
Movement and Genomic Methods Reveal Mechanisms Promoting Connectivity in a Declining Shorebird: The Lesser Yellowlegs

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
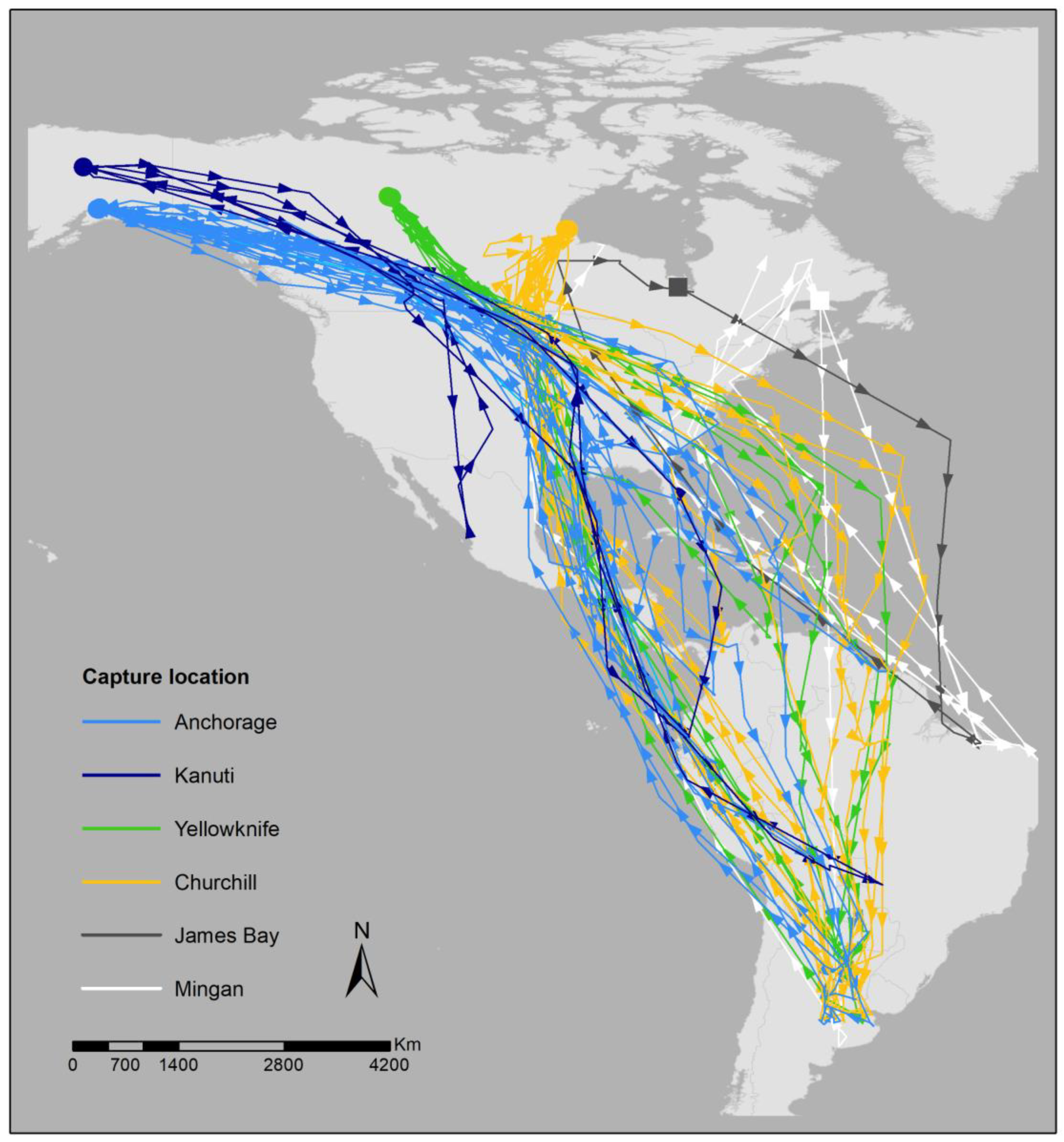
2.1. Breeding Site Fidelity via GPS
2.2. Samples and DNA Extraction
2.3. ddRAD Library Preparation and Read Assembly
2.4. mtDNA Sequencing
2.5. Genomic Diversity and Divergence
2.6. Population Genetic Structure
2.7. Spatial Genetic Structure
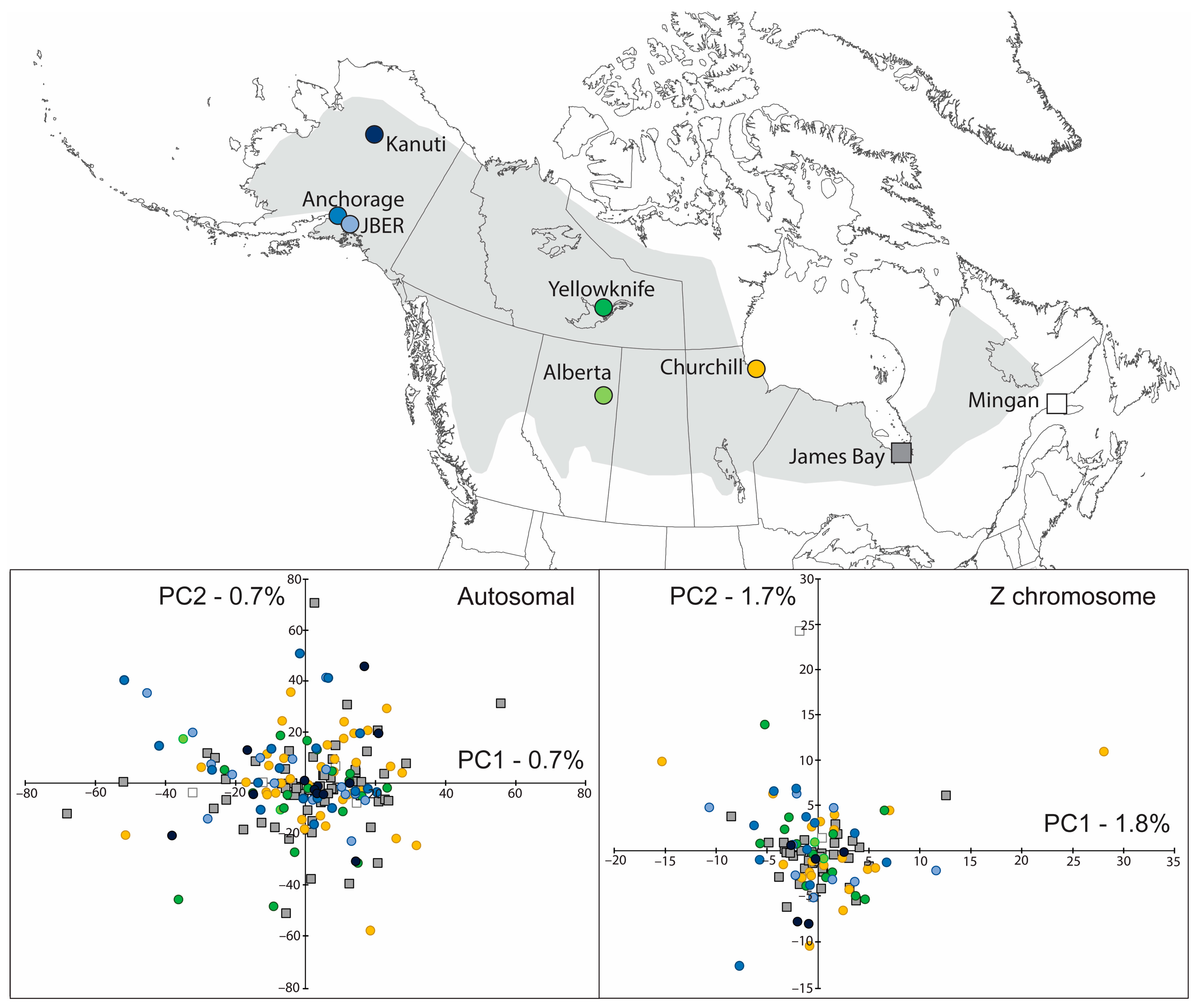
3. Results
3.1. Breeding Site Fidelity via GPS
3.2. Read Assembly and Loci Identification
3.3. Genomic Diversity and Divergence
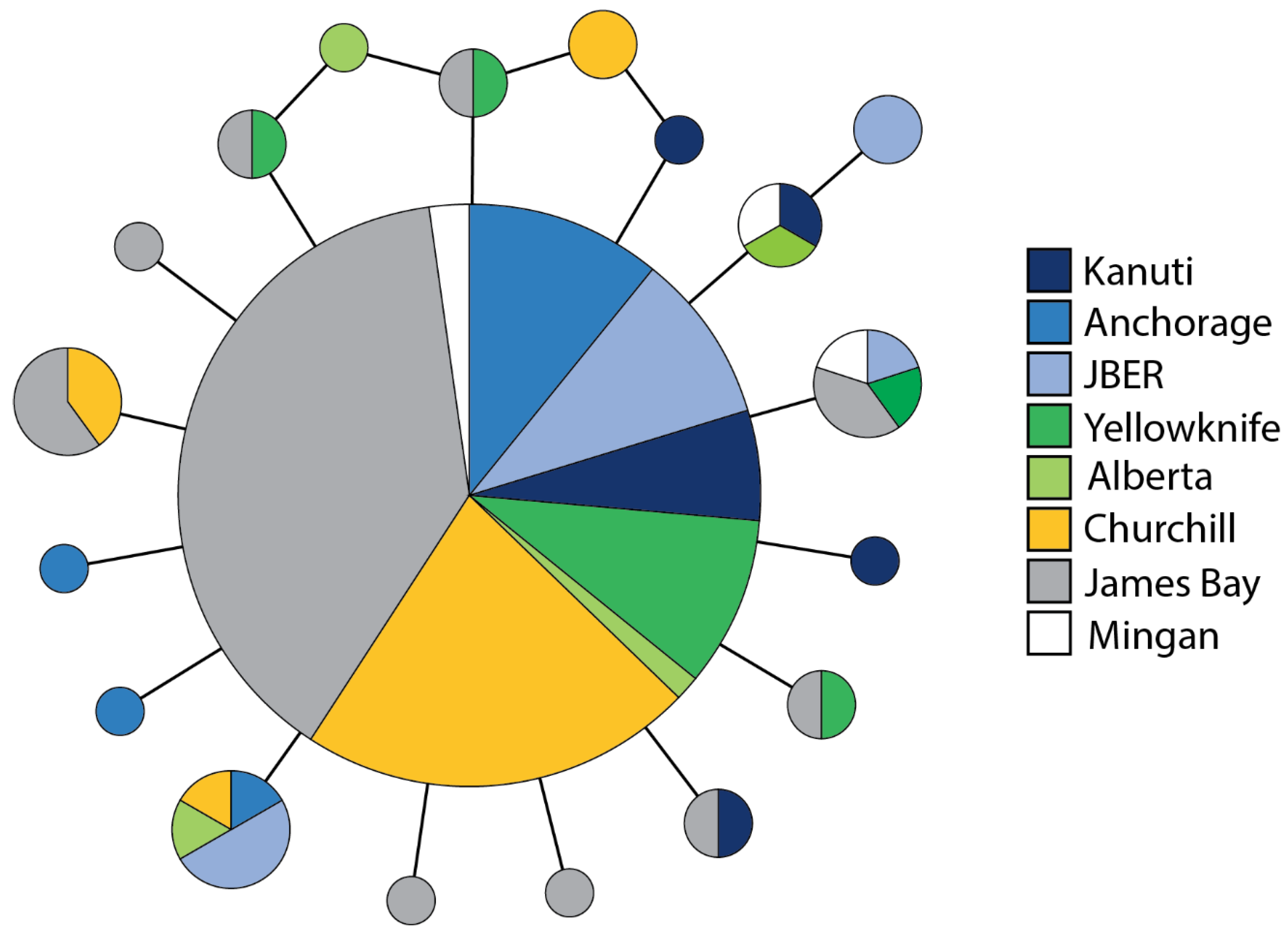
3.4. Population Genetic Structure
3.5. Spatial Genetic Structure
4. Discussion
4.1. Key Findings
4.2. Mechanisms
4.3. Comparisons with Boreal Avifauna
4.4. Conservation Implications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saccheri, I.; Kuussaari, M.; Kankare, M.; Vikman, P.; Hanski, I. Inbreeding and Extinction in a Butterfly Metapopulation. Nature 1998, 392, 491–494. [Google Scholar] [CrossRef]
- Kinley, T.A.; Apps, C.D. Mortality Patterns in a Subpopulation of Endangered Mountain Caribou. Wildl. Soc. Bull. 2001, 29, 158–164. [Google Scholar] [CrossRef]
