Genome-Wide Loss of Diversity in the Critically Endangered Hawaiian Monk Seal

Abstract
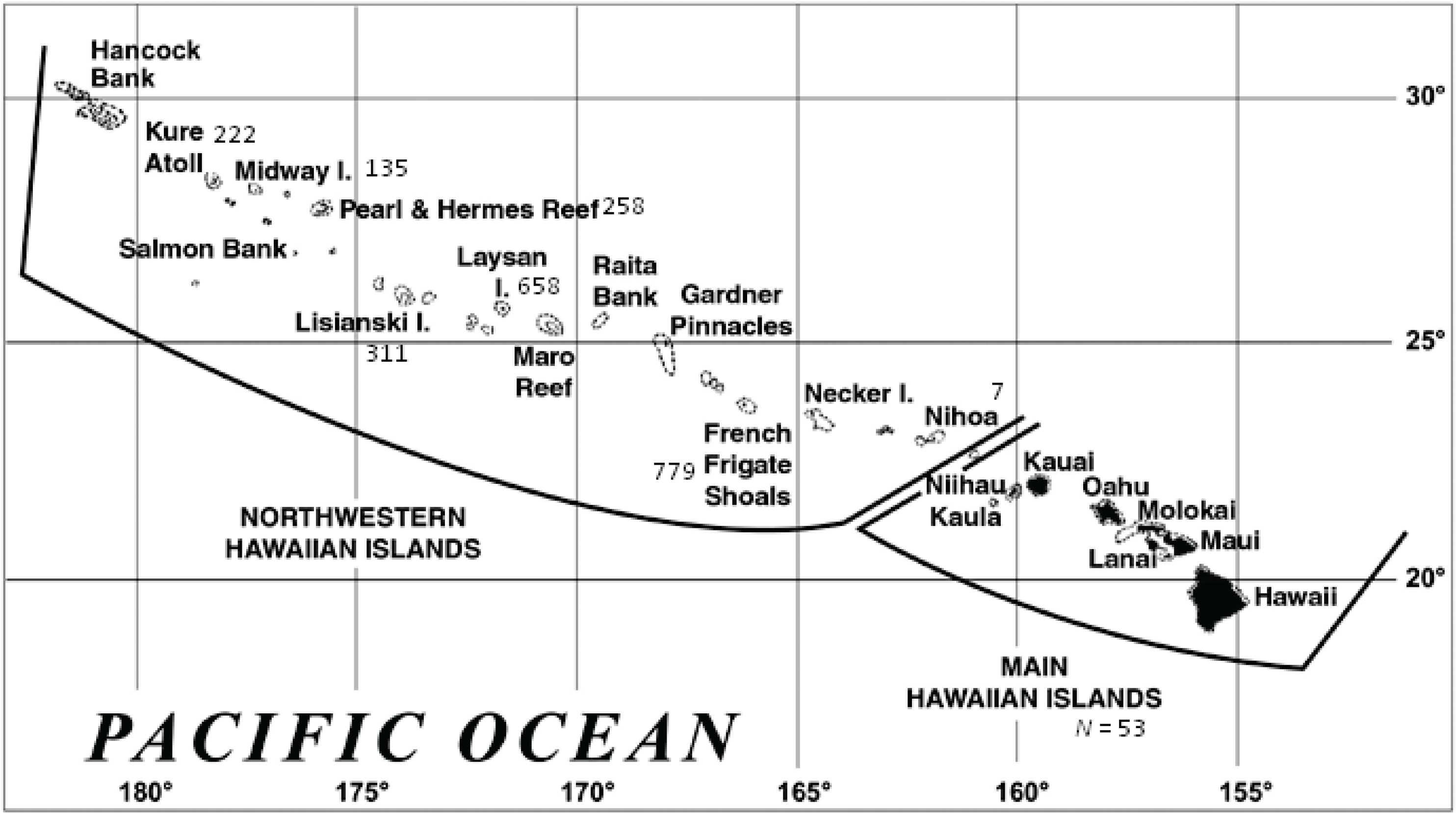
:1. Introduction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Site | Vessel | Event | Seals taken |
|---|---|---|---|---|
| 1805 | Lisianski | Neva | Tour | 4 |
| 182? | NWHI | General Gates | Sealing | “all” |
| 1842 | Kure | Parker | Wrecked | ~60 |
| 1850 | Lisianski | Holder Borden | Wrecked | ? |
| 1850 | Pearl Hermes Reef | Rodolf | Tour | 10–12 |
| 1859 | French Frigate Shoals | Gambia | Sealing | 150 |
| 1859 | NWHI | Gambia | Sealing | 1,500 |
| 1870 | Midway, Kure | Saginaw | Wrecked | > 60 |
| 1886 | Laysan | General Siegel | Shark fishing | ? |
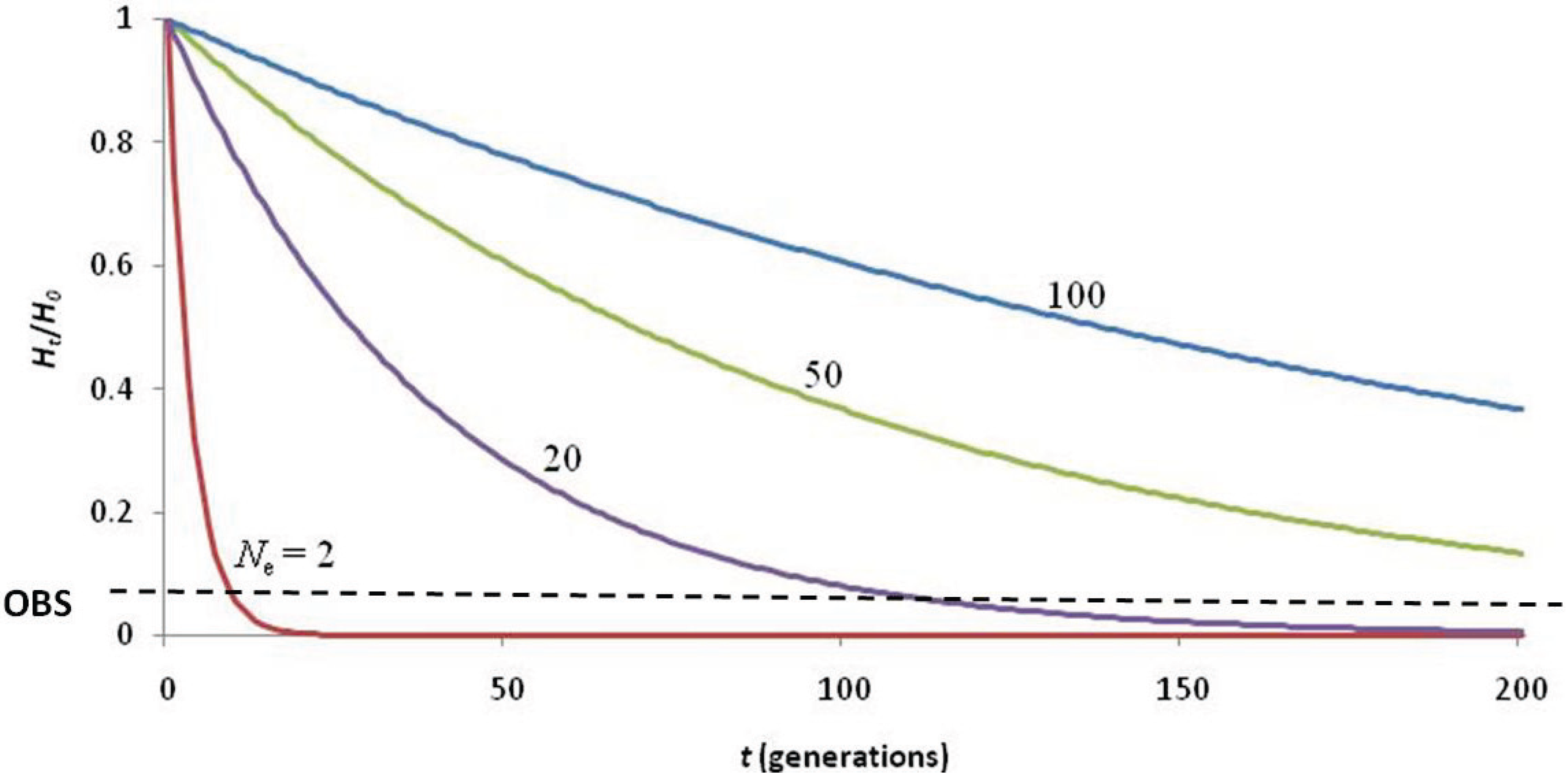
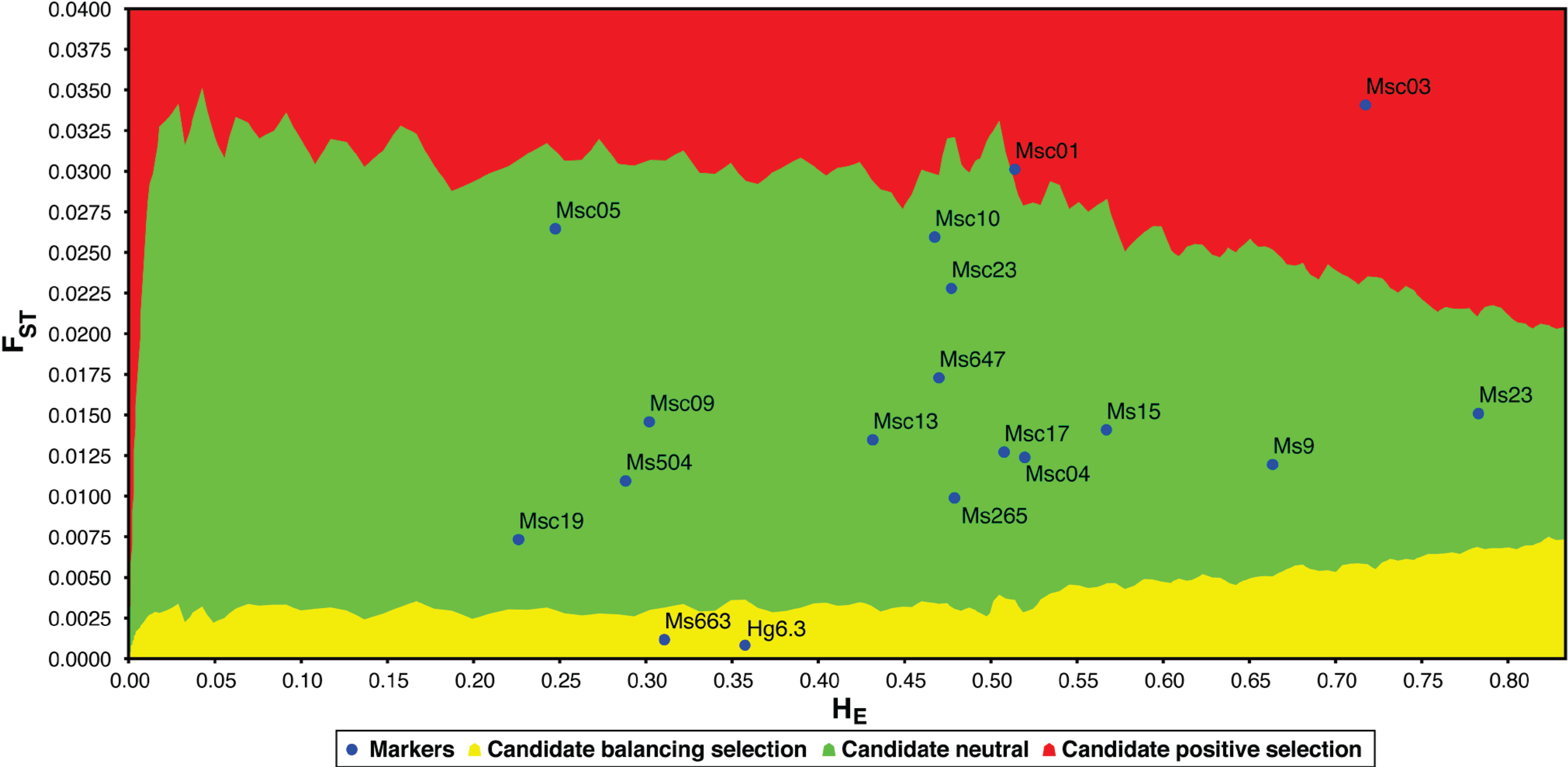
2. Results and Discussion

| Locus | Reference | Genbank # | Repeat motif | Primers | Size | HO | HE | A | DH /sd | CI | P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Msc01 | This study | GU206362 | (AC)18 | F: ATTTTAATTATGGGTTACTTTGAACC R: TCACCATTTAATGCATATGAGC | 161–167 | 0.47 | 0.49 | 3 | 0.13 | 0.004–0.033 | 0.962 |
| Msc03 | This study | GU206363 | (TG)21 | F: TGGTCTTTCTTAAGGCCAAG R: ATATGGAAGCAGCCCAAGTG | 126–136 | 0.70 | 0.71 | 5 | 1.14 | 0.007–0.025 | 0.999 |
| Msc04 | This study | GU206364 | (TATC)14 | F: CTTTAGTTTCCGGTGTTCAGTG R: CTCAGGGTTGTGAGTTCAAGC | 159–167 | 0.52 | 0.52 | 4 | 0.29 | 0.005–0.029 | 0.445 |
| Msc05 | This study | GU206365 | (GATA)11 | F: TGGTCTCAAGTTGGAGGATTG R: AAGCCATTGAGTGTGATGGAC | 198– 206 | 0.27 | 0.27 | 3 | −0.41 | 0.004–0.034 | 0.922 |
| Msc09 | This study | GU206366 | (CTAT)13 | F: GCCTGATTTGCCTCTTCTTC R: GCGTCAGAAAGACACAGGAG | 208–216 | 0.28 | 0.29 | 3 | −1.26 | 0.004–0.032 | 0.579 |
