


Spatial Pattern of Genetic Diversity in the Blood Fluke Aporocotyle argentinensis (Digenea, Aporocotylidae) from South American Hakes (Pisces: Merluccidae)

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
Samples
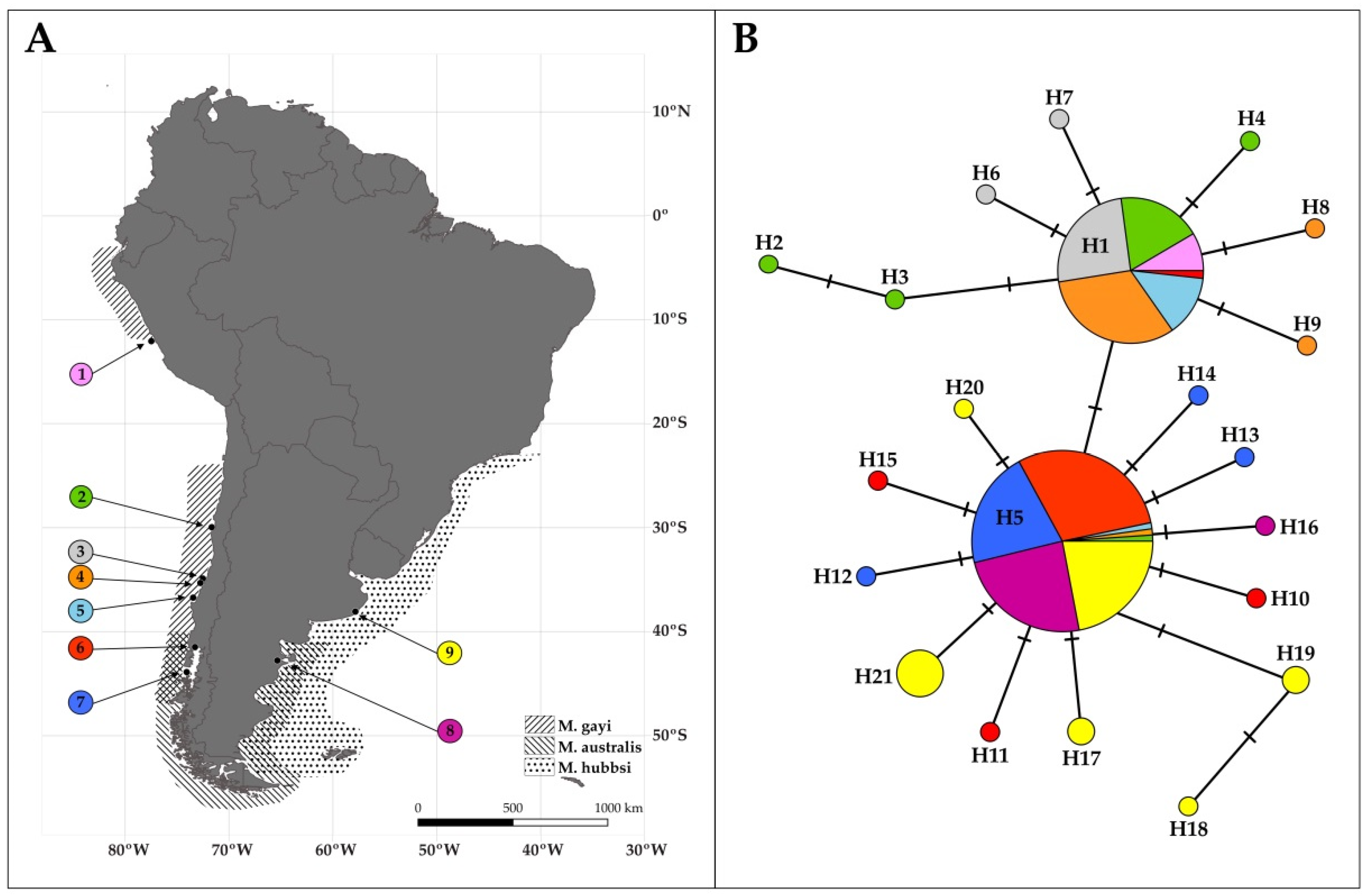
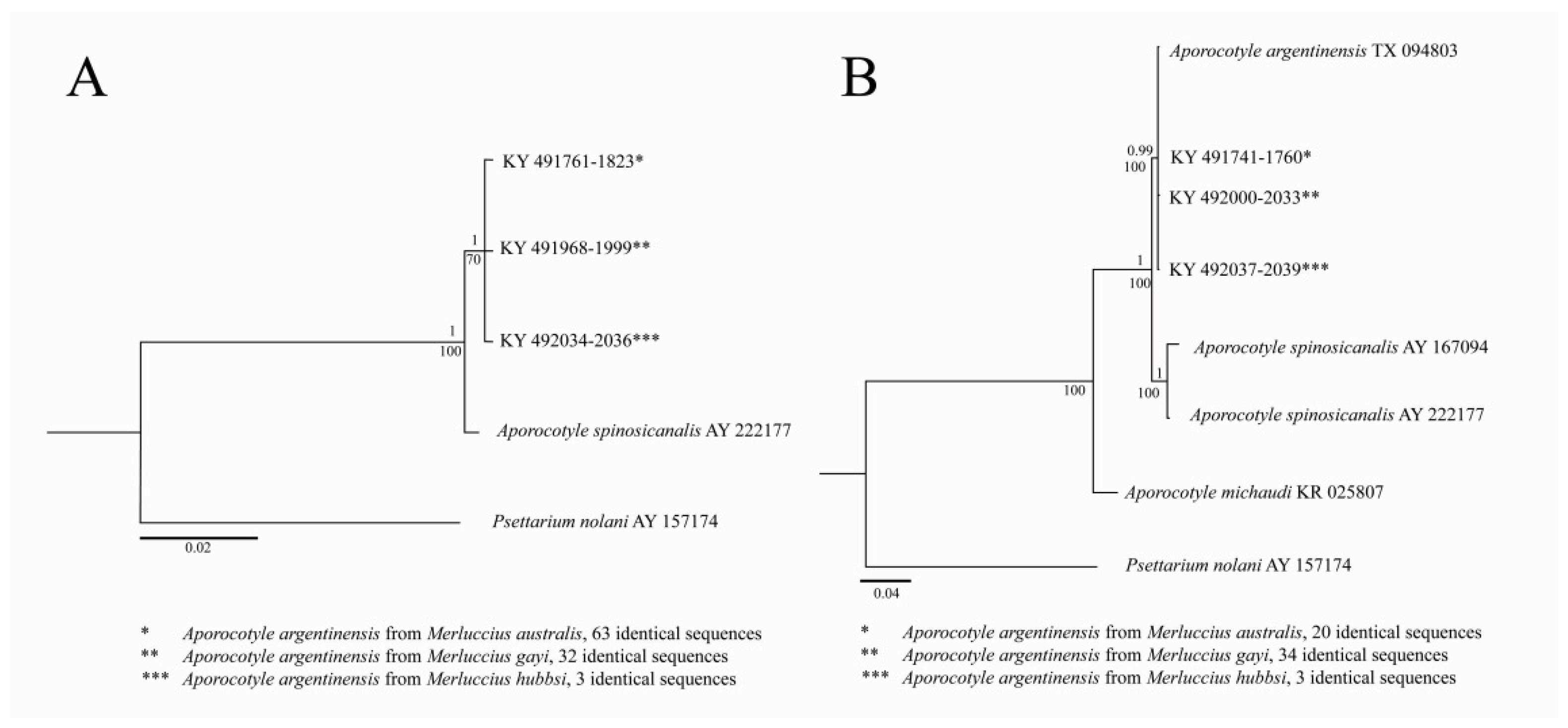
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perkins, S.L.; Martinsen, E.S.; Falk, B.G. Do molecules matter more than morphology? Promises and pitfalls in parasites. Parasitology 2011, 138, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Criscione, C.D.; Poulin, R.; Blouin, M.S. Molecular ecology of parasites: Elucidating ecological and microevolutionary processes. Mol. Ecol. 2005, 14, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Nadler, S.A.; Pérez-Ponce de León, G. Integrating molecular and morphological approaches for characterizing parasite cryptic species: Implications for parasitology. Parasitology 2011, 138, 1688–1709. [Google Scholar] [CrossRef]
- Martínez-Aquino, A.; Ceccarelli, F.S.; Pérez-Ponce de León, G. Molecular phylogeny of the genus Margotrema (Digenea: Allocreadiidae), parasitic flatworms of goodeid freshwater fishes across central Mexico: Species boundaries, host-specificity, and geographical congruence. Zool. J. Linn. Soc. 2013, 168, 1–16. [Google Scholar] [CrossRef]
- Presswell, B.; Bennett, J. Galactosomum otepotiense n.sp. (Trematoda: Heterophydae) infecting four different species of fish-eating birds in New Zealand: Genetically identical but morphologically variable. J. Helminthol. 2020, 94, e86. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Orts, J.S.; Hernández- Mena, D.L.; Alama-Bermejo, G.; Kuchta, R.; Jacobson, K.C. Morphological and molecular characterization of Aporocotyle margolisi Smith, 1967 (Digenea: Aporocotylidae) from the North Pacific hake Merluccius productus (Ayres) (Gadiformes: Merluccidae) off Oregon, USA. Syst. Parasitol. 2017, 94, 819–829. [Google Scholar] [CrossRef]
- Smith, J.W. On Aporocotyle argentinensis n. sp. (Digenea: Sanguinicolidae) from Merluccius hubbsi, and the phylogeny of Aporocotyle Odhner, 1900 in Hake. J. Helminthol. 1969, 43, 371–382. [Google Scholar] [CrossRef]