- Currey, R.J.C.; Dawson, S.M.; Slooten, E. An Approach for Regional Threat Assessment under IUCN Red List Criteria That Is Robust to Uncertainty: The Fiordland Bottlenose Dolphins Are Critically Endangered. Biol. Conserv. 2009, 142, 1570–1579. [Google Scholar] [CrossRef]
- Norman, S.A.; Hobbs, R.C.; Goertz, C.E.C.; Burek-Huntington, K.A.; Shelden, K.E.W.; Smith, W.A.; Beckett, L.A. Potential Natural and Anthropogenic Impediments to the Conservation and Recovery of Cook Inlet Beluga Whales, Delphinapterus leucas. Mar. Fish. Rev. 2016, 77, 89–105. [Google Scholar] [CrossRef]
- Kraus, R.H.S.; Van Hooft, P.; Megens, H.J.; Tsvey, A.; Fokin, S.Y.; Ydenberg, R.C.; Prins, H.H.T. Global Lack of Flyway Structure in a Cosmopolitan Bird Revealed by a Genome Wide Survey of Single Nucleotide Polymorphisms. Mol. Ecol. 2013, 22, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Ruegg, K.C.; Anderson, E.C.; Paxton, K.L.; Apkenas, V.; Lao, S.; Siegel, R.B.; Desante, D.F.; Moore, F.; Smith, T.B. Mapping Migration in a Songbird Using High-Resolution Genetic Markers. Mol. Ecol. 2014, 23, 5726–5739. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.E.; Sonsthagen, S.A.; Dacosta, J.M.; Sorenson, M.D.; Fox, A.D.; Weaver, M.; Skalos, D.; Kondratyev, A.V.; Scribner, K.T.; Walsh, A.; et al. As the Goose Flies: Migration Routes and Timing Influence Patterns of Genetic Diversity in a Circumpolar Migratory Herbivore. Diversity 2022, 14, 1067. [Google Scholar] [CrossRef]
- Funk, C.W.; McKay, J.K.; Hohenlohe, P.A.; Allendorf, F. Harnessing Genomics for Delineating Conservation Units. Trends Ecol. Evol. 2012, 27, 489–496. [Google Scholar] [CrossRef]
- Klicka, L.B.; Kus, B.E.; Title, P.O.; Burns, K.J. Conservation Genomics Reveals Multiple Evolutionary Units within Bell’s Vireo (Vireo bellii). Conserv. Genet. 2016, 17, 455–471. [Google Scholar] [CrossRef]
- Waples, R.S.; Lindley, S.T. Genomics and Conservation Units: The Genetic Basis of Adult Migration Timing in Pacific Salmonids. Evol. Appl. 2018, 11, 1518–1526. [Google Scholar] [CrossRef]
- Forester, B.R.; Murphy, M.; Mellison, C.; Petersen, J.; Pilliod, D.S.; Van Horne, R.; Harvey, J.; Funk, W.C. Genomics-informed Delineation of Conservation Units in a Desert Amphibian. Mol. Ecol. 2022, 31, 5249–5269. [Google Scholar] [CrossRef] [PubMed]
- Tibbitts, T.L.; Moskoff, W. Lesser Yellowlegs, Tringa flavipes. In Birds of the World; Poole, A., Ed.; Cornell Lab of Ornithology: Ithaca, NY, USA, 2020. [Google Scholar]