| Msc10 | This study | GU206367 | (TATC)13 | F: CCTCCATCGCTACCATCTTC R: TGAACGCAAGTGGATGAGTC | 141–149 | 0.47 | 0.48 | 3 | 0.07 | 0.004–0.033 | 0.903 |
| Msc13 | This study | GU206368 | (CA)20 | F: CACCTTTGGCTTCCAGTGTC R: ATTCGGTGGTGGCTTTTATG | 194–202 | 0.40 | 0.40 | 3 | −0.48 | 0.004–0.031 | 0.488 |
| Msc17 | This study | GU206370 | (GT)24 | F: GCAAGGAGAAGCTACAGAAGG R: CCCTGCTAACTGGTTTCTGC | 115– 119 | 0.50 | 0.51 | 3 | 0.93 | 0.003–0.036 | 0.483 |
| Msc19 | This study | GU206371 | (CTAT)11 | F: GGCTATTGGCCAACTGGTAG R: TTGGCCTGCTCCAATAAGAC | 118–138 | 0.24 | 0.24 | 4 | −3.68 | 0.004–0.035 | 0.166 |
| Msc23 | This study | GU206372 | (GATA)2 (GAT) (GATA)2 (GAT) (GATA)15 | F: GCTTCTCTGTTTCTATCTCAAATAAAT R: CTCCTTCCTGGCTGCTTATG | 160–168 | 0.48 | 0.50 | 3 | 0.14 | 0.004–0.031 | 0.841 |
| Ms9 | Schultz et al. 2009 | EU913766 | (GAAA)18 | F: CCAAAGCCTATTTCTTTCAATCC R: AGCAGAGGCCCTAAGACAGG | 297–317 | 0.67 | 0.68 | 6 | 0.42 | 0.006–0.027 | 0.334 |
| Ms15 | Schultz et al. 2009 | EU913767 | (CCTT)6 CCCT (CCTT)6 | F: CTGAATTCATGCTGTATCTTGG R: GTGCTTGGGACATGATGG | 203–315 | 0.54 | 0.56 | 3 | 1.27 | 0.006–0.030 | 0.514 |
| Ms23 | Schultz et al. 2009 | EU913768 | (GAAA)9 GGAA (GAAA)8 | F: CGCTTAGTGTGGAGTCACTTAGG R: GTGAGATGAATGCCCTTTGG | 340–370 | 0.76 | 0.78 | 9 | 0.43 | 0.008–0.024 | 0.584 |
| Ms265 | Schultz et al. 2009 | EU913769 | (GT)13 | F: GACTGGTAATTTACGCCCTACC R: AAGTGTTGGGTTGAAAATTGG | 158,162 | 0.50 | 0.49 | 2 | 0.80 | 0.004–0.032 | 0.311 |
| Ms504 | Schultz et al. 2009 | EU913763 | (AAG)24 | F: ATCAGCTATCAGGGGTAGGG R: GTCATTCCCTAGTGGTAAAGACTC | 308,326 | 0.28 | 0.29 | 2 | 0.94 | 0.004–0.033 | 0.394 |
| Ms647 | Schultz et al. 2009 | EU913765 | (TG)14 | F: GAACTCCAAACAGCCATTCC R: CCTGCTCCTTCTTTCTGATCC | 115,117 | 0.44 | 0.46 | 2 | 1.97 | 0.004–0.033 | 0.665 |
