
Comparing the Utility of Microsatellites and Single Nucleotide Polymorphisms in Conservation Genetics: Insights from a Study on Two Freshwater Fish Species in France




Abstract
:1. Introduction
2. Materials and Methods
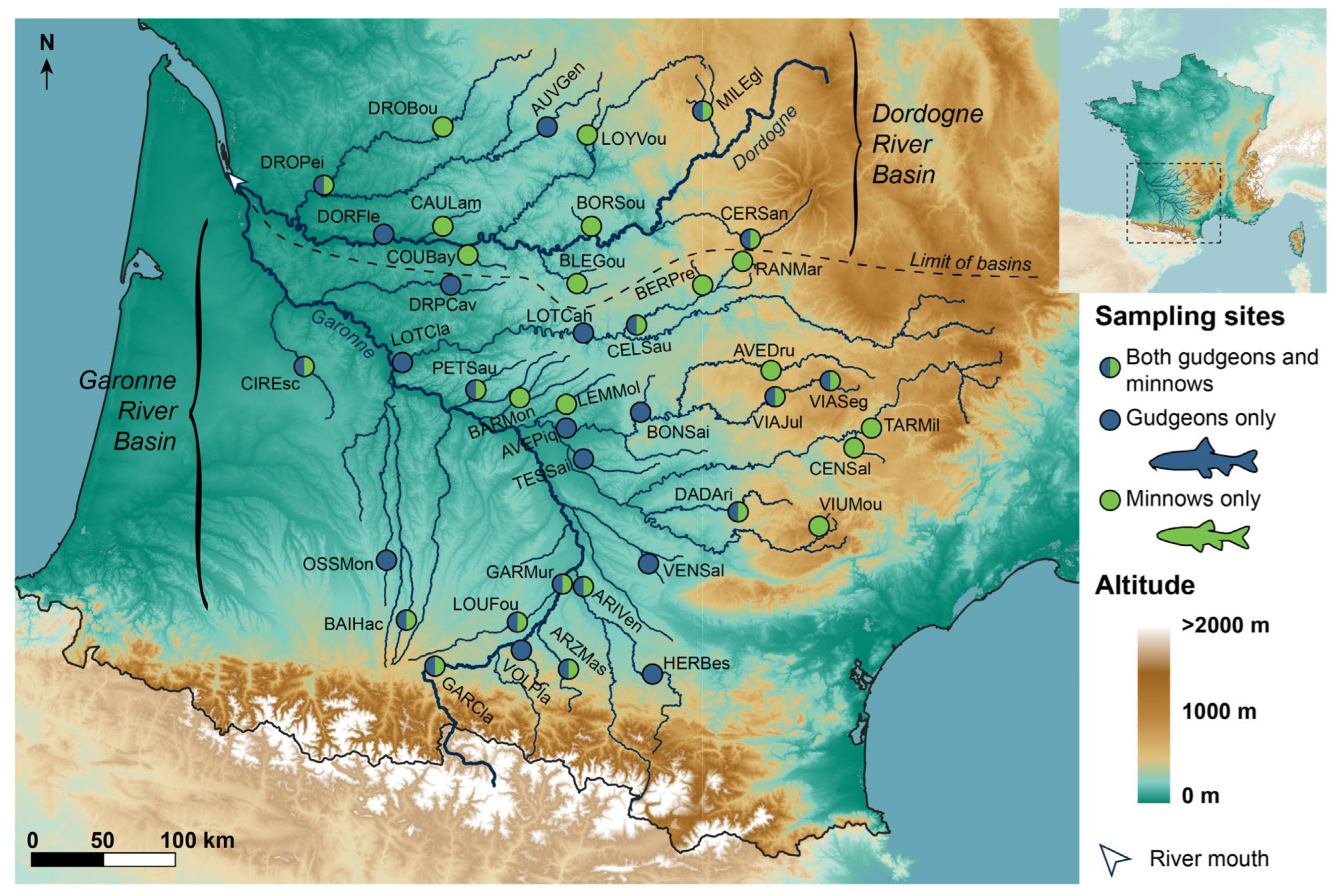
2.1. Study Area, Biological Models and Molecular Data
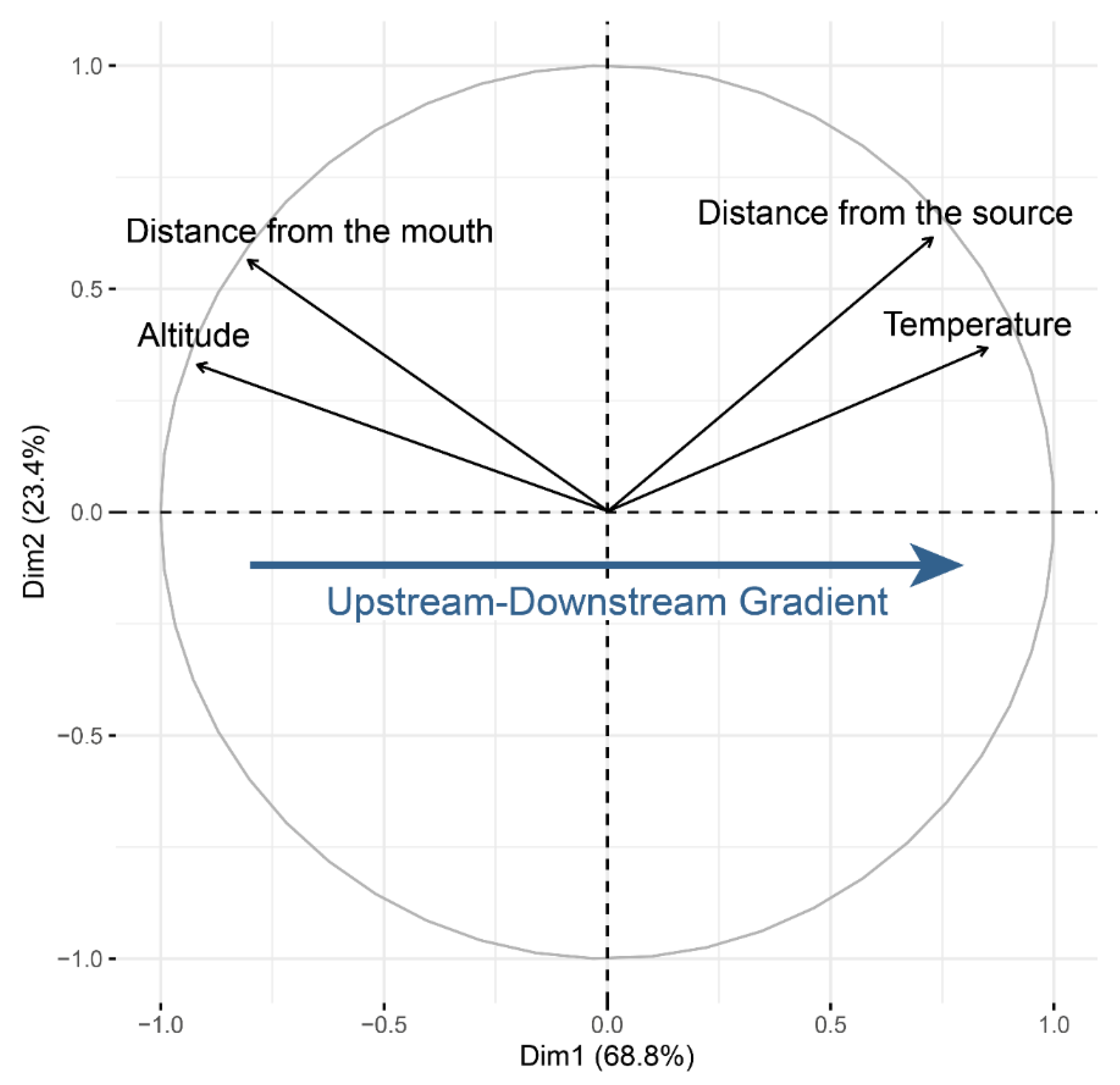
2.2. Environmental Data
2.3. Genetic Diversity and Spatial Patterns in Genetic Diversity
2.4. Genetic Differentiation and Isolation-by-Distance
2.5. Genetic Structures
3. Results
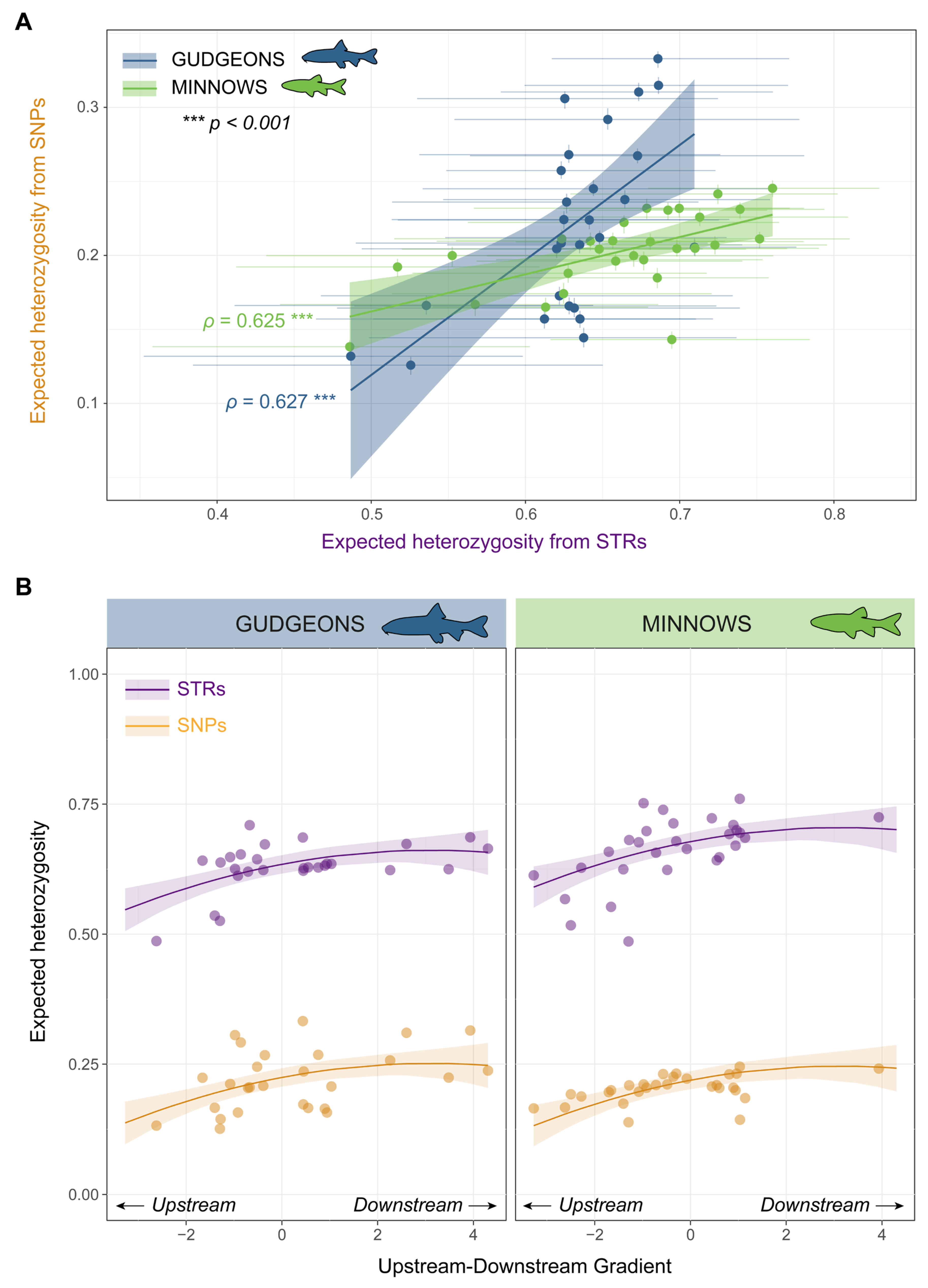
3.1. Genetic Diversity and Spatial Patterns in Genetic Diversity
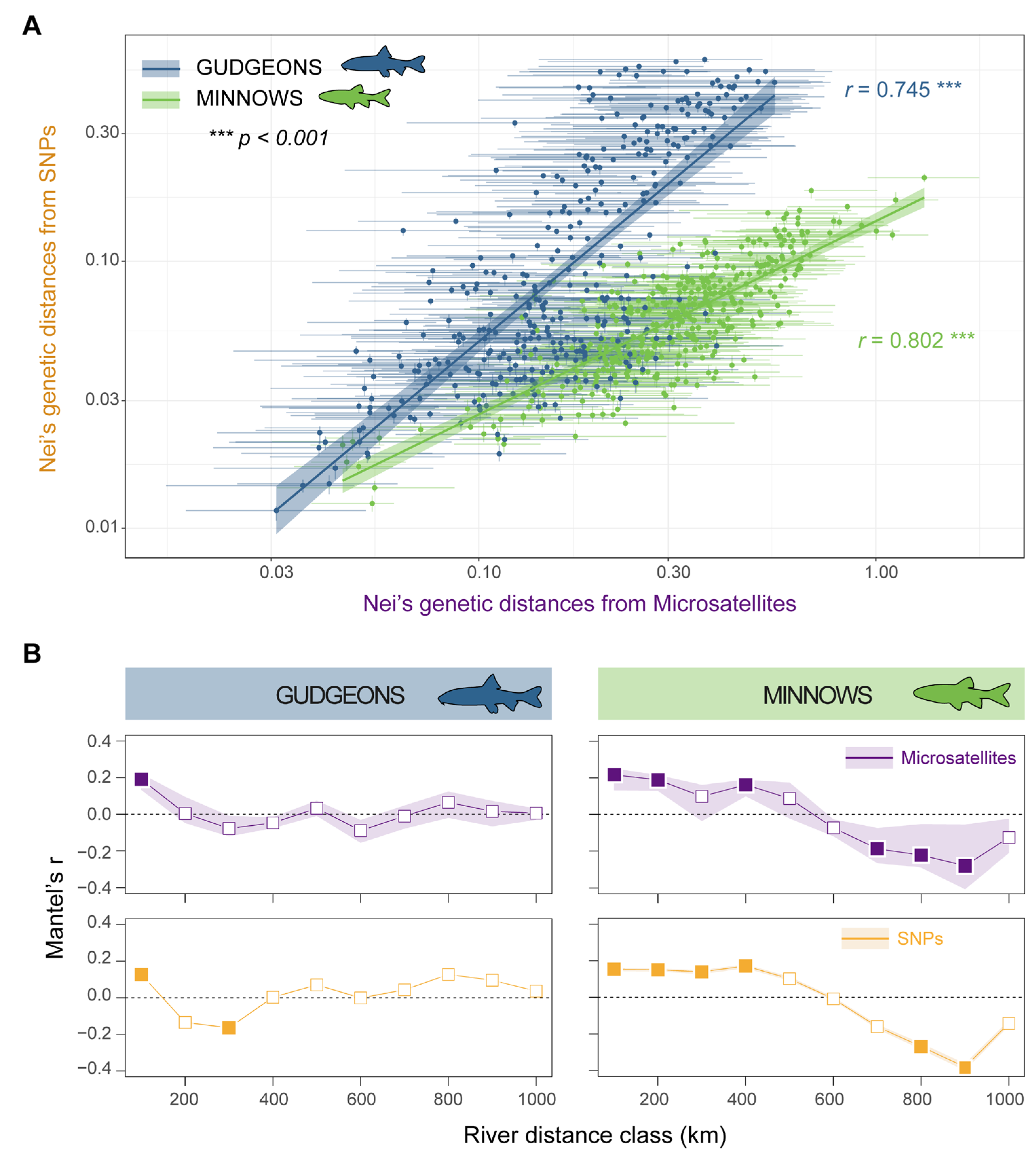
3.2. Genetic Differentiation and Isolation-by-Distance
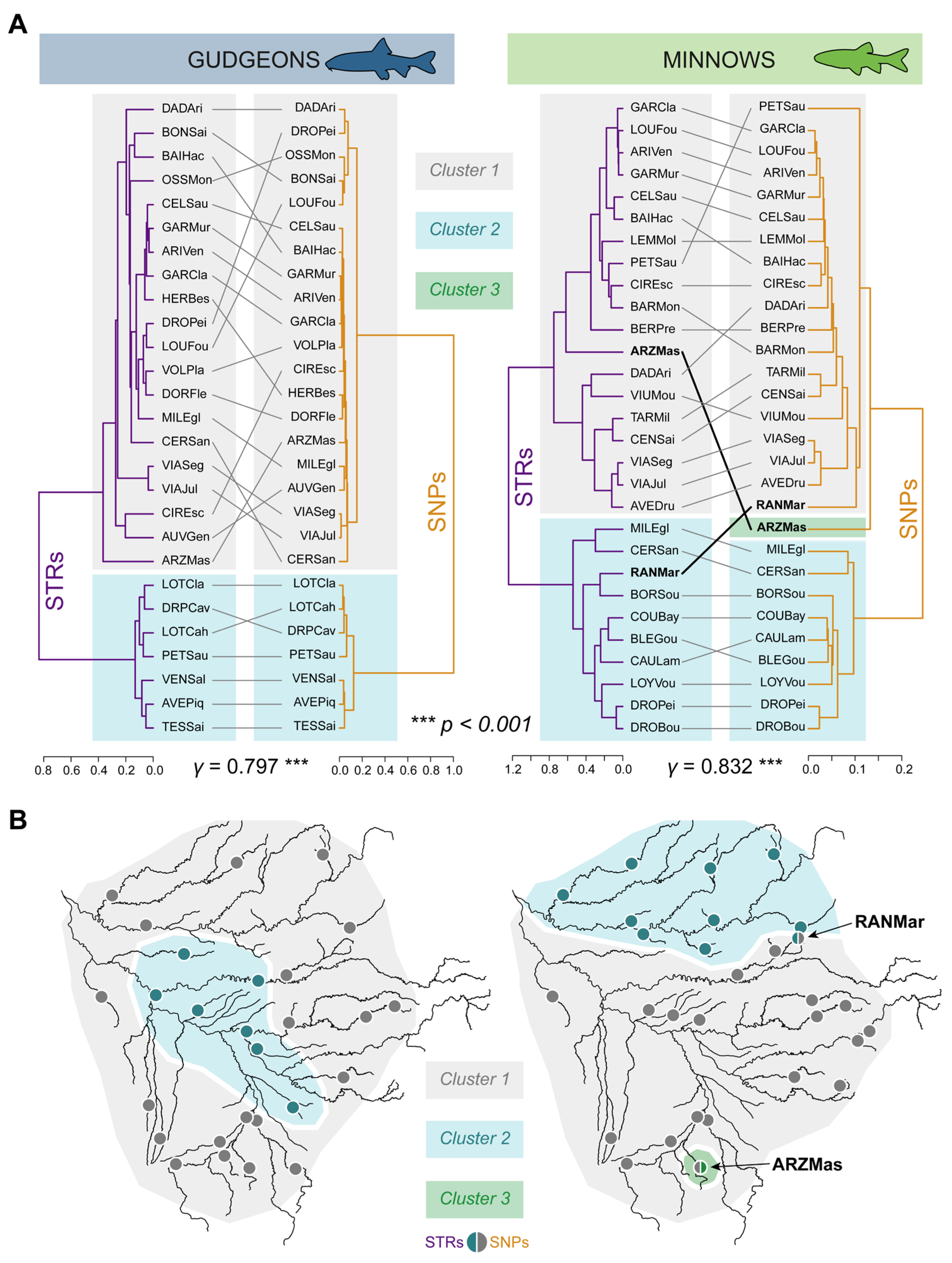
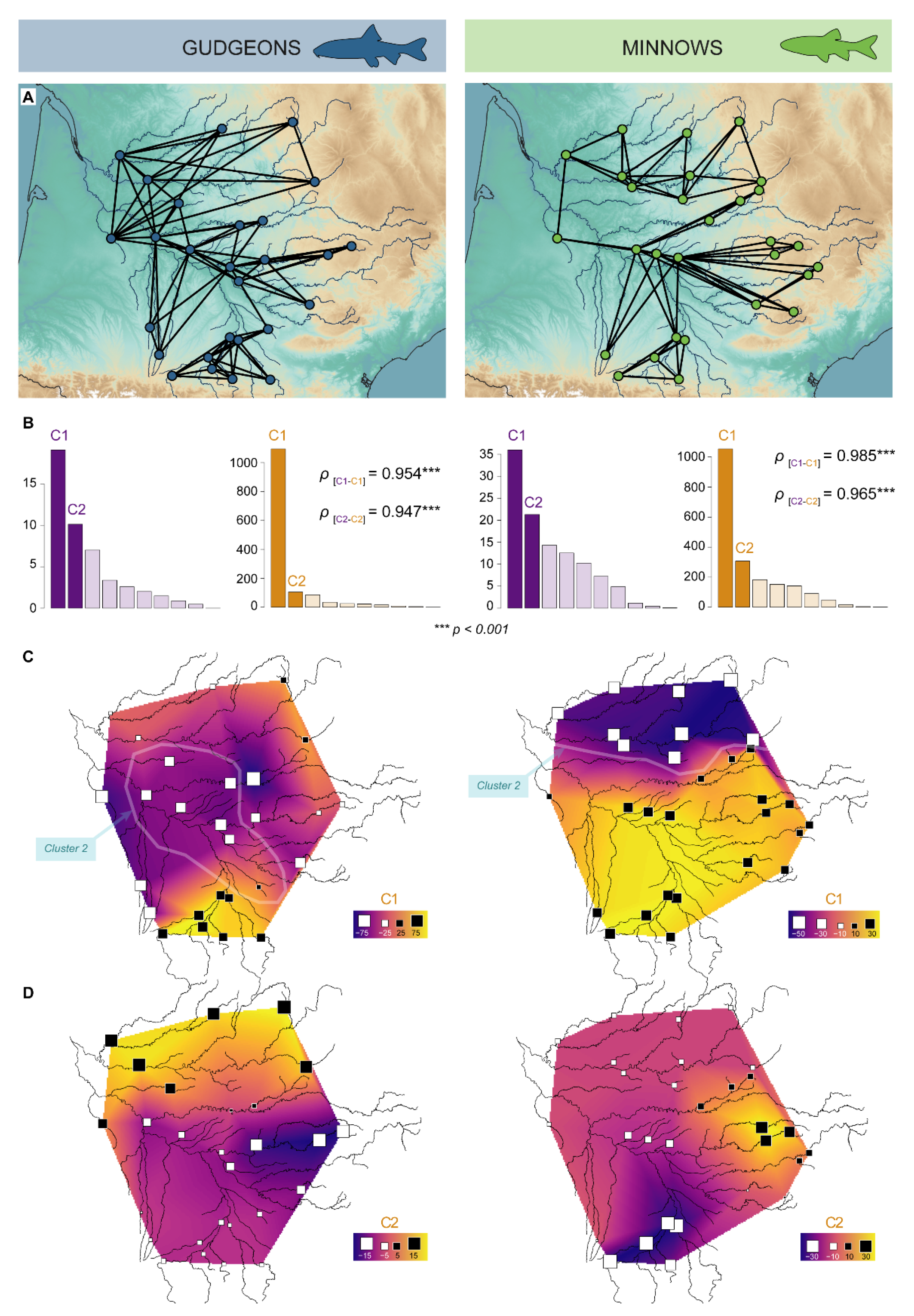
3.3. Genetic Structures
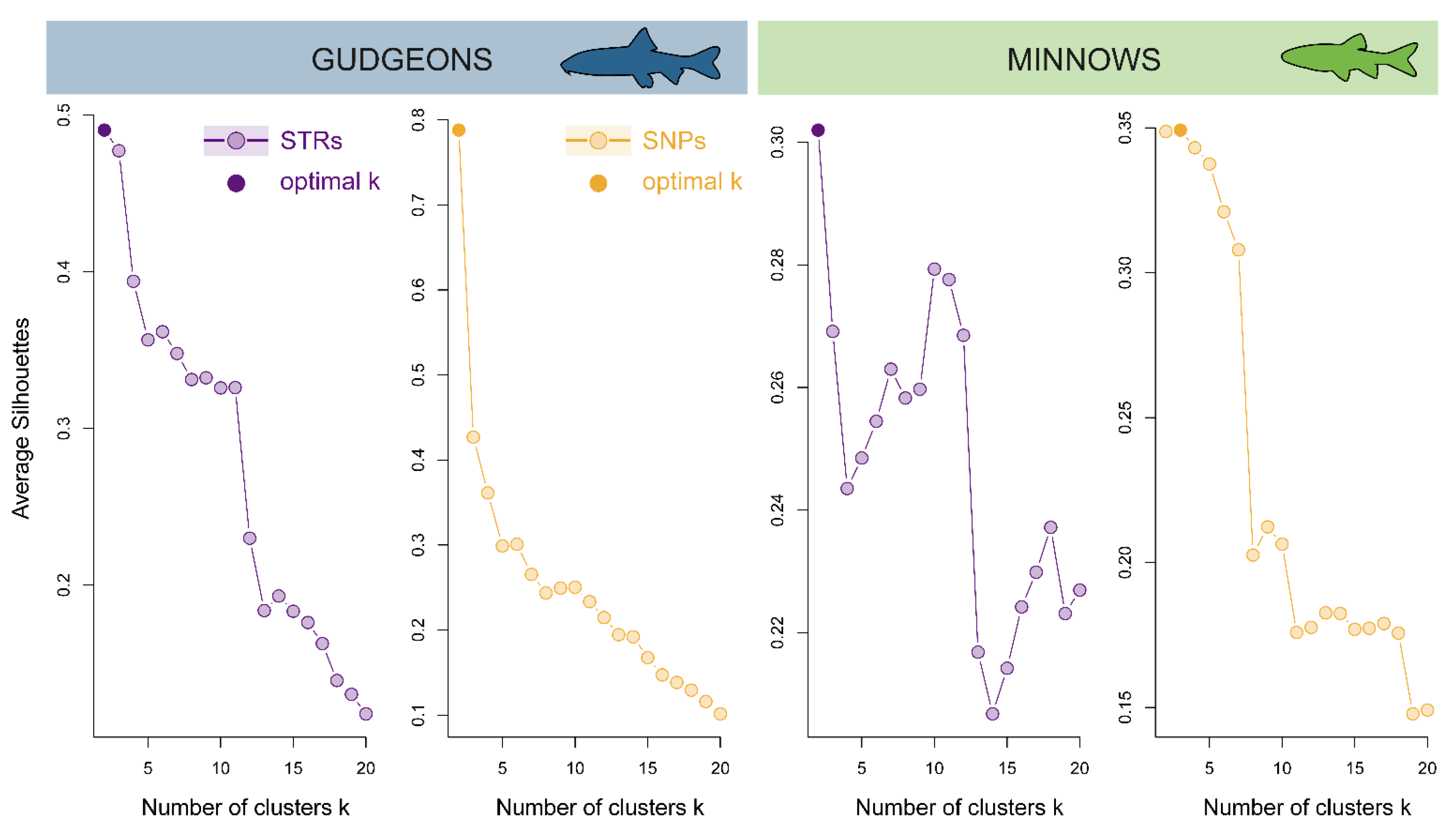
3.3.1. Hierarchical Clustering
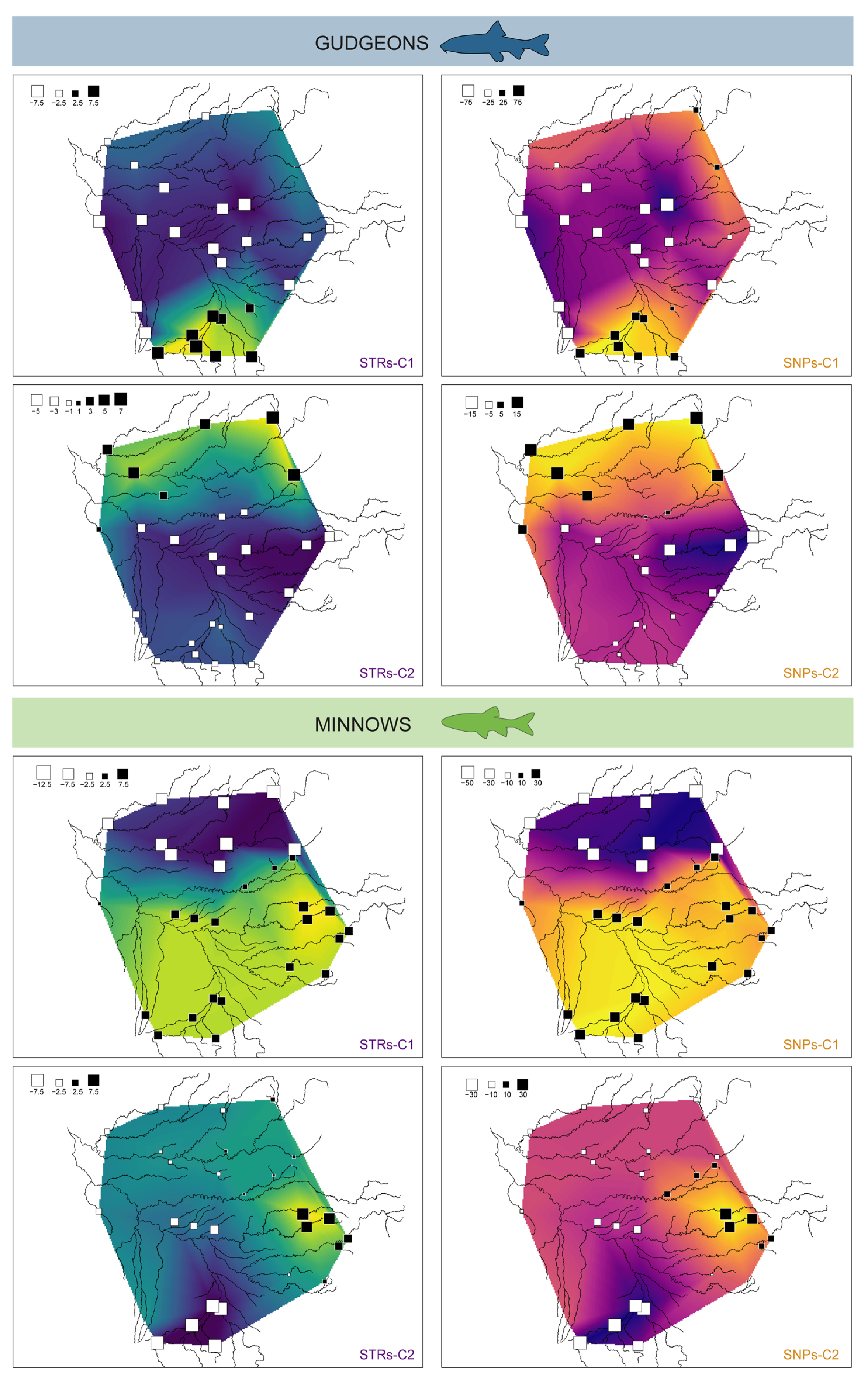
3.3.2. Spatial Principal Component Analyses
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Genome Assembly
| Phoxinus dragarum | Gobio occitaniae | |
| Localization of specimens (Lat. Long.) | 42.958 N 1.085 E | 42.921 N 1.898 E |
| Accession number | JARPMJ000000000 | JARQWZ000000000 |
| Assembly name | CNRS_Phodra_1.0 | CNRS_Gobocc_1.0 |
| Assembly size (Mb) | 968.1 | 1721.8 |
| % missing bases | 0 | 0 |
| % GC | 39.14 | 39.99 |
| Number of contigs | 10,137 | 10,985 |
| Number of contigs > 100 kb | 3100 | 4833 |
| N50 contig length (kb) | 128.19 | 315.98 |
| Shortest contig | 13,920 | 8919 |
| Longest contig | 1,089,874 | 2,163,237 |
| Complete BUSCOs | 3195 (87.8%) | 3394 (93.2%) |
| Complete and single-copy BUSCOs | 3050 (83.8%) | 2666 (73.2%) |
| Complete and duplicated BUSCOs | 145 (4%) | 728 (20.0%) |
| Fragmented BUSCOs | 117 (3.2%) | 86 (2.4%) |
| Missing BUSCOs | 328 (9%) | 160 (4.4%) |
| Total BUSCO groups searched | 3640 (100%) | 3640 (100%) |
Appendix B. Production of SNP Allelic Frequencies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In-Text Reference | Resources | Functions/Scripts | Reference |
|---|---|---|---|
| a | R-adegenet | as.genpop, Hs, dist.genpop, spca | [65] |
| b | R-factoMineR | PCA | [66] |
| c | R-missMDA | imputePCA | [67] |
| d | R-riverdist | riverdistancemat | [68] |