- Tantaléan, M.; Martínez, R. Aporocotyte garciai (Digenea Sanguinicolidae), parasito de Genypterus sp. de la costa Peruana. Parasit. al Dia 1990, 14, 67–69. [Google Scholar]
- Kamegai, S.; Machida, M.; Kuramochi, T. Two Blood Flukes from Deep-sea Fishes of Suruga Bay. Bull. Natl. Sci. Mus. Tokyo Ser. A 2002, 28, 29–34. [Google Scholar]
- Pitcher, T.J.; Alheit, J. What Makes a Hake? A Review of the Critical Biological Features That Sustain Global Hake Fisheries. In Hake: Fisheries, Ecology and Markets; Pitcher, T.J., Alheit, J., Eds.; Chapman & Hall: Salisbury, UK, 1995; pp. 1–14. [Google Scholar]
- Hall, K.A.; Cribb, T.H.; Barker, S.C. V4 region of small subunit rDNA indicates polyphyly of the Fellodistomidae (Digenea) which is supported by morphology and life-cycle data. Syst. Parasitol. 1999, 43, 81–92. [Google Scholar] [CrossRef]
- Chisholm, L.; Morgan, J.; Adlard, R.; Whittington, I. Phylogenetic analysis of the Monocotylidae (Monogenea) inferred from 28S rDNA sequences. Int. J. Parasitol. 2001, 31, 1537–1547. [Google Scholar] [CrossRef]
- Leung, T.; Poulin, R.; Keeney, D. Accumulation of diverse parasite genotypes within the bivalve second intermediate host of the digenean Gymnophallus sp. Int. J. Parasitol. 2009, 39, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetic Analysis. V. 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, H. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Polzin, T.; Daneschmand, S.V. On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 2003, 31, 12–20. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its applications. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Blasco-Costa, I.; Cutmore, S.C.; Miller, T.L.; Nolan, M.J. Molecular approaches to trematode systematics: ‘best practice’ and implications for future study. Syst. Parasitol. 2016, 93, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.; Zegers, J. Variaciones intraespecíficas del adulto de Proctoeces lintoni Siddiqi & Cable (Trematoda: Fellodistomidae) en hospedadores vertebrados e invertebrados. Stud. Neotrop. Fauna Environ. 1988, 23, 189–195. [Google Scholar]