| Ms663 | Schultz et al. 2009 | EU913764 | (TC)11 | F: TCAACTTCTCAATTTAGGATTCACA R: GCAAAAAGGGATGAGCCATA | 290,294 | 0.31 | 0.31 | 2 | 1.13 | 0.004–0.033 | 0.002 |
| Hg6.3 | Allen et al. 1995 | G02092 | (GT)18 | F: CAGGGGACCTGAGTGCTTATG R: GACCCAGCATCAGAACTCAAG | 227,237 | 0.36 | 0.36 | 2 | 1.43 | 0.004–0.031 | 0.000006 |


3. Experimental Section
4. Conclusions
Acknowledgments
References
- Frankham, R. Conservation genetics. Annu. Rev. Genet. 1995, 29, 305–327. [Google Scholar] [CrossRef]
- Spielman, D.; Brook, B.W.; Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA 2004, 101, 15261–15264. [Google Scholar] [CrossRef]
- Garner, A.; Rachlow, J.L.; Hicks, J.F. Patterns of genetic diversity and its loss in mammalian populations. Conserv. Biol. 2005, 19, 1215–1221. [Google Scholar] [CrossRef]
- Evans, S.R.; Sheldon, B.C. Interspecific patterns of genetic diversity in birds: correlations with extinction risk. Conserv. Biol. 2008, 22, 1016–1025. [Google Scholar] [CrossRef]
- Frankham, R.; Lees, K.; Montgomery, M.E.; England, P.R.; Lowe, E.H.; Briscoe, D.A. Do population size bottlenecks reduce evolutionary potential? Anim. Conserv. 1999, 2, 255–260. [Google Scholar]
- Frankham, R. Genetics and conservation biology. Comptes Rendus Biologies 2003, 326, 22–29. [Google Scholar] [CrossRef]
- Gaggiotti, O.E. Genetic threats to population persistence. Ann. Zool. Fennici. 2003, 40, 155–168. [Google Scholar]
- Shaffer, M.L. Minimum population sizes for species conservation. BioScience 1981, 31, 131–134. [Google Scholar] [CrossRef]
- Jackson, J.B.; Kirby, M.X.; Berger, W.H.; Bjorndal, K.A.; Botsford, L.W.; Bourque, B.J.; Bradbury, R.H.; Cooke, R.; Erlandson, J.; Estes, J.A.; Hughes, T.P.; Kidwell, S.; Lange, C.B.; Lenihan, H.S.; Pandolfi, J.M.; Peterson, C.H.; Steneck, R.S.; Tegner, M.J.; Warner, R.R. Historical overfishing and the recent collapse of coastal ecosystems. Science 2001, 293, 629–637. [Google Scholar] [CrossRef]