- Rosenberg, K.V.; Dokter, A.M.; Blancher, P.J.; Sauer, J.R.; Smith, A.C.; Smith, P.A.; Stanton, J.C.; Panjabi, A.; Helft, L.; Parr, M.; et al. Decline of the North American Avifauna. Science 2019, 366, 120–124. [Google Scholar] [CrossRef]
- Handel, C.M.; Sauer, J.R. Combined Analysis of Roadside and Off-Road Breeding Bird Survey Data to Assess Population Change in Alaska. Condor 2017, 119, 557–575. [Google Scholar] [CrossRef]
- COSEWIC. COSEWIC Assessment and Status Report on the Lesser Yellowlegs Tringa flavipes in Canada; Committee on the Status of Endangered Wildlife in Canada, Environment and Climate Change Canada: Ottawa, ON, Canada, 2020. [Google Scholar]
- Clay, R.; Lesterhuis, A.; Centron, S. Conservation Plan for the Lesser Yellowlegs; Manomet Center for Conservation Sciences: Manomet, MA, USA, 2012. [Google Scholar]
- Atlantic Flyway Shorebird Initiative Harvest Working Group. A Plan to Address the Sustainability of Shorebird Harvest in the Western Atlantic Flyway; Atlantic Flyway Shorebird Harvest Group: Hadley, MA, USA, 2016; Available online: https://www.shorebirdplan.org/wp-content/uploads/2018/08/Shorebird-Harvest-Atlantic-Flyway-Plan-May-2016.pdf (accessed on 20 April 2023).