- Palm, H.W.; Waeschenbach, A.; Littlewood, D.T.J. Genetic diversity in the trypanorhynch cestode Tentacularia coryphaenae Bosc, 1797: Evidence for a cosmopolitan distribution and low host specificity in the teleost intermediate host. Parasitol. Res. 2007, 101, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Aiken, H.M.; Bott, N.J.; Mladineo, I.; Montero, F.E.; Nowak, B.; Hayward, C.J. Molecular evidence for cosmopolitan distribution of platyhelminth parasites of tunas (Thunnus spp.). Fish Fish. 2007, 8, 167–180. [Google Scholar] [CrossRef]
- Lo, C.M.; Morgan, J.A.T.; Galzin, R.; Cribb, T.H. Identical digeneans in coral reef fishes from French Polynesia and the Great Barrier Reef (Australia) demonstrated by morphology and molecules. Int. J. Parasitol. 2001, 31, 1573–1578. [Google Scholar] [CrossRef]
- Valdivia, I.M.; Cárdenas, L.; González, K.; Jofré, D.; George-Nascimento, M.; Guiñez, R.; Oliva, M.E. Molecular evidence confirms that Proctoeces humboldti and Proctoeces chilensis (Digenea: Fellodistomidae) are the same species. J. Helminthol. 2010, 84, 341–347. [Google Scholar] [CrossRef]
- Villalba, J.; Fernández, C. Tres nuevas especies de Aporocotyle Odhner, 1900 (Digenea: Sanguinicolidae) parasitas de Genypterus spp. en Chile (Pisces. Ophiididae). Rev. Biol. Mar. 1986, 22, 125–139. [Google Scholar]
- Thulin, J. A redescription of the fish blood-fluke Aporocotyle simplex Odhner, 1900 (Digenea, Sanguinicolidae) with comments on its biology. Sarsia 1980, 65, 35–48. [Google Scholar] [CrossRef]
- Bray, R.B.; Cutmore, S.C.; Cribb, T.H. A paradigm for the recognition of cryptic trematode species in tropical Indo-West Pacific fishes: The problematic genus Preptetos (Trematoda: Lepocreadiidae). Int. J. Parasitol. 2022, 52, 169–203. [Google Scholar] [CrossRef]
- Huyse, T.; Audenaert, V.; Volckaert, F.A.M. Speciation and host-parasite relationships in parasite genus Gyrodactylus (Monogenea, Platyhelminthes) infecting gobies of the genus Pomatoschistus (Gobiidae, Teleostei). Int. J. Parasitol. 2003, 33, 1679–1689. [Google Scholar] [CrossRef]