- Frankham, R. Relationship of genetic variation to population size in wildlife. Conserv. Biol. 1996, 10, 1500–1508. [Google Scholar]
- Nei, M.; Maruyama, T.; Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution 1975, 29, 1–10. [Google Scholar]
- Watterson, G.A. Allele frequencies after a bottleneck. Theor. Popul. Biol. 1984, 26, 387–407. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar]
- Amos, W.; Harwood, J. Factors affecting levels of genetic diversity in natural populations. Philos. Trans. R. Soc. London, Ser. B 1998, 353, 177–186. [Google Scholar] [CrossRef]
- Smith, J.M.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974, 23, 23–35. [Google Scholar] [CrossRef]
- Cavalli-Sforza, L.L. Population structure and human evolution. Proc. R. Soc. London, Ser. B 1966, 164, 362–379. [Google Scholar] [CrossRef]
- Aldridge, B.M.; Bowen, L.; Smith, B.R.; Antonelis, G.A.; Gulland, F.; Stott, J.L. Paucity of class I MHC gene heterogeneity between individuals in the endangered Hawaiian monk seal population. Immunogenetics 2006, 58, 203–215. [Google Scholar] [CrossRef]
- Zinkernagel, R.M. Associations between major histocompatibility antigens and susceptibility to disease. Annu. Rev. Microbiol. 1979, 33, 201–213. [Google Scholar] [CrossRef]
- Hedrick, P.W. Pathogen resistance and genetic variation at MHC loci. Evolution 2002, 56, 1902–1908. [Google Scholar] [CrossRef]
- Kretzmann, M.B.; Gilmartin, W.G.; Meyer, A.; Zegers, G.P.; Fain, R.; Taylor, B.F.; Costa, D.P. Low genetic variability in the Hawaiian monk seal. Conserv. Biol. 1997, 11, 482–490. [Google Scholar]