- Studds, C.E.; Kendall, B.E.; Murray, N.J.; Wilson, H.B.; Rogers, D.I.; Clemens, R.S.; Gosbell, K.; Hassell, C.J.; Jessop, R.; Melville, D.S.; et al. Rapid Population Decline in Migratory Shorebirds Relying on Yellow Sea Tidal Mudflats as Stopover Sites. Nat. Commun. 2017, 8, 14895. [Google Scholar] [CrossRef] [PubMed]
- Kleijn, D.; Schekkerman, H.; Dimmers, W.J.; Van Kats, R.J.M.; Melman, D.; Teunissen, W.A. Adverse Effects of Agricultural Intensification and Climate Change on Breeding Habitat Quality of Black-Tailed Godwits Limosa l. limosa in the Netherlands. Ibis 2010, 152, 475–486. [Google Scholar] [CrossRef]
- Strum, K.M.; Hooper, M.J.; Johnson, K.A.; Lanctot, R.B.; Zaccagnini, M.E.; Sandercock, B.K. Exposure of Nonbreeding Migratory Shorebirds to Cholinesterase-inhibiting Contaminants in the Western Hemisphere. Condor 2010, 112, 15–28. [Google Scholar] [CrossRef]
- Galbraith, H.; DesRochers, D.W.; Brown, S.; Reed, J.M. Predicting Vulnerabilities of North American Shorebirds to Climate Change. PLoS ONE 2014, 9, e108899. [Google Scholar] [CrossRef]
- Atlantic Flyway Shorebird Initiative Harvest Working Group. Actions for the Atlantic Flyway Shorebird Initiative’s Shorebird Harvest Working Group 2020–2025. 2020, pp. 1–16. Available online: https://www.shorebirdplan.org/wp-content/uploads/2020/09/AFSI-Shorebird-Harvest-Action-Plan-2020_25-April-2020.pdf (accessed on 20 April 2023).
- Wege, D.C.; Burke, W.; Reed, E.T. Migratory Shorebirds in Barbados: Hunting, Management and Conservation; BirdLife International: Cambridge, UK, 2014. [Google Scholar]
- Andres, B.A.; Moore, L.; Cox, A.R.; Frei, B.; Roy, C. A Preliminary Assessment of Shorebird Harvest in Coastal Guyana. Wader Study 2022, 129, 1–9. [Google Scholar] [CrossRef]
- Watts, B.D.; Turrin, C. Assessing Hunting Policies for Migratory Shorebirds throughout the Western Hemisphere. Wader Study 2016, 123, 6–15. [Google Scholar] [CrossRef]
- McDuffie, L.A.; Christie, K.S.; Harrison, A.-L.; Taylor, A.R.; Andres, B.A.; Laliberté, B.; Johnson, J.A. Eastern-Breeding Lesser Yellowlegs Are More Likely than Western-Breeding Birds to Visit Areas with High Shorebird Hunting during Southward Migration. Ornithol. Appl. 2022, 124, 1–16. [Google Scholar] [CrossRef]
- McDuffie, L.A.; Christie, K.S.; Taylor, A.R.; Nol, E.; Friis, C.; Harwood, C.M.; Rausch, J.; Laliberte, B.; Gesmundo, C.; Wright, J.R.; et al. Flyway-scale GPS Tracking Reveals Migratory Routes and Key Stopover and Non-breeding Locations of Lesser Yellowlegs. Ecol. Evol. 2022, 12, e9495. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, P.J.; Harvey, P.H. The Natal and Breeding Dispersal of Birds. Ann. Rev. Ecol. Syst. 1982, 13, 1–21. [Google Scholar] [CrossRef]
- Kwon, E.; Valcu, M.; Cragnolini, M.; Bulla, M.; Lyon, B.; Kempenaers, B. Breeding Site Fidelity Is Lower in Polygamous Shorebirds and Male-Biased in Monogamous Species. Behav. Ecol. 2022, 33, 592–605. [Google Scholar] [CrossRef]
- Kempenaers, B.; Valcu, M. Breeding Site Sampling across the Arctic by Individual Males of a Polygynous Shorebird. Nature 2017, 541, 528–531. [Google Scholar] [CrossRef]
- Adkins-Regan, E.; Tomaszycki, M. Monogamy on the Fast Track. Biol. Lett. 2007, 3, 617–619. [Google Scholar] [CrossRef]
- Brown, J.I.; Lavretsky, P.; Wilson, R.E.; Haughey, C.L.; Boyd, W.S.; Esler, D.; Talbot, S.L.; Sonsthagen, S.A. High Site Fidelity Does Not Equate to Population Genetic Structure for Common Goldeneye and Barrow’s Goldeneye in North America. J. Avian Biol. 2020, 51, e02600. [Google Scholar] [CrossRef]
- Rappole, J.H.; Tipton, A.R. New Harness Design for Attachment of Radio Transmitters to Small Passerines. J. F. Ornithol. 1991, 62, 335–337. [Google Scholar]
- McDuffie, L.A. Migration Ecology and Harvest Exposure Risk of Lesser Yellowlegs. Ph.D. Thesis, University of Alaska Anchorage, Anchorage, AK, USA, 2021. [Google Scholar]
- ESRI Environmental Systems Research Institute. ArcMap 10.5.1, Redlands, CA, USA. 2011. Available online: https://www.esri.com/en-us/arcgis/products/arcgis-desktop/resources (accessed on 20 April 2023).