- Johnson, K.P.; Adams, R.J.; Page, R.D.M.; Clayton, D.H. When Do Parasites Fail to Speciate in Response to Host Speciation? Syst. Biol. 2003, 52, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Alemany, D.; Acha, E.M.; Iribarne, O. The relationship between fronts and fish diversity in the Patagonian Shelf Ecosystem. J. Biogeogr. 2009, 36, 2111–2124. [Google Scholar] [CrossRef]
- Wieters, E.A.; McQuaid, C.; Palomo, G.; Pappalardo, P.; Navarrete, S.A. Biogeographical Boundaries, Functional Group Structure and Diversity of Rocky Shore Communities along the Argentinean Coast. PLoS ONE 2012, 7, e49725. [Google Scholar] [CrossRef] [PubMed]
- Cribb, T.H.; Adlard, R.D.; Hayward, C.J.; Botte, N.J.; Ellis, D.; Evans, D.; Nowak, B.F. The life cycle of Cardicola forsteri (Trematoda: Aporocotylidae), a pathogen of ranched southern bluefin tuna, Thunnus maccoyi. Int. J. Parasitol. 2011, 41, 861–870. [Google Scholar] [CrossRef]
- Koei, M. The redia, cercaria and early stages of Aporocotyle simplex Odhner, 1900 (Sanguinicolidae)—A digenetic trematode which has a polychaete annelid as the only intermediate host. Copeia 1982, 21, 115–145. [Google Scholar]
- Cribb, T.H.; Bray, R.A.; Littlewood, D.T.J.; Pichelin, S.P.; Herniou, E.A. The Digenea. In Interrelationships of the Platyhelminthes; Littleworod, D.T.J., Bray, R.A., Eds.; Taylor and Francis: London, UK, 2001; pp. 168–185. [Google Scholar]
- Olson, P.D.; Cribb, T.H.; Tkach, V.V.; Bray, R.A.; Littlewood, D.T.J. Phylogeny and classification of the Digenea (Platyhelmithes: Trematoda). Int. J. Parasitol. 2003, 33, 733–755. [Google Scholar] [CrossRef]
- Snyder, S.D.; Loker, E.S. Evolutionary relationships among the Schistosomatidae (Platyhelminthes: Digenea) and an Asian origin for Schistosoma. J. Parasitol. 2000, 86, 283–288. [Google Scholar] [CrossRef]
- Santoro, M.; Cipriani, P.; Pankov, P.; Lawton, S.P. Aporocotyle michaudi n. sp. (Digenea: Aporocotylidae) from the emerald rock cod, Trematomus bernacchii (Teleostei: Perciformes) in Antarctica. Parasitol. Int. 2015, 64, 324–329. [Google Scholar] [CrossRef]