- Gemmell, N.J.; Allen, P.J.; Goodman, S.J.; Reed, J.Z. Interspecific microsatellite markers for the study of pinniped populations. Mol. Ecol. 1997, 6, 661–666. [Google Scholar]
- Schultz, J.K.; Baker, J.D.; Toonen, R.J.; Bowen, B.W. Extremely low genetic diversity in the endangered Hawaiian monk seal (Monachus schauinslandi). J. Hered. 2009, 100, 25–33. [Google Scholar]
- Fyler, C.A.; Reeder, T.W.; Berta, A.; Antonelis, G.; Aguilar, A.; Androukaki, E. Historical biogeography and phylogeny of monachine seals (Pinnipedia: Phocidae) based on mitochondrial and nuclear DNA data. J. Biogeog. 2005, 32, 1267–1279. [Google Scholar] [CrossRef]
- Arnason, U.; Gullberg, A.; Janke, A.; Kullberg, M.; Lehman, N.; Petrov, E.A.; Vainola, R. Pinniped phylogeny and a new hypothesis for their origin and dispersal. Mol. Phyl. Evol. 2006, 41, 345–354. [Google Scholar] [CrossRef]
- Briggs, J.C. Operation of zoogeographic barriers. Syst. Zool. 1974, 23, 248–256. [Google Scholar] [CrossRef]
- Stannard, D.E. Before the Horror: the Population of Hawai'i on the Eve of Western Contact; University of Hawaii Press: Honolulu, HI, USA, 1989. [Google Scholar]
- Svihla, A. Notes on the Hawaiian monk seal. J. Mammal. 1959, 40, 226–229. [Google Scholar] [CrossRef]
- Liliuokalani, Q. The Kumulipo: an Account of the Creation of the World according to Hawaiian Tradition; Lee and Shepard: Boston, MA, USA, 1897. [Google Scholar]
- Beckwith, M.W. The Kumulipo: a Hawaiian Creation Chant; University of Hawaii Press: Honolulu, HI, USA, 1981. [Google Scholar]
- Rosendahl, P.H. Aboriginal Hawaiian structural remains and settlement patterns in the upland archeological zone at Lapakahi, Island of Hawaii. Hawaiian Archaeol. 1994, 3, 14–70. [Google Scholar]