| e | R-stats | cor.test, AIC, hclust | [69] |
| f | R-glmmTMB | glmmTMB | [70] |
| g | R-DHARMa | simulateResiduals | [71] |
| h | R-sjPlot | plot_model | [72] |
| i | R-vegan | mantel | [73] |
| j | R-mpmcorrelogram | mpmcorrelogram | [74] |
| k | R-dendextend | cor_bakers_gamma, untangle, tantelgram, sample.dendrogram | [75] |
| l | R-factoextra | hcut | [76] |
| m | R-cluster | silhouette | [77] |
| n | R-evclust | knn.dist | [78] |
| o | R-interp | interp | [79] |
| p | github-PacificBiosciences | ccs | [58] |
| q | conda-bedtools | [80] | |
| r | github-marl | canu | [81] |
| s | conda-SeqKit | seq, grep | [82] |
| t | conda-purge_haplotigs | hist, cov, purge | [83] |
| u | conda-BUSCO | busco | [84] |



References
- Steffen, W.; Grinevald, J.; Crutzen, P.; McNeill, J. The Anthropocene: Conceptual and historical perspectives. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2011, 369, 842–867. [Google Scholar] [CrossRef] [PubMed]
- Miraldo, A.; Li, S.; Borregaard, M.K.; Flórez-Rodríguez, A.; Gopalakrishnan, S.; Rizvanovic, M.; Wang, Z.; Rahbek, C.; Marske, K.A.; Nogués-Bravo, D. An Anthropocene map of genetic diversity. Science 2016, 353, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Margules, C.R.; Pressey, R.L. Systematic conservation planning. Nature 2000, 405, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Diniz-Filho, J.A.F.; Melo, D.B.; de Oliveira, G.; Collevatti, R.G.; Soares, T.N.; Nabout, J.C.; de Souza Lima, J.; Dobrovolski, R.; Chaves, L.J.; Naves, R.V.; et al. Planning for optimal conservation of geographical genetic variability within species. Conserv. Genet. 2012, 13, 1085–1093. [Google Scholar] [CrossRef]
- Paz-Vinas, I.; Loot, G.; Hermoso, V.; Veyssiere, C.; Poulet, N.; Grenouillet, G.; Blanchet, S. Systematic conservation planning for intraspecific genetic diversity. bioRxiv 2018. bioRxiv:105544. [Google Scholar] [CrossRef]
- Comte, L.; Olden, J.D. Fish dispersal in flowing waters: A synthesis of movement- and genetic-based studies. Fish Fish. 2018, 19, 1063–1077. [Google Scholar] [CrossRef]
- Pertoldi, C.; Bijlsma, R.; Loeschcke, V. Conservation genetics in a globally changing environment: Present problems, paradoxes and future challenges. Biodivers. Conserv. 2007, 16, 4147–4163. [Google Scholar] [CrossRef]