- Lockyer, A.E.; Olson, P.D.; Littlewood, D.T.J. Utility of complete large and small subunit rRNA genes in resolving the phylogeny of the Neodermata (Platyhelminthes): Implications and a review of the cercomer theory. Biol. J. Linn. Soc. Lond. 2003, 78, 155–171. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Host | Locality | S | W | N |
|---|---|---|---|---|
| Merluccius gayi | Callao (1) | 12°03′50″ | 77°08′58″ | 10 |
| Coquimbo (2) | 29°57′37″ | 71°20′19″ | 20 | |
| Duao (2) | 34°53′58″ | 72°10′59″ | 15 | |
| Constitución (2) | 35°18′59″ | 72°25′43″ | 20 | |
| Talcahuano (2) | 36°43′52″ | 73°07′39″ | 110 | |
| Merluccius australis | Puerto Montt (2) | 41°28′39″ | 72°56′44″ | 45 |
| Guaitecas Island (2) | 43°52′43″ | 73°44′55″ | 38 | |
| Puerto Madryn (3) | 42°46′10″ | 65°01′18″ | 20 | |
| Merluccius hubbsi | Mar del Plata (3) | 38°04′25″ | 57°30′50″ | 5 |
| Falkland Islands /Islas Malvinas (4) | 51°40′58″ | 57°40′44″ | 12 |
| Host | Location | Np | Nhap | S | He ± SD | π ± SD | k ± SD |
|---|---|---|---|---|---|---|---|
| M. gayi | Callao | 5 | 1 | 0 | 0 | 0 | 0 |
| Coquimbo | 15 | 5 | 4 | 0.48 ± 0.15 | 0.00115 ± 0.00099 | 0.7809 ± 0.6012 | |
| Duao | 17 | 3 | 2 | 0.23 ± 0.13 | 0.00035 ± 0.00047 | 0.2353 ± 0.2857 | |
| Constitución | 22 | 4 | 3 | 0.26 ± 0.12 | 0.00040 ± 0.00051 | 0.2727 ± 0.3076 | |
| Talcahuano | 9 | 2 | 1 | 0.22 ± 0.17 | 0.000328 ± 0.00048 | 0.2222 ± 0.2880 | |
| Total | 68 | 9 | 9 | 0.27 ± 0.07 | 0.00052 ± 0.00057 | 0.3443 ± 0.34692 | |
| M. australis | Puerto Montt | 31 | 5 | 4 | 0.25 ± 0.10 | 0.00038 ± 0.00049 | 0.2581 ± 0.2948 |
| Guaitecas Islands | 22 | 4 | 3 | 0.26 ± 0.12 | 0.0004 ± 0.00051 | 0.2727 ± 0.3076 | |
| Puerto Madryn | 23 | 2 | 1 | 0.09 ± 0.08 | 0.00013 ± 0.00027 | 0.0869± 0.1640 | |
| Total | 76 | 9 | 8 | 0.20 ± 0.06 | 0.00031 ± 0.00042 | 0.2105 ± 0.2586 | |
| M. hubbsi | Mar del Plata | 32 | 6 | 4 | 0.58 ± 0.09 | 0.00108 ± 0.00092 | 0.7278 ± 0.5571 |