- Ragen, T. Human activities affecting the population trends of the Hawaiian monk seal. Am. Fish. Soc. Symp. 1999, 23, 183–194. [Google Scholar]
- Baker, J.D.; Johanos, T.C. Abundance of the Hawaiian monk seal in the main Hawaiian Islands. Biol. Conserv. 2004, 116, 103–110. [Google Scholar] [CrossRef]
- Lisiansky, U. A Voyage around the World in the Years 1803–1806 in the Ship Neva; John Booth: London, UK, 1814. [Google Scholar]
- Cobb, J.N. Commercial Fisheries of the Hawaii Islands; US Commission of Fish and Fisheries: Honolulu, HI, USA, 1902. [Google Scholar]
- Carretta, J.V.; Forney, K.A.; Lowry, M.S.; Barlow, J.; Baker, J.; Johnston, D.; Hanson, B.; Brownell, R.L.; Robbins, J.; Mattila, D.K.; Ralls, K.; Muto, M.M.; Lynch, D.; Carswell, L. Pacific Marine Mammal Stock Assessments: 2009; NOAA Technical Memorandum NMFS-SWFSC-414: LaJolla, CA, USA, 2009. [Google Scholar]
- Allen, P.J.; Amos, W.; Pomeroy, P.P.; Twiss, S.D. Microsatellite variation in grey seals (Halichoerus grypus) shows evidence of genetic differentiation between two British breeding colonies. Mol. Ecol. 1995, 4, 653–662. [Google Scholar] [CrossRef]
- Morrell, B. A Narrative of Four Voyages to the South Sea, North and South Pacific Ocean, Chinese Sea, Ethiopic and Southern Atlantic Ocean, Indian, and Antarctic Ocean, from the Year 1822 to 1831; J. & J. Harper: New York, NY, USA, 1832; pp. 215–219. [Google Scholar]
- Paty, J. Arrival of the Manuokawai—interesting account of her explorations. The Polynesian 1897, 388, 838–839. [Google Scholar]
- Brooks, N.C. Islands and reefs west-north-west of the Sandwich Islands, Pacific. Nautical Magazine 1860, 29, 499–504. [Google Scholar]
- Hiruki, L.M.; Ragen, T.J. A Compilation of Historical (Monachus Schauinslandi) Monk Seal Counts; NOAA Technical Memorandum, US Department of Commerce, 1992.