- DaCosta, J.M.; Sorenson, M.D. Amplification Biases and Consistent Recovery of Loci in a Double-Digest RAD-Seq Protocol. PLoS ONE 2014, 9, e106713. [Google Scholar] [CrossRef] [PubMed]
- Lavretsky, P.; Dacosta, J.M.; Hernández-Baños, B.E.; Engilis, A.; Sorenson, M.D.; Peters, J.L. Speciation Genomics and a Role for the Z Chromosome in the Early Stages of Divergence between Mexican Ducks and Mallards. Mol. Ecol. 2015, 24, 5364–5378. [Google Scholar] [CrossRef] [PubMed]
- Sonsthagen, S.A.; Talbot, S.L.; White, C.M. Gene Flow and Genetic Characterization of Northern Goshawks Breeding in Utah. Condor 2004, 106, 826–836. [Google Scholar] [CrossRef]
- Schneider, S.; Roessli, D.; Excofier, L. Arlequin: A Software for Population Genetic Data Analysis. 2000. Available online: http://cmpg.unibe.ch/software/arlequin3/ (accessed on 20 April 2023).
- Fu, Y.X. Statistical Tests of Neutrality of Mutations against Population Growth, Hitchhiking and Background Selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Tajima, F. The Effect of Change in Population Size on DNA Polymorphism. Genetics 1989, 123, 597–601. [Google Scholar] [CrossRef]
- Fluxus Technology Ltd. NETWORK 10.1. 2022. Available online: www.fluxus-engineering.com (accessed on 20 April 2023).
- Bandelt, H.J.; Forster, P.; Sykes, B.C.; Richards, M.B. Mitochondrial Portraits of Human Populations Using Median Networks. Genetics 1995, 141, 743–753. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Jones, O.; Wang, J. COLONY: A Program for Parentage and Sibship Inference from Multilocus Genotype Data. Mol. Ecol. Resour. 2010, 10, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE Algorithm for Individual Ancestry Estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Alexander, D.; November, J.; Lange, K. Admixture 1.22 Software Manual 2012. Available online: https://dalexander.github.io/admixture/download.html (accessed on 20 April 2023).
- Janes, J.K.; Miller, J.M.; Dupuis, J.R.; Malenfant, R.M.; Gorrell, J.C.; Cullingham, C.I.; Andrew, R.L. The K = 2 Conundrum. Mol. Ecol. 2017, 26, 3594–3602. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A Cluster Matching and Permutation Program for Dealing with Label Switching and Multimodality in Analysis of Population Structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Malinsky, M.; Trucchi, E.; Lawson, D.J.; Falush, D. RADpainter and FineRADstructure: Population Inference from RADseq Data. Mol. Biol. Evol. 2018, 35, 1284–1290. [Google Scholar] [CrossRef]
- Petkova, D.; Novembre, J.; Stephens, M. Visualizing Spatial Population Structure with Estimated Effective Migration Surfaces. Nat. Genet. 2016, 48, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Weatherhead, P.J.; Forbes, M.R.L. Natal Philopatry in Passerine Birds: Genetic or Ecological Influences? Behav. Ecol. 1994, 5, 426–433. [Google Scholar] [CrossRef]
- Webster, K.L.; Mclaughlin, J.W. ScienceDirect Application of a Bayesian Belief Network for Assessing the Vulnerability of Permafrost to Thaw and Implications for Greenhouse Gas Production and Climate Feedback. Environ. Sci. Policy 2013, 38, 28–44. [Google Scholar] [CrossRef]
- Procházka, P.; Stokke, B.G.; Jensen, H.; Fainová, D.; Bellinvia, E.; Fossøy, F.; Vikan, J.R.; Bryja, J.; Soler, M. Low genetic differentiation among Reed Warbler Acrocephalus scirpaceus populations across Europe. J. Avian Biol. 2011, 42, 103–113. [Google Scholar] [CrossRef]
- Rönkä, N.; Pakanen, V.M.; Pauliny, A.; Thomson, R.L.; Nuotio, K.; Pehlak, H.; Thorup, O.; Lehikoinen, P.; Rönkä, A.; Blomqvist, D.; et al. Genetic Differentiation in an Endangered and Strongly Philopatric, Migrant Shorebird. BMC Ecol. Evol. 2021, 21, 125. [Google Scholar] [CrossRef] [PubMed]