- Sarre, S.D.; Georges, A. Genetics in conservation and wildlife management: A revolution since Caughley. Wildl. Res. 2009, 36, 70. [Google Scholar] [CrossRef]
- Schlötterer, C. The evolution of molecular markers—Just a matter of fashion? Nat. Rev. Genet. 2004, 5, 63–69. [Google Scholar] [CrossRef]
- Hauser, S.S.; Athrey, G.; Leberg, P.L. Waste not, want not: Microsatellites remain an economical and informative technology for conservation genetics. Ecol. Evol. 2021, 11, 15800–15814. [Google Scholar] [CrossRef]
- Putman, A.I.; Carbone, I. Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol. Evol. 2014, 4, 4399–4428. [Google Scholar] [CrossRef]
- Morin, P.A.; Luikart, G.; Wayne, R.K.; the SNP Workshop Group. SNPs in ecology, evolution and conservation. Trends Ecol. Evol. 2004, 19, 208–216. [Google Scholar] [CrossRef]
- Zimmerman, S.J.; Aldridge, C.L.; Oyler-McCance, S.J. An empirical comparison of population genetic analyses using microsatellite and SNP data for a species of conservation concern. BMC Genom. 2020, 21, 382. [Google Scholar] [CrossRef] [PubMed]
- Brumfield, R.T.; Beerli, P.; Nickerson, D.A.; Edwards, S.V. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol. 2003, 18, 249–256. [Google Scholar] [CrossRef]
- Seddon, J.M.; Parker, H.G.; Ostrander, E.A.; Ellegren, H. SNPs in ecological and conservation studies: A test in the Scandinavian wolf population. Mol. Ecol. 2005, 14, 503–511. [Google Scholar] [CrossRef]
- Puckett, E.E. Variability in total project and per sample genotyping costs under varying study designs including with microsatellites or SNPs to answer conservation genetic questions. Conserv. Genet. Resour. 2017, 9, 289–304. [Google Scholar] [CrossRef]
- Schlötterer, C.; Tobler, R.; Kofler, R.; Nolte, V. Sequencing pools of individuals—Mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014, 15, 749–763. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.J.; Teeling, E.C.; Höglund, J. How and why should we implement genomics into conservation? Evol. Appl. 2014, 7, 999–1007. [Google Scholar] [CrossRef]
- Muñoz, I.; Henriques, D.; Jara, L.; Johnston, J.S.; Chávez-Galarza, J.; De La Rúa, P.; Pinto, M.A. SNPs selected by information content outperform randomly selected microsatellite loci for delineating genetic identification and introgression in the endangered dark European honeybee (Apis mellifera mellifera). Mol. Ecol. Resour. 2017, 17, 783–795. [Google Scholar] [CrossRef]