- Pastor, T.; Garza, J.C.; Allen, P.; Amos, W.; Aguilar, A. Low genetic variability in the highly endangered Mediterranean monk seal. J. Hered. 2004, 95, 291–300. [Google Scholar] [CrossRef]
- Kenyon, K.W.; Rice, D.W. Life history of the Hawaiian monk seal. Pac. Sci. 1959, 13, 215–252. [Google Scholar]
- Dill, H.R. Report of an expedition to Laysan Island in 1911. In Biological Survey Bulletin; US Department of Agriculture: Washington, DC, USA, 1912; Volume 42, p. 9. [Google Scholar]
- Franklin, I.R. Evolutionary change in small populations. In Conservation Biology: An Evolutionary-Ecological Perspective; Soule, M.E., Wilcox, B.A., Eds.; Sinauer Associates: Sunderland, MA, USA, 1980; pp. 135–150. [Google Scholar]
- Franklin, I.R.; Frankham, R. How large must populations be to retain evolutionary potential? Anim. Conserv. 1998, 1, 69–71. [Google Scholar] [CrossRef]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja-Pereira, A.; Beja-pereira, A.; Luikart, G. LOSITAN: workbench to detect molecular adaptation based on a FST-outlier method. BMC Bioinformatic 2008, 9, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Bean, K.; Amos, W.; Pomeroy, P.P.; Twiss, S.D.; Coulson, T.N.; Boyd, I.L. Patterns of parental relatedness and pup survival in the grey seal (Halichoerus grypus). Mol. Ecol. 2004, 13, 2365–2370. [Google Scholar] [CrossRef]
- Kretzmann, M.; Mentzer, L.; DiGiovanni, R.; Leslie, M.S.; Amato, G. Microsatellite diversity and fitness in stranded juvenile harp seals (Phoca groenlandica). J. Hered. 2006, 97, 555–560. [Google Scholar] [CrossRef]
- Baker, J.D.; Thompson, P.M. Temporal and spatial variation in age-specific survival rates of a long-lived mammal, the Hawaiian monk seal. Proc. R. Soc. London Ser. B 2007, 274, 407–415. [Google Scholar] [CrossRef]
- Glenn, T.C.; Schable, N.A. Isolating microsatellite DNA loci. Methods Enzymol. 2005, 395, 202–222. [Google Scholar] [CrossRef]
- Armour, J.A.; Neumann, R.; Gobert, S.; Jeffreys, A.J. Isolation of human simple repeat loci by hybridization selection. Hum. Mol. Genet. 1994, 3, 599–605. [Google Scholar] [CrossRef]
- Gautschi, B.; Tenzer, I.; Mueller, J.P.; Schmid, B. Isolation and characterization of microsatellite loci in the bearded vulture (Gypaetus barbatus) and cross amplification in three Old World vulture species. Mol. Ecol. 2000, 9, 2193–2195. [Google Scholar] [CrossRef]
- Gautschi, B.; Widmer, A.; Koella, J. Isolation and characterization of microsatellite loci in the dice snake (Natrix tessellata). Mol. Ecol. 2000, 9, 2191–2193. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.; Shipley, P. Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (version 1.2): a computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar]
- Marshall, A.J.; Schultz, J.K.; Kennedy, M.A.; Slate, J.; Gemmell, N.J. Development of a predicted microsatellite map of the pinniped genome, with wider applicability to the Carnivora. Mol. Ecol. Res. 2010. in review. [Google Scholar]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open 3.0. 1996–2004.