| Whole data set | 176 | 21 | 18 | 0.62 ± 0.03 | 0.04369 ± 0.03532 | 0.7865 ± 0.5745 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.524 | 0.999 | 0.999 | 0.999 | 0.000 | 0.000 | 0.000 | 0.000 | |
| 2 | −0.014 | 0.394 | 0.418 | 0.762 | 0.000 | 0.000 | 0.000 | 0.000 | |
| 3 | −0.078 | 0.005 | 0.999 | 0.999 | 0.000 | 0.000 | 0.000 | 0.000 | |
| 4 | −0.071 | −0.006 | −0.025 | 0.999 | 0.000 | 0.000 | 0.000 | 0.000 | |
| 5 | −0.078 | −0.039 | −0.044 | −0.064 | 0.000 | 0.000 | 0.000 | 0.000 | |
| 6 | 0.792 | 0.638 | 0.754 | 0.730 | 0.727 | 0.999 | 0.389 | 0.006 | |
| 7 | 0.796 | 0.625 | 0.754 | 0.730 | 0.726 | −0.018 | 0.363 | 0.031 | |
| 8 | 0.928 | 0.735 | 0.852 | 0.821 | 0.858 | −0.0004 | 0.004 | 0.003 | |
| 9 | 0.561 | 0.437 | 0.561 | 0.548 | 0.502 | 0.091 | 0.075 | 0.146 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, M.E.; Cárdenas, L.; Valdivia, I.M.; Bruning, P.; Figueroa-Fabrega, L.; Escribano, R. Spatial Pattern of Genetic Diversity in the Blood Fluke Aporocotyle argentinensis (Digenea, Aporocotylidae) from South American Hakes (Pisces: Merluccidae). Diversity 2022, 14, 772. https://doi.org/10.3390/d14090772
Oliva ME, Cárdenas L, Valdivia IM, Bruning P, Figueroa-Fabrega L, Escribano R. Spatial Pattern of Genetic Diversity in the Blood Fluke Aporocotyle argentinensis (Digenea, Aporocotylidae) from South American Hakes (Pisces: Merluccidae). Diversity. 2022; 14(9):772. https://doi.org/10.3390/d14090772
Chicago/Turabian StyleOliva, Marcelo E., Leyla Cárdenas, Isabel M. Valdivia, Paulina Bruning, Luis Figueroa-Fabrega, and Rubén Escribano. 2022. "Spatial Pattern of Genetic Diversity in the Blood Fluke Aporocotyle argentinensis (Digenea, Aporocotylidae) from South American Hakes (Pisces: Merluccidae)" Diversity 14, no. 9: 772. https://doi.org/10.3390/d14090772
APA StyleOliva, M. E., Cárdenas, L., Valdivia, I. M., Bruning, P., Figueroa-Fabrega, L., & Escribano, R. (2022). Spatial Pattern of Genetic Diversity in the Blood Fluke Aporocotyle argentinensis (Digenea, Aporocotylidae) from South American Hakes (Pisces: Merluccidae). Diversity, 14(9), 772. https://doi.org/10.3390/d14090772