Molecular Characterization of the Archaeal Community in an Amazonian Wetland Soil and Culture-Dependent Isolation of Methanogenic Archaea


Abstract
:1. Introduction

2. Materials and Methods
2.1. Study Sites and Soil Samples

2.2. Construction of Archaeal 16S rRNA Gene Clone Libraries
2.3. Data Analysis
2.4. Culture-Dependent Isolation of Methanogenic Archaea
2.5. Morphological Characterization of Methanogenic Cultures
3. Results and Discussion
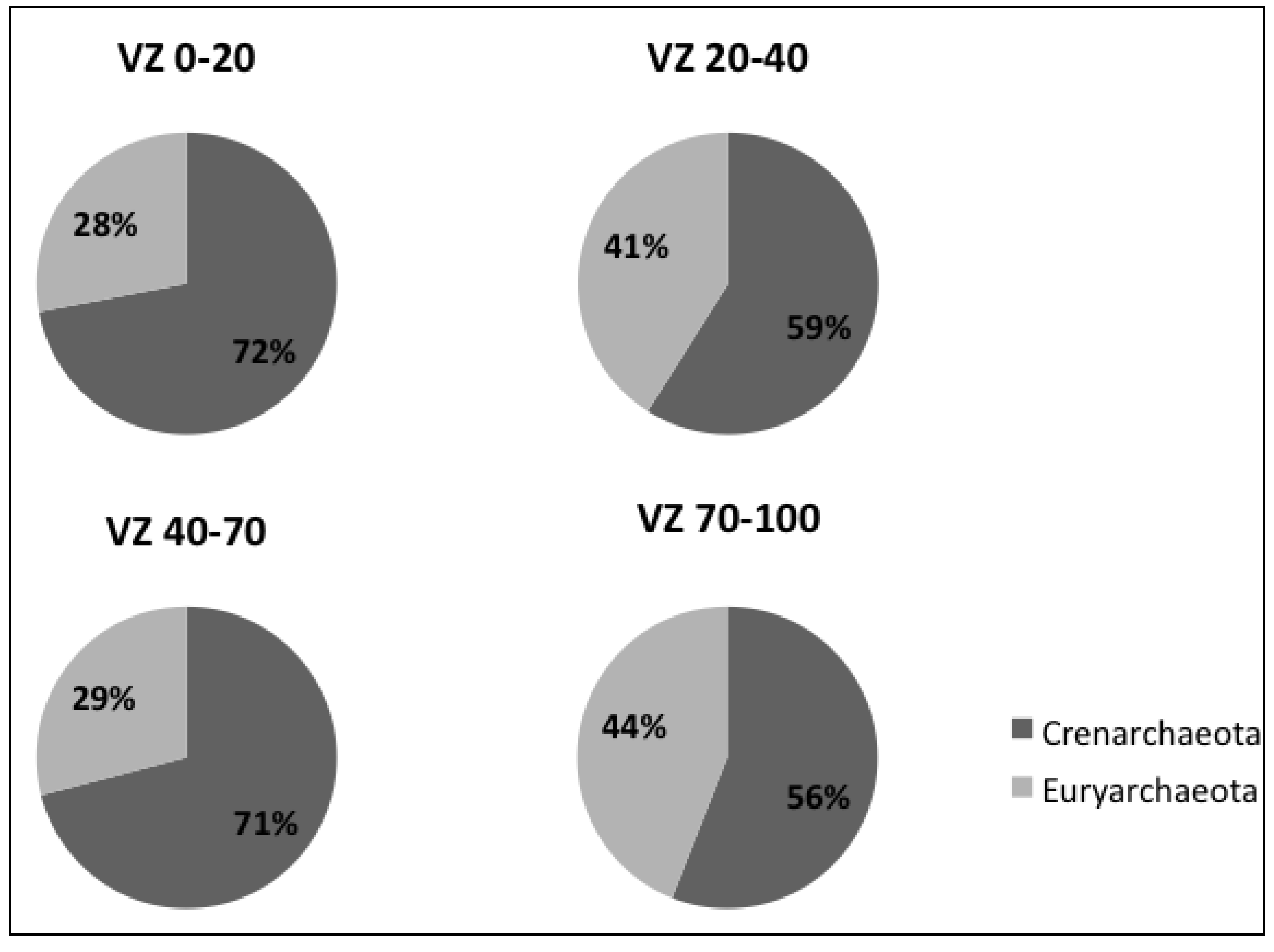
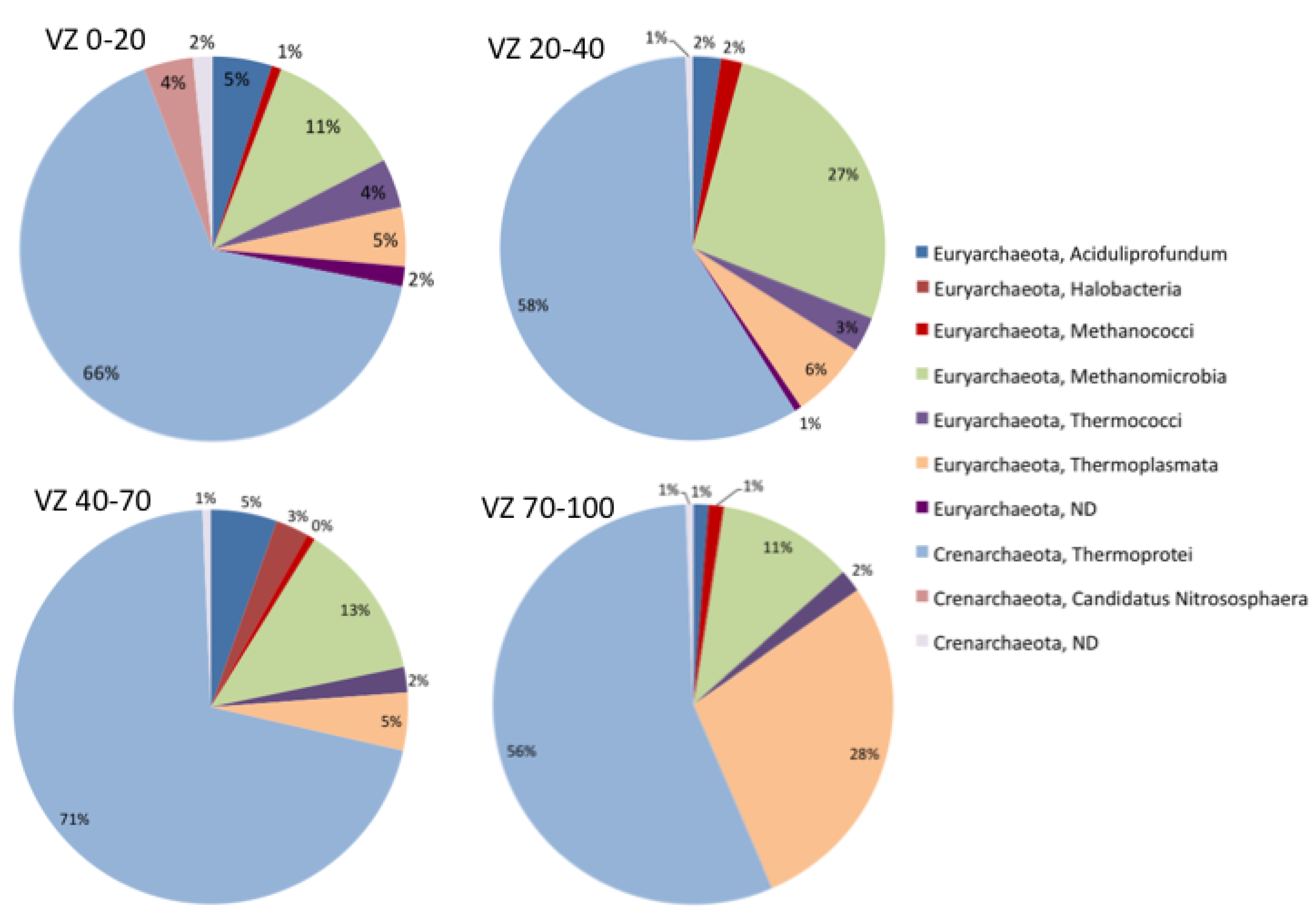
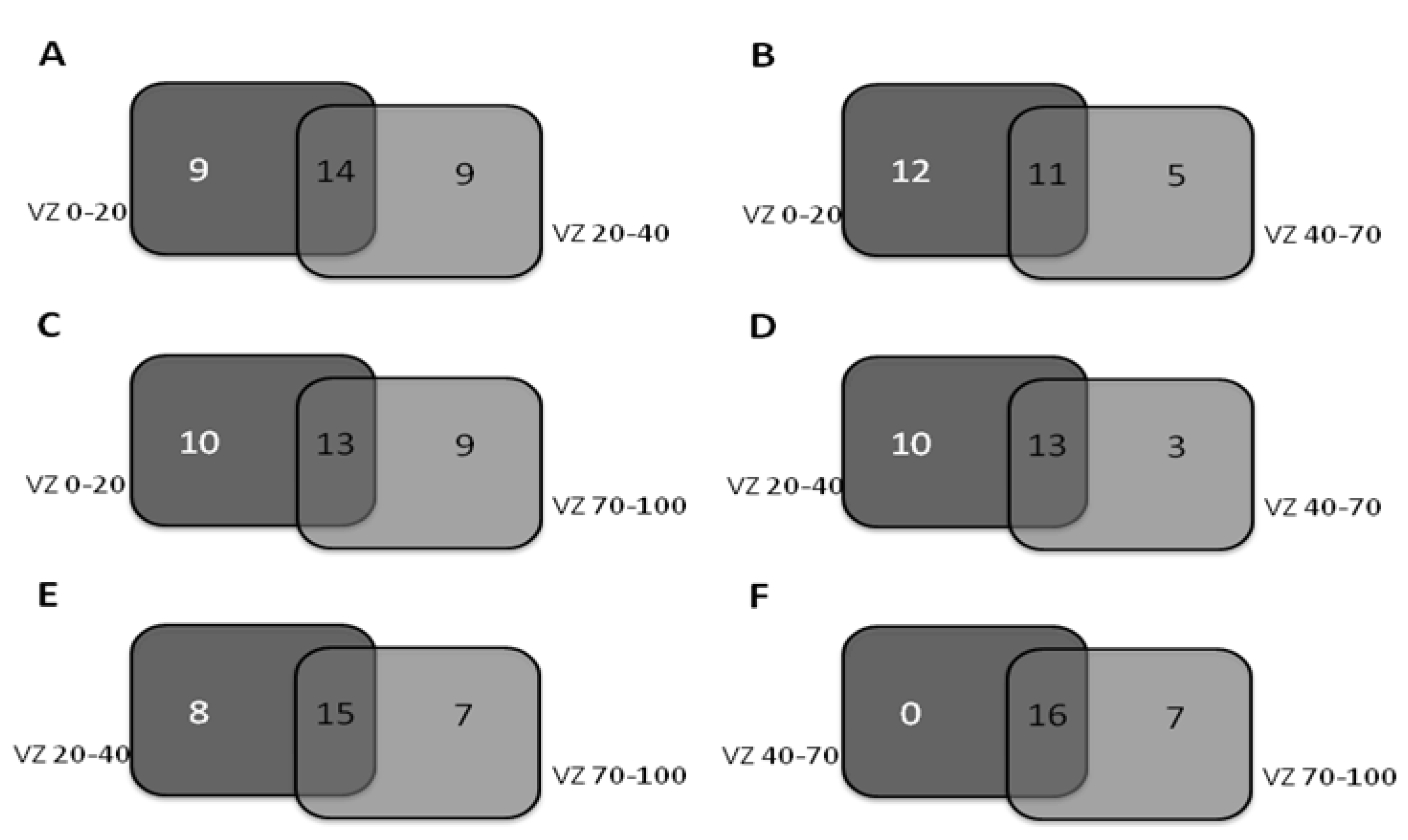
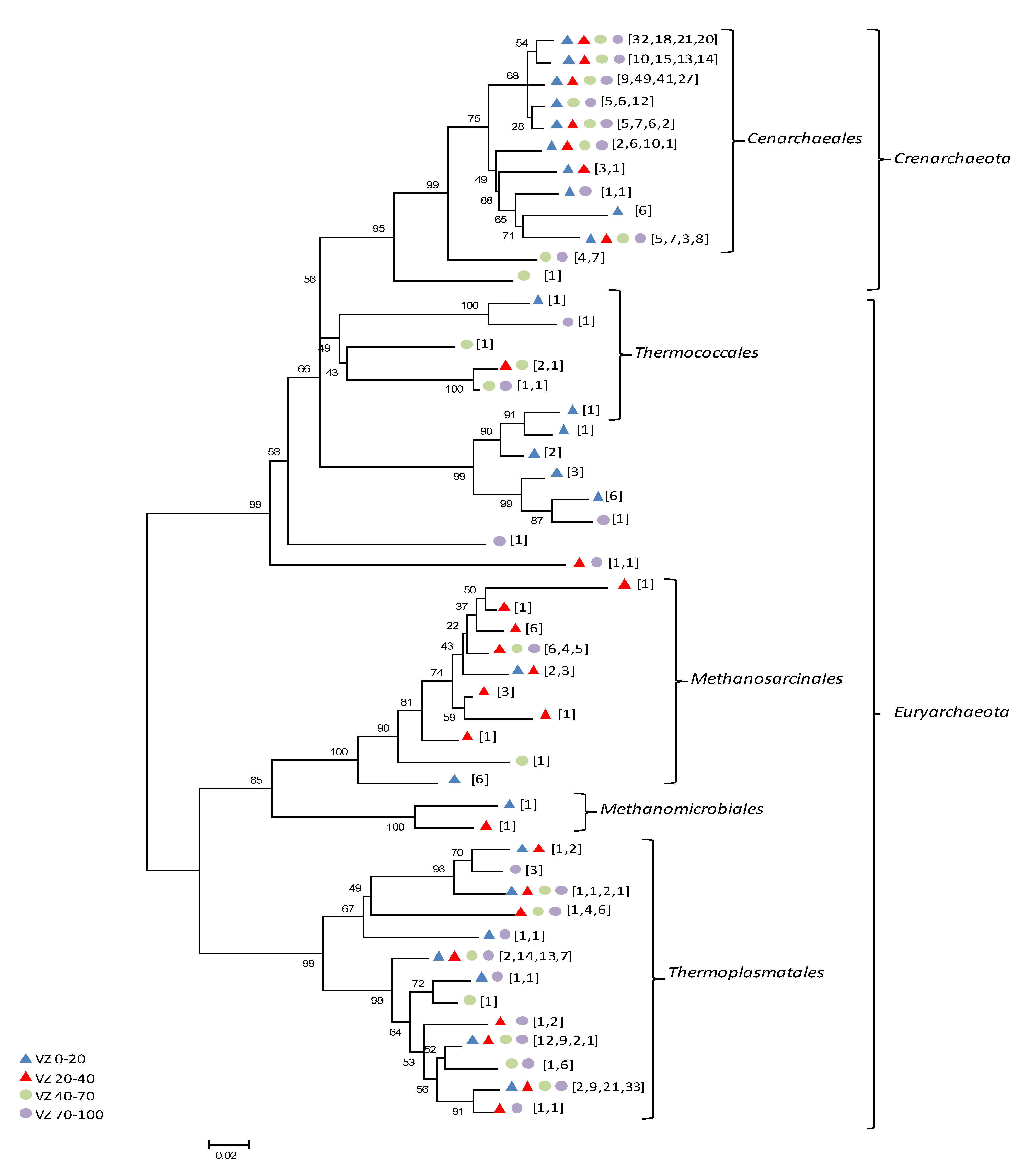
3.1. Diversity of the Archaeal Community

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attributes | Site | |||