- Dawson, D.A.; Burke, T.; Hansson, B.; Pandhal, J.; Hale, M.C.; Hinten, G.N.; Slate, J. A predicted microsatellite map of the passerine genome based on chicken-passerine sequence similarity. Mol. Ecol. 2006, 15, 1299–1320. [Google Scholar] [CrossRef]
- Park, S.D.E. The Excel Microsatellite Toolkit; University of Dublin: Dublin, Ireland, 2001. [Google Scholar]
- Waple, R.S.; Do, C. LDNe: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Res. 2008, 8, 753–756. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J. Heredity 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar]
- Luikart, G.; Cornuet. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv. Biol. 1998, 12, 228–237. [Google Scholar] [CrossRef]
- Di Rienzo, A.; Peterson, A.C.; Garza, J.C.; Valdes, A.M.; Slatkin, M.; Freimer, N.B. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 1994, 91, 3166–3170. [Google Scholar]
- Lewontin, R.C.; Krakauer, J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 1973, 74, 175–195. [Google Scholar]
- Beaumont, M.A.; Nichols, R.A. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. London Ser. B 1996, 263, 1619–1626. [Google Scholar] [CrossRef]
- Beaumont, M.A. Adaptation and speciation: what can FST tell us? Trends Ecol. Evol. 2005, 20, 435–440. [Google Scholar] [CrossRef]
- Watterson, G.A. The homozygosity test of neutrality. Genetics 1978, 88, 405–417. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genetics 2003, 4, 981–994. [Google Scholar]
- Vitalis, R. DetSel 1.0: a computer program to detect markers responding to selection. J. Hered. 2003, 94, 429–431. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- International Union for Conservation of Nature and Natural Resources. IUCN Red List of Threatened Species. Version 2009.2. Available online: http://www.iucnredlist.org (accessed August 2009).
- Baker, J.D.; Polovina, J.J.; Howell, E.A. Effect of variable oceanic productivity on the survival of an upper trophic predator, the Hawaiian monk seal, Monachus schauinslandi. Mar. Ecol. Prog. Ser. 2007, 346, 277–283. [Google Scholar] [CrossRef]
- Baker, J.D.; Littnan, C.L.; Johnston, D.W. Potential effects of sea level rise on the terrestrial habitats of endangered and endemic megafauna in the Northwestern Hawaiian Islands. Endang. Species Res. 2006, 4, 1–10. [Google Scholar]
- Littnan, C.L.; Stewart, B.S.; Yochem, P.K.; Braun, R. Survey for selected pathogens and evaluation of disease risk factors for endangered Hawaiian monk seals in the main Hawaiian Islands. EcoHealth 2006, 3, 232–244. [Google Scholar]
- Nachman, M.W.; Hoekstra, H.E.; D’Agostino, S.L. The genetic basis of adaptive melanism in pocket mice. Proc. Natl. Acad. Sci. USA 2003, 100, 5268–5273. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schultz, J.K.; Marshall, A.J.; Pfunder, M. Genome-Wide Loss of Diversity in the Critically Endangered Hawaiian Monk Seal. Diversity 2010, 2, 863-880. https://doi.org/10.3390/d2060863
Schultz JK, Marshall AJ, Pfunder M. Genome-Wide Loss of Diversity in the Critically Endangered Hawaiian Monk Seal. Diversity. 2010; 2(6):863-880. https://doi.org/10.3390/d2060863
Chicago/Turabian StyleSchultz, Jennifer K., Amy J. Marshall, and Monika Pfunder. 2010. "Genome-Wide Loss of Diversity in the Critically Endangered Hawaiian Monk Seal" Diversity 2, no. 6: 863-880. https://doi.org/10.3390/d2060863
APA StyleSchultz, J. K., Marshall, A. J., & Pfunder, M. (2010). Genome-Wide Loss of Diversity in the Critically Endangered Hawaiian Monk Seal. Diversity, 2(6), 863-880. https://doi.org/10.3390/d2060863