- Dziech, A. Identification of Wolf-Dog Hybrids in Europe—An Overview of Genetic Studies. Front. Ecol. Evol. 2021, 9, 760160. [Google Scholar] [CrossRef]
- Flanagan, S.P.; Jones, A.G. The future of parentage analysis: From microsatellites to SNPs and beyond. Mol. Ecol. 2019, 28, 544–567. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef]
- Shafer, A.B.A.; Wolf, J.B.W.; Alves, P.C.; Bergström, L.; Bruford, M.W.; Brännström, I.; Colling, G.; Dalén, L.; De Meester, L.; Ekblom, R.; et al. Genomics and the challenging translation into conservation practice. Trends Ecol. Evol. 2015, 30, 78–87. [Google Scholar] [CrossRef]
- Theissinger, K.; Fernandes, C.; Formenti, G.; Bista, I.; Berg, P.R.; Bleidorn, C.; Bombarely, A.; Crottini, A.; Gallo, G.R.; Godoy, J.A.; et al. How genomics can help biodiversity conservation. Trends Genet. 2023, S0168952523000203. [Google Scholar] [CrossRef] [PubMed]
- Meybeck, M. Global analysis of river systems: From Earth system controls to Anthropocene syndromes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 1935–1955. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.J.; Carlson, A.K.; Creed, I.F.; Eliason, E.J.; Gell, P.A.; Johnson, P.T.J.; Kidd, K.A.; MacCormack, T.J.; Olden, J.D.; Ormerod, S.J.; et al. Emerging threats and persistent conservation challenges for freshwater biodiversity. Biol. Rev. 2018, 94, 849–873. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.D.; Epps, C.W.; Flitcroft, R.L.; Banks, M.A. Refining and defining riverscape genetics: How rivers influence population genetic structure. Wiley Interdiscip. Rev. Water 2018, 5, e1269. [Google Scholar] [CrossRef]
- Blanchet, S.; Prunier, J.G.; Paz-Vinas, I.; Saint-Pé, K.; Rey, O.; Raffard, A.; Mathieu-Bégné, E.; Loot, G.; Fourtune, L.; Dubut, V. A river runs through it: The causes, consequences, and management of intraspecific diversity in river networks. Evol. Appl. 2020, 13, 1195–1213. [Google Scholar] [CrossRef]
- Morgan, T.D.; Graham, C.F.; McArthur, A.G.; Raphenya, A.R.; Boreham, D.R.; Manzon, R.G.; Wilson, J.Y.; Lance, S.L.; Howland, K.L.; Patrick, P.H.; et al. Genetic population structure of the round whitefish (Prosopium cylindraceum) in North America: Multiple markers reveal glacial refugia and regional subdivision. Can. J. Fish. Aquat. Sci. 2018, 75, 836–849. [Google Scholar] [CrossRef]