|---|---|---|---|---|
| VZ 0–20 | VZ 20–40 | VZ 40–70 | VZ 70–100 | |
| Physical | ||||
| Sand (%) | 22 | 20 | 17 | 7 |
| Silt (%) | 58 | 60 | 65 | 65 |
| Clay (%) | 20 | 20 | 18 | 28 |
| Chemical | ||||
| pH (CaCl2) | 5.7 | 6.0 | 5.7 | 6.1 |
| O.M. (g.dm−3) | 12 | 10 | 9 | 11 |
| P (mg.dm−3) | 11 | 13 | 11 | 11 |
| K (mmolc.dm−3) | 10.9 | 1.2 | 0.8 | 0.8 |
| Ca (mmolc.dm−3) | 71 | 75 | 76 | 70 |
| Mg (mmolc.dm−3) | 30 | 33 | 32 | 31 |
| H+Al (mmolc.dm−3) | 12 | 10 | 10 | 11 |
| SB (mmolc.dm−3) | 111.9 | 109.2 | 108.8 | 101.8 |
| CEC (mmolc.dm−3) | 123.9 | 119.2 | 118.8 | 112.8 |
| BS (%) | 90 | 92 | 92 | 90 |
| B (mg.dm−3) | 0.48 | 0.21 | 0.44 | 0.43 |