- Oring, L.W.; Lank, D.B. Breeding Area Fidelity, Natal Philopatry, and the Social Systems of Sandpipers. In Shorebirds: Breeding Behavior and Populations; Burger, J., Olla, B.L., Eds.; Plenum Publishing Corporation: Ann Arbor, MI, USA, 1984. [Google Scholar]
- Lecomte, N.; Gauthier, G.; Giroux, J.F.; Milot, E.; Bernatchez, L. Tug of War between Continental Gene Flow and Rearing Site Philopatry in a Migratory Bird: The Sex-Biased Dispersal Paradigm Reconsidered. Mol. Ecol. 2009, 18, 593–602. [Google Scholar] [CrossRef]
- Nol, E.; Williams, S.; Sandercock, B.K. Natal Philopatry and Apparent Survival of Juvenile Semipalmated Plovers. Wilson J. Ornithol. 2010, 122, 23–28. [Google Scholar] [CrossRef]
- Pakanen, V.M.; Hagstedt, R.; Pauliny, A.; Blomqvist, D. Survival during the Pre-fledging Period Rather than during Post-fledging Drives Variation in Local Recruitment of an Endangered Migratory Shorebird, the Southern Dunlin Calidris alpina schinzii. J. Ornithol. 2021, 162, 119–124. [Google Scholar] [CrossRef]
- Avise, J. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Koopman, M.E.; Hayward, G.D.; McDonald, D.B. High Connectivity and Minimal Genetic Structure among North American Boreal Owl (Aegolius funereus) Populations, Regardless of Habitat Matrix. Auk 2007, 124, 690–704. [Google Scholar] [CrossRef]
- Wilson, R.E.; Matsuoka, S.M.; Powell, L.L.; Johnson, J.A.; Demarest, D.W.; Stralberg, D.; Sonsthagen, S.A. Implications of Historical and Contemporary Processes on Genetic Differentiation of a Declining Boreal Songbird: The Rusty Blackbird. Diversity 2021, 13, 103. [Google Scholar] [CrossRef]
- Ralston, J.; FitzGerald, A.M.; Burg, T.M.; Starkloff, N.C.; Warkentin, I.G.; Kirchman, J.J. Comparative Phylogeographic Analysis Suggests a Shared History among Eastern North American Boreal Forest Birds. Ornithology 2021, 138, 1–16. [Google Scholar] [CrossRef]
- Ramírez, O.; Gómez-Díaz, E.; Olalde, I.; Illera, J.C.; Rando, J.C.; González-Solís, J.; Lalueza-Fox, C. Population Connectivity Buffers Genetic Diversity Loss in a Seabird. Front. Zool. 2013, 10, 1–5. [Google Scholar] [CrossRef]
- Rehfeldt, G.E.; Crookston, N.L.; Sáenz-Romero, C.; Campbell, E.M. North American Vegetation Model for Land-use Planning in a Changing Climate: A Solution to Large Classification Problems. Ecol. Appl. 2012, 22, 119–141. [Google Scholar] [CrossRef]
- Bateman, B.L.; Taylor, L.; Wilsey, C.; Wu, J.; LeBaron, G.S.; Langham, G. Risk to North American Birds from Climate Change-related Threats. Conserv. Sci. Pract. 2020, 2, 1–15. [Google Scholar] [CrossRef]
- Bateman, B.L.; Wilsey, C.; Taylor, L.; Wu, J.; LeBaron, G.S.; Langham, G. North American Birds Require Mitigation and Adaptation to Reduce Vulnerability to Climate Change. Conserv. Sci. Pract. 2020, 2, 1–18. [Google Scholar] [CrossRef]
- Stralberg, D.; Berteaux, D.; Drever, C.R.; Drever, M.; Lewis, I.N.; Schmiegelow, F.K.A.; Tremblay, J.A. Conservation Planning for Boreal Birds in a Changing Climate: A Framework for Action. Avian Conserv. Ecol. 2019, 14, 13. [Google Scholar] [CrossRef]
- Seaborn, T.; Griffith, D.; Kliskey, A.; Caudill, C.C. Building a Bridge between Adaptive Capacity and Adaptive Potential to Understand Responses to Environmental Change. Glob. Chang. Biol. 2021, 27, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, T.; Possingham, H.P.; Chadès, I.; Minton, C.; Murray, N.J.; Rogers, D.I.; Treml, E.A.; Fuller, R.A. Migratory Connectivity Magnifies the Consequences of Habitat Loss from Sea-level Rise for Shorebird Populations. Tohoku J. Exp. Med. 2013, 230, 1–8. [Google Scholar] [CrossRef]