- Dufresnes, C.; Dutoit, L.; Brelsford, A.; Goldstein-Witsenburg, F.; Clément, L.; López-Baucells, A.; Palmeirim, J.; Pavlinić, I.; Scaravelli, D.; Ševčík, M.; et al. Inferring genetic structure when there is little: Population genetics versus genomics of the threatened bat Miniopterus schreibersii across Europe. Sci. Rep. 2023, 13, 1523. [Google Scholar] [CrossRef]
- Denys, G.P.J.; Dettai, A.; Persat, H.; Daszkiewicz, P.; Hautecœur, M.; Keith, P. Revision of Phoxinus in France with the description of two new species (Teleostei, Leuciscidae). Cybium 2020, 44, 205–237. [Google Scholar] [CrossRef]
- Kottelat, M.; Persat, H. The genus Gobio in France, with redescription of G. gobio and description of two new species (Teleostei: Cyprinidae). Cybium 2005, 29, 211–234. [Google Scholar]
- Paz-Vinas, I.; Blanchet, S. Dendritic connectivity shapes spatial patterns of genetic diversity: A simulation-based study. J. Evol. Biol. 2015, 28, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Aljanabi, S.M.; Martinez, I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef] [PubMed]
- Prunier, J.G.; Chevalier, M.; Raffard, A.; Loot, G.; Poulet, N.; Blanchet, S. Genetic erosion reduces biomass temporal stability in wild fish populations. bioRxiv 2023. [Google Scholar] [CrossRef]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef]
- Prunier, J.G.; Dubut, V.; Loot, G.; Tudesque, L.; Blanchet, S. The relative contribution of river network structure and anthropogenic stressors to spatial patterns of genetic diversity in two freshwater fishes: A multiple-stressors approach. Freshw. Biol. 2018, 63, 6–21. [Google Scholar] [CrossRef]
- Nei, M. Genetic Distance between Populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Sokal, R.R.; Smouse, P.E.; Neel, J.V. The genetic structure of a tribal population, the Yanomama Indians. XV. Patterns inferred by autocorrelation analysis. Genetics 1986, 114, 259–287. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Dufour, A.B.; Pontier, D. Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity 2008, 101, 92–103. [Google Scholar] [CrossRef]