| Cu (mg.dm−3) | 2.4 | 1.7 | 2.0 | 2.0 |
| Fe (mg.dm−3) | 57 | 27 | 27 | 30 |
| Mn (mg.dm−3) | 15.1 | 8.4 | 6.6 | 7.7 |
| Zn (mg.dm−3) | 1.3 | 0.9 | 0.8 | 0.9 |



| Sample | Reads | OTU a | Chao1 b | Jackknife | Shannon | Simpson |
| VZ 0–20 | 121 | 23 | 30.2 ± 5.6 | 32.0 ± 8.3 | 2.40 ± 0.22 | 0.142 |
| VZ 20–40 | 168 | 23 | 41.3 ± 13.8 | 45.2 ± 17.7 | 2.24 ± 0.19 | 0.167 |
| VZ 40–70 | 147 | 16 | 21.0 ± 4.2 | 21.0 ± 6.2 | 1.94 ± 0.18 | 0.207 |
| VZ 70–100 | 163 | 22 | 44.5 ± 17.2 | 51.4 ± 23.3 | 2.36 ± 0.17 | 0.132 |


3.2. Culture-Dependent Isolation of Methanogenic Archaea

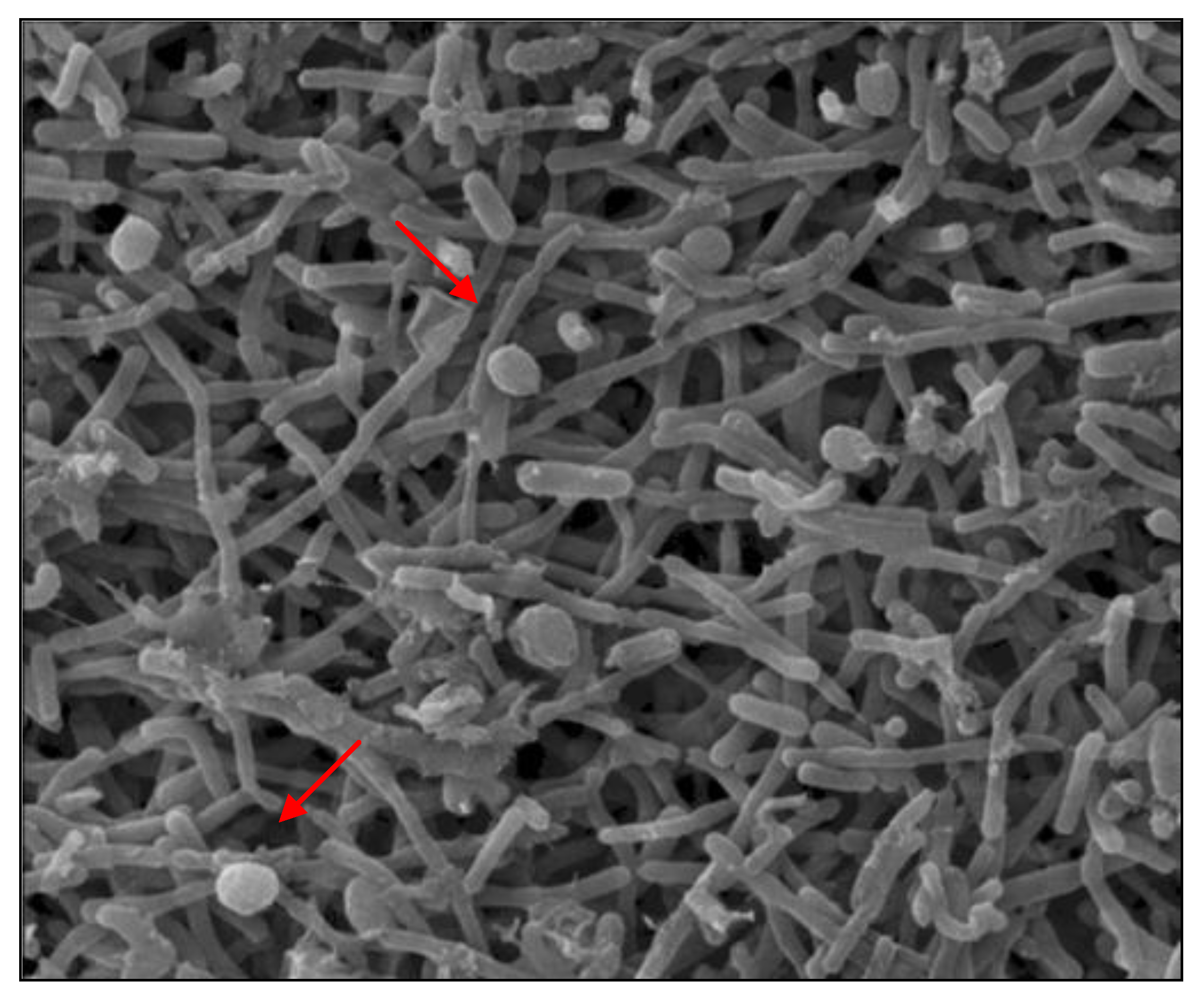
3.3. Morphological Characterization of Methanogenic Cultures


4. Conclusions
Acknowledgements
References
