14,741 | 76.0 | |
| Lycosidae | Lycosa shansia | OK032619 | 14,638 | 79.3 | |
| Lycosidae | Lycosa singoriensis | OK032620 | 13,686 | 75.1 | |
| Hypochilidae | Hypochilus thorelli | EU523753.1 | 13,991 | 70.3 | |
| Cheiracanthiidae | Cheiracanthium triviale | MN334527.1 | 14,595 | 77.9 | |
| Sicariidae | Loxosceles similis | MK425700.1 | 14,683 | 72.8 | |
| Pholcidae | Mesabolivar sp | MH643812.1 | 14,941 | 70.6 | |
| Pholcidae | Pholcus sp | KJ782458.1 | 14,279 | 65.8 | |
| Pholcidae | Pholcus phalangioides | JQ407804.1 | 14,459 | 65.9 | |
| Dysderidae | Parachtes romandiolae | MN052923.1 | 14,220 | 71.4 | |
| Mygalomorphae | Dipluridae | Phyxioschema suthepium | JQ407802.1 | 13,931 | 67.4 |
| Atypidae | Atypus karschi | MT832081.1 | 14,149 | 73.7 | |
| Nemesiidae | Calisoga longitarsis | EU523754.1 | 14,070 | 64.0 | |
| Theraphosidae | Cyriopagopus hainanus | MN877932.1 | 13,874 | 69.6 | |
| Theraphosidae | Ornithoctonus huwena | AY309259.1 | 13,874 | 69.8 | |
| Mesothelae | Liphistiidae | Songthela hangzhouensis | AY309258.1 | 14,215 | 72.2 |
| Liphistiidae | Liphistius erawan | JQ407803.1 | 14,197 | 67.7 |
| Name | Location | Size (bp) | Intergenic Nucleotides | Codon | Strand | ||
|---|---|---|---|---|---|---|---|
| From | To | Start | Stop | ||||
| trnM | 1\1 | 66\63 | 66\63 | 0\0 | J | ||
| ND2 | 66\67 | 1013\1009 | 948\943 | −1\3 | ATA\ATT | TAA\T | J |
| trnW | 1012\1010 | 1058\1068 | 47\59 | −2\0 | J | ||
| trnY | 1045\1041 | 1103\1097 | 59\57 | −14\28 | N | ||
| trnC | 1109\1086 | 1150\1140 | 42\55 | 5\−12 | N | ||
| COI | 1160\1142 | 2701\2680 | 1542\1539 | 9\1 | ATA\ATA | TAA\TAA | J |
| COII | 2728\2689 | 3375\3355 | 648\667 | 26\8 | ATA\TTG | TAA\T | J |
| trnK | 3376\3356 | 3422\3415 | 47\60 | 0\0 | J | ||
| trnD | 3422\3399 | 3486\3455 | 65\57 | −1\−17 | J | ||
| ATP8 | 3472\3448 | 3622\3606 | 151\159 | −15\−8 | ATA\ATT | T\TAA | J |
| ATP6 | 3624\3606 | 4289\4268 | 666\663 | 1\−1 | ATA\ATA | TAA\TAA | J |
| COIII | 4293\4272 | 5078\5057 | 786\786 | 3\3 | TTG\TTG | TAA\TAG | J |
| trnG | 5096\5071 | 5146\5125 | 51\55 | 17\13 | J | ||
| ND3 | 5150\5129 | 5503\5473 | 354\345 | 3\3 | ATT\ATT | TAA\TAG | J |
| trnL2(UUR) | 5504\5473 | 5561\5532 | 58\60 | 0\−1 | N | ||
| trnN | 5563\5532 | 5622\5584 | 60\53 | 1\−1 | J | ||
| trnA | 5600\5567 | 5658\5633 | 59\67 | −23\−18 | J | ||
| trnS1(AGN) | 5653\5631 | 5706\5683 | 54\53 | −6\−3 | J | ||
| trnR | 5709\5679 | 5776\5733 | 68\55 | 2\−5 | J | ||
| trnE | 5754\5721 | 5812\5775 | 59\55 | −23\−13 | J | ||
| trnF | 5788\5768 | 5845\5812 | 58\45 | −25\−8 | N | ||
| ND5 | 5844\5813 | 7475\7436 | 1632\1624 | −2\0 | ATA\ATA | TAA\T | N |
| trnH | 7481\7438 | 7535\7502 | 55\65 | 5\1 | N | ||
| ND4 | 7536\7503 | 8817\8726 | 1282\1224 | 0\0 | TTG\ATT | T\TAA | N |
| ND4L | 8818\8724 | 9085\9048 | 268\325 | 0\−3 | ATT\ATA | T\T | N |
| trnP | 9078\9052 | 9134\9103 | 57\52 | −8\3 | N | ||
| ND6 | 9138\9109 | 9569\9538 | 432\430 | 3\5 | TTG\TTG | TAA\T | J |
| trnI | 9568\9539 | 9632\9604 | 65\66 | −2\0 | J | ||
| CYTB | 9624\9620 | 10754\10721 | 1131\1102 | −9\15 | ATT\ATT | TAA\T | J |
| trnS2(UCN) | 10755\10722 | 10806\10775 | 52\54 | 0\0 | J | ||
| trnT | 10814\10781 | 10854\10824 | 41\44 | 7\5 | J | ||
| ND1 | 10856\10833 | 11765\11733 | 910\901 | 1\8 | ATA\ATA | T\T | N |
| trnL1(CUN) | 11756\11737 | 11821\11788 | 66\52 | 10\3 | N | ||
| rrnL | 11822\11789 | 12836\12806 | 1015\1018 | 0\0 | N | ||
| trnV | 12837\12807 | 12897\12862 | 61\56 | 0\0 | N | ||
| rrnS | 12898\12861 | 13590\13566 | 693\706 | 0\2 | N | ||
| trnQ | 13591\13567 | 13654\13613 | 64\47 | 0\0 | N | ||
| CR | 13655\13614 | 14638\13686 | 984\73 | 0\0 | J | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, W.; Wang, J.; Zhao, X.; Liu, H.; Zhu, S. Mitochondrial Genomes of two Lycosa spiders (Araneae, Lycosidae): Genome Description and Phylogenetic Implications. Diversity 2022, 14, 538. https://doi.org/10.3390/d14070538
Ye W, Wang J, Zhao X, Liu H, Zhu S. Mitochondrial Genomes of two Lycosa spiders (Araneae, Lycosidae): Genome Description and Phylogenetic Implications. Diversity. 2022; 14(7):538. https://doi.org/10.3390/d14070538
Chicago/Turabian StyleYe, Wentao, Jiachen Wang, Xinyi Zhao, Hongyi Liu, and Sheng Zhu. 2022. "Mitochondrial Genomes of two Lycosa spiders (Araneae, Lycosidae): Genome Description and Phylogenetic Implications" Diversity 14, no. 7: 538. https://doi.org/10.3390/d14070538
APA StyleYe, W., Wang, J., Zhao, X., Liu, H., & Zhu, S. (2022). Mitochondrial Genomes of two Lycosa spiders (Araneae, Lycosidae): Genome Description and Phylogenetic Implications. Diversity, 14(7), 538. https://doi.org/10.3390/d14070538