- Ward, J.H. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Hutchison, D.W.; Templeton, A.R. Correlation of Pairwise Genetic and Geographic Distance Measures: Inferring the Relative Influences of Gene Flow and Drift on the Distribution of Genetic Variability. Evolution 1999, 53, 1898. [Google Scholar] [CrossRef] [PubMed]
- van Strien, M.J.; Holderegger, R.; Van Heck, H.J. Isolation-by-distance in landscapes: Considerations for landscape genetics. Heredity 2015, 114, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Paz-Vinas, I.; Loot, G.; Hermoso, V.; Veyssière, C.; Poulet, N.; Grenouillet, G.; Blanchet, S. Systematic conservation planning for intraspecific genetic diversity. Proc. R. Soc. B Biol. Sci. 2018, 285, 20172746. [Google Scholar] [CrossRef] [PubMed]
- Paetkau, D. Using Genetics to Identify Intraspecific Conservation Units: A Critique of Current Methods. Conserv. Biol. 1999, 13, 1507–1509. [Google Scholar] [CrossRef]
- Finn, D.S.; Bonada, N.; Múrria, C.; Hughes, J.M. Small but mighty: Headwaters are vital to stream network biodiversity at two levels of organization. J. North Am. Benthol. Soc. 2011, 30, 963–980. [Google Scholar] [CrossRef]
- Saint-Pé, K.; Blanchet, S.; Tissot, L.; Poulet, N.; Plasseraud, O.; Loot, G.; Veyssière, C.; Prunier, J.G. Genetic admixture between captive-bred and wild individuals affects patterns of dispersal in a brown trout (Salmo trutta) population. Conserv. Genet. 2018, 19, 1269–1279. [Google Scholar] [CrossRef]
- Diana, M.J.; Wahl, D.H. Growth and Survival of Four Sizes of Stocked Largemouth Bass. North Am. J. Fish. Manag. 2009, 29, 1653–1663. [Google Scholar] [CrossRef]
- Prunier, J.G.; Saint-Pé, K.; Tissot, L.; Poulet, N.; Marselli, G.; Veyssière, C.; Blanchet, S. Captive-bred ancestry affects spatial patterns of genetic diversity and differentiation in brown trout (Salmo trutta) populations. Aquat. Conserv. Mar. Freshw. Ecosyst. 2022, 32, 1529–1543. [Google Scholar] [CrossRef]
- Narum, S.R.; Banks, M.; Beacham, T.D.; Bellinger, M.R.; Campbell, M.R.; Dekoning, J.; Elz, A.; Guthrieiii, C.M.; Kozfkay, C.; Miller, K.M.; et al. Differentiating salmon populations at broad and fine geographical scales with microsatellites and single nucleotide polymorphisms. Mol. Ecol. 2008, 17, 3464–3477. [Google Scholar] [CrossRef]