- Xu, Y.; Si, Y.; Wang, Y.; Zhang, Y.; Prins, H.H.T.; Cao, L.; de Boer, W.F. Loss of Functional Connectivity in Migration Networks Induces Population Decline in Migratory Birds. Ecol. Appl. 2019, 29, 1–10. [Google Scholar] [CrossRef]
- Wells, J.V.; Dawson, N.; Culver, N.; Reid, F.A.; Morgan Siegers, S. The State of Conservation in North America’s Boreal Forest: Issues and Opportunities. Front. For. Glob. Chang. 2020, 3, 1–18. [Google Scholar] [CrossRef]
- Medan, D.; Torretta, J.P.; Hodara, K.; de la Fuente, E.B.; Montaldo, N.H. Effects of Agriculture Expansion and Intensification on the Vertebrate and Invertebrate Diversity in the Pampas of Argentina. Biodivers. Conserv. 2011, 20, 3077–3100. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ddRAD | mtDNA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Location | Type | π–A | π–Z | n | H | HR | π | h | Fs | D | n |
| Kanuti | B | 0.0060 | 0.0038 | 12 | 5 | 3.1 | 0.0012 (0.0011) | 0.538 (0.161) | −3.1 | −1.8 | 13 |
| Anchorage | B | 0.0061 | 0.0037 | 20 | 4 | 2.1 | 0.0006 (0.0007) | 0.298 (0.133) | −2.7 | −1.7 | 19 |
| JBER | B | 0.0061 | 0.0037 | 20 | 4 | 2.7 | 0.0015 (0.0013) | 0.500 (0.122) | −1.0 | −0.7 | 20 |
| Yellowknife | B | 0.0061 | 0.0037 | 18 | 6 | 2.9 | 0.0010 (0.0010) | 0.468 (0.140) | −4.5 | −2.0 | 19 |
| Alberta | B | 0.0053 | 0.0029 | 3 | 3 | 3.0 | 0.0020 (0.0019) | 0.833 (0.222) | −0.9 | −0.7 | 4 |
| Churchill | B | 0.0062 | 0.0038 | 37 | 4 | 1.9 | 0.0007 (0.0008) | 0.252 (0.092) | −1.7 | −1.5 | 37 |
| James Bay | NB | 0.0062 | 0.0038 | 66 | 10 | 2.4 | 0.0008 (0.0008) | 0.331 (0.075) | −11.1 | −2.0 | 66 |
| Mingan | NB | 0.0057 | 0.0036 | 5 | 3 | 2.6 | 0.0016 (0.0016) | 0.700 (0.218) | −0.8 | −1.0 | 5 |
| Overall | – | 0.0063 | 0.0038 | 181 | 19 | – | – | – | – | – | 183 |
| Deployment Location | Location Type | Minimum Distance (m) | Maximum Distance (m) | Median Distance (m) | n |
|---|---|---|---|---|---|
| Anchorage | Breeding area | 200.2 | 17,821.8 | 776.9 | 11 |
| Kanuti | Breeding area | 341.1 | 1101.6 | 753.5 | 3 |
| Yellowknife | Breeding area | 88.6 | 405.5 | 246.1 | 5 |
| Churchill | Breeding area | 212.5 | 423,143.6 | 547.1 | 8 |
| All breeding | Breeding area | 88.6 | 423,143.6 | 629.2 | 27 |
| James Bay | Migratory stopover | 235,722.2 | 235,722.2 | 235,722.2 | 1 |
| Mingan | Migratory stopover | 297,447.0 | 1,849,575.8 | 489,060.1 | 5 |
| All non-breeding | Migratory stopover | 235,722.21 | 1,849,575.80 | 413,628.23 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christie, K.; Wilson, R.E.; Johnson, J.A.; Friis, C.; Harwood, C.M.; McDuffie, L.A.; Nol, E.; Sonsthagen, S.A. Movement and Genomic Methods Reveal Mechanisms Promoting Connectivity in a Declining Shorebird: The Lesser Yellowlegs. Diversity 2023, 15, 595. https://doi.org/10.3390/d15050595
Christie K, Wilson RE, Johnson JA, Friis C, Harwood CM, McDuffie LA, Nol E, Sonsthagen SA. Movement and Genomic Methods Reveal Mechanisms Promoting Connectivity in a Declining Shorebird: The Lesser Yellowlegs. Diversity. 2023; 15(5):595. https://doi.org/10.3390/d15050595
Chicago/Turabian StyleChristie, Katherine, Robert E. Wilson, James A. Johnson, Christian Friis, Christopher M. Harwood, Laura A. McDuffie, Erica Nol, and Sarah A. Sonsthagen. 2023. "Movement and Genomic Methods Reveal Mechanisms Promoting Connectivity in a Declining Shorebird: The Lesser Yellowlegs" Diversity 15, no. 5: 595. https://doi.org/10.3390/d15050595
APA StyleChristie, K., Wilson, R. E., Johnson, J. A., Friis, C., Harwood, C. M., McDuffie, L. A., Nol, E., & Sonsthagen, S. A. (2023). Movement and Genomic Methods Reveal Mechanisms Promoting Connectivity in a Declining Shorebird: The Lesser Yellowlegs. Diversity, 15(5), 595. https://doi.org/10.3390/d15050595