- Roques, S.; Chancerel, E.; Boury, C.; Pierre, M.; Acolas, M. From microsatellites to single nucleotide polymorphisms for the genetic monitoring of a critically endangered sturgeon. Ecol. Evol. 2019, 9, 7017–7029. [Google Scholar] [CrossRef] [PubMed]
- Saint-Pé, K.; Leitwein, M.; Tissot, L.; Poulet, N.; Guinand, B.; Berrebi, P.; Marselli, G.; Lascaux, J.-M.; Gagnaire, P.-A.; Blanchet, S. Development of a large SNPs resource and a low-density SNP array for brown trout (Salmo trutta) population genetics. BMC Genom. 2019, 20, 582. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, S.J.; Puritz, J.B.; Willis, S.C.; Hollenbeck, C.M.; Portnoy, D.S. These aren’t the loci you’e looking for: Principles of effective SNP filtering for molecular ecologists. Mol. Ecol. 2018, 27, 3193–3206. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Signorovitch, J. Correcting for ascertainment biases when analyzing SNP data: Applications to the estimation of linkage disequilibrium. Theor. Popul. Biol. 2003, 63, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.L.; Jasper, M.; Weeks, A.R.; Hoffmann, A.A. Unbiased population heterozygosity estimates from genome-wide sequence data. Methods Ecol. Evol. 2021, 12, 1888–1898. [Google Scholar] [CrossRef]
- Dokan, K.; Kawamura, S.; Teshima, K.M. Effects of single nucleotide polymorphism ascertainment on population structure inferences. G3 GenesGenomesGenetics 2021, 11, jkab128. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Gregory, T.R. Animal Genome Size Database. 2002. Available online: http://www.genomesize.com (accessed on 1 October 2021).
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Stromberg, M.P.; Marth, G.T. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011, 27, 1691–1692. [Google Scholar] [CrossRef]
- Kofler, R.; Pandey, R.V.; Schlötterer, C. PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 2011, 27, 3435–3436. [Google Scholar] [CrossRef]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Josse, J.; Husson, F. missMDA: A Package for Handling Missing Values in Multivariate Data Analysis. J. Stat. Softw. 2016, 70, 1–31. [Google Scholar] [CrossRef]
- Tyers, M. Riverdist: River Network Distance Computation and Applications; R Package Version 0.14. 0; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing, Vienna, Austria. 2022. Available online: https://www.R-project.org/ (accessed on 17 May 2023).
- Brooks, M.E.; Kristensen, K.; Benthem, K.J.; van Magnusson, A.; Berg, C.W.; Nielsen, A.; Skaug, H.J.; Mächler, M.; Bolker, B.M. glmmTMB Balances Speed and Flexibility Among Packages for Zero-inflated Generalized Linear Mixed Modeling. R J. 2017, 9, 378. [Google Scholar] [CrossRef]
- Hartig, F. DHARMa: Residual Diagnostics for Hierarchical (Multi-Level/Mixed) Regression Models_; R Package Version 0.4.6; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Lüdecke, D. sjPlot: Data Visualization for Statistics in Social Science; R Package Version 2.8.12; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package; R Package Version 25-7; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Matesanz, S.; Gimeno, T.E.; de la Cruz, M.; Escudero, A.; Valladares, F. Competition may explain the fine-scale spatial patterns and genetic structure of two co-occurring plant congeners: Spatial genetic structure of congeneric plants. J. Ecol. 2011, 99, 838–848. [Google Scholar] [CrossRef]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses; R Package Version 1.0.7; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster: Cluster Analysis Basics and Extensions; R Package Version 2.1; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Denoeux, T. Evclust: Evidential Clustering; R Package Version 2.0.2; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Gebhardt, A.; Bivand, R.; Sinclair, D. Interp: Interpolation Methods; R Package Version 1.1-3; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Walenz, B.P.; Rhie, A.; Vollger, M.R.; Logsdon, G.A.; Grothe, R.; Miga, K.H.; Eichler, E.E.; Phillippy, A.M.; Koren, S. HiCanu: Accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 2020, 30, 1291–1305. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]





| Basin | Site | Number of Species | Latitude | Longitude | Sample Sizes | |||
|---|---|---|---|---|---|---|---|---|
| P. dragarum | G. occitaniae | |||||||
| STRs | SNPs | STRs | SNPs | |||||
| Dordogne | AUVGen | 1 | 45.3439517 | 1.1738551 | 24 | 24 | ||
| BLEGou | 1 | 44.7062117 | 1.3764714 | 29 | 29 | |||
| BORSou | 1 | 44.9207676 | 1.4612556 | 30 | 30 | |||
| CAULam | 1 | 44.8990195 | 0.6001853 | 30 | 30 | |||
| CERSan | 2 | 44.8769731 | 2.3688147 | 30 | 30 | 30 | 28 | |
| COUBay | 1 | 44.8047176 | 0.7292281 | 30 | 30 | |||
| DORFle | 1 | 44.8624623 | 0.2432444 | 30 | 25 | |||
| DROBou | 1 | 45.3229357 | 0.5851939 | 30 | 30 | |||
| DROPei | 2 | 45.0745045 | −0.121676 | 30 | 30 | 30 | 30 | |
| LOUFou | 2 | 43.2743574 | 1.0686578 | 30 | 30 | 30 | 30 | |
| MILEgl | 2 | 45.4151425 | 2.0796179 | 29 | 30 | 30 | 30 | |
| Garonne | ARIVen | 2 | 43.4371547 | 1.4376488 | 30 | 30 | 24 | 24 |
| ARZMas | 2 | 43.0843932 | 1.3737039 | 30 | 30 | 30 | 30 | |
| AVEDru | 1 | 44.3367647 | 2.4914351 | 30 | 30 | |||
| AVEPiq | 1 | 44.0968569 | 1.3163485 | 29 | 29 | |||
| BAIHac | 2 | 43.2859682 | 0.4610215 | 30 | 30 | 30 | 30 | |
| BARMon | 1 | 44.2097195 | 1.0612774 | 29 | 30 | |||
| BERPre | 1 | 44.6998674 | 2.1039632 | 30 | 30 | |||
| BONSai | 1 | 44.1671669 | 1.7498205 | 26 | 26 | |||
| CELSau | 2 | 44.5194144 | 1.7162116 | 29 | 30 | 30 | 30 | |
| CENSai | 1 | 44.0367039 | 2.9641243 | 30 | 30 | |||
| CIREsc | 2 | 44.3196088 | −0.1896798 | 30 | 30 | 30 | 30 | |
| DADAri | 2 | 43.766423 | 2.3169348 | 29 | 29 | 30 | 30 | |
| DRPCav | 1 | 44.6590784 | 0.6481635 | 30 | 30 | |||
| GARCla | 2 | 43.0997996 | 0.6294647 | 30 | 30 | 28 | 28 | |
| GARMur | 2 | 43.4601354 | 1.3313024 | 30 | 30 | 30 | 30 | |
| HERBes | 1 | 43.0842176 | 1.8400499 | 30 | 25 | |||
| LEMMol | 1 | 44.1795074 | 1.3338616 | 30 | 30 | |||
| LOTCah | 1 | 44.4740653 | 1.4252254 | 30 | 30 | |||
| LOTCla | 1 | 44.3472466 | 0.369653 | 30 | 29 | |||
| LOYVou | 1 | 45.3037878 | 1.4134422 | 30 | 30 | |||
| OSSMon | 1 | 43.5300669 | 0.335614 | 30 | 30 | |||
| PETSau | 2 | 44.2439564 | 0.8077916 | 28 | 28 | 30 | 30 | |
| RANMar | 1 | 44.7966506 | 2.3406886 | 30 | 29 | |||
| TARMil | 1 | 44.1082554 | 3.085726 | 30 | 25 | |||
| TESSai | 1 | 43.9686527 | 1.4284642 | 30 | 30 | |||
| VENSal | 1 | 43.5395467 | 1.8041663 | 30 | 30 | |||
| VIAJul | 2 | 44.2170222 | 2.5434064 | 28 | 30 | 30 | 30 | |
| VIASeg | 2 | 44.2967126 | 2.8388392 | 30 | 30 | 30 | 30 | |
| VIUMou | 1 | 43.7039452 | 2.7827139 | 30 | 30 | |||
| VOLPla | 1 | 43.1711731 | 1.1186909 | 30 | 30 | |||
| Estimate | 2.5% | 97.5% | p-Value | ||
|---|---|---|---|---|---|
| Intercept (STRs in gudgeons) | 0.634 | 0.617 | 0.652 | 4947.91 | <0.0001 |
| SNP | −0.410 | −0.428 | −0.391 | 1856.42 | <0.0001 |
| Minnows | 0.044 | 0.024 | 0.064 | 18.35 | <0.0001 |
| UDG | 0.017 | 0.010 | 0.24 | 23.94 | <0.0001 |
| UDG² | −0.003 | −0.005 | −0.0001 | 4.22 | 0.0399 |
| SNP: Minnows | −0.049 | −0.075 | −0.023 | 13.72 | 0.0002 |
| Random effect | 0.026 | 0.018 | 0.037 | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prunier, J.G.; Veyssière, C.; Loot, G.; Blanchet, S. Comparing the Utility of Microsatellites and Single Nucleotide Polymorphisms in Conservation Genetics: Insights from a Study on Two Freshwater Fish Species in France. Diversity 2023, 15, 681. https://doi.org/10.3390/d15050681
Prunier JG, Veyssière C, Loot G, Blanchet S. Comparing the Utility of Microsatellites and Single Nucleotide Polymorphisms in Conservation Genetics: Insights from a Study on Two Freshwater Fish Species in France. Diversity. 2023; 15(5):681. https://doi.org/10.3390/d15050681
Chicago/Turabian StylePrunier, Jérôme G., Charlotte Veyssière, Géraldine Loot, and Simon Blanchet. 2023. "Comparing the Utility of Microsatellites and Single Nucleotide Polymorphisms in Conservation Genetics: Insights from a Study on Two Freshwater Fish Species in France" Diversity 15, no. 5: 681. https://doi.org/10.3390/d15050681
APA StylePrunier, J. G., Veyssière, C., Loot, G., & Blanchet, S. (2023). Comparing the Utility of Microsatellites and Single Nucleotide Polymorphisms in Conservation Genetics: Insights from a Study on Two Freshwater Fish Species in France. Diversity, 15(5), 681. https://doi.org/10.3390/d15050681