- Bartlett, K.B.; Harris, R.C. Review and Assessment of Methane Emission from Wetlands. Chemosphere 1993, 26, 261–320. [Google Scholar] [CrossRef]
- Junk, W.J. The Central Amazon Floodpalin: Ecology of a Pulsing System; Springer: Berlin, Germany, 1997. [Google Scholar]
- Devol, A.H.; Richey, J.E.; Forsberg, B.R.; Martinelli, L.A. Seasonal Dynamics in Methane Emissions from the Amazon River Floodplain to the Troposphere. J. Geophys. Res. 1990, 95, 16417–16426. [Google Scholar] [CrossRef]
- Fearnside, P.M. Agriculture in Amazonia. In Key Environments: Amazonia; Prance, G.T., Lovejoy, T.E., Eds.; Pergam Press: Oxford, UK, 1985. [Google Scholar]
- Zinder, S.H. Physiological Ecology of Methanogens. In Methanogenesis: Ecology, Physiology, Biochemistry & Genetics; Ferry, J.G., Ed.; Chapman & Hall: New York, NY, USA, 1983. [Google Scholar]
- Houghton, J. The Climate and the Latest Scientific Understanding of Climate Change. Renewable Energy 1994, 5, 1–4. [Google Scholar] [CrossRef]
- Alvalá, P.C.; Kirchhoff, W.V.J.H.; Pavão, H.G. Metano na Atmosfera. Biotecnol. Ciência e Desenv. 1999, 7, 40–43. [Google Scholar]
- Koschorreck, M. Methane Tournover in Exposed Sediments of an Amazon Floodplain Lake. Biogeochemistry 2000, 50, 195–206. [Google Scholar] [CrossRef]
- Melack, J.M.; Hess, L.L.; Gastil, M.; Forsberf, B.R.; Hamilton, S.K.; Lima, I.B.T.; Novo, E.M.L.M. Regionalization of Methane Emissions in the Amazon Basin with Microwave Remote Sensing. Glob. Change Biol. 2004, 10, 530–544. [Google Scholar] [CrossRef]
- Davidson, E.A.; Ishida, F.Y.; Nepstad, D.C. Effects of an Experimental Drought on Soil Emissions of Carbon Dioxide, Methane, Nitrous Oxide, and Nitric Oxide in a Moist Tropical Forest. Glob. Change Biol. 2004, 10, 718–730. [Google Scholar] [CrossRef]
- Conrad, R.; Klose, M.; Claus, P.; Enrich-Prast, A. Methanogenic Pathway, 13C Isotope Fractionation, and Archaeal Community Composition in the Sediment of Two Clear–Water Lakes of Amazonia. Limnol. Oceanogr. 2010, 55, 689–702. [Google Scholar] [CrossRef]
- de Oliveira Júnior, R.C.; Corrêa, J.R.V. Caracterização dos Solos do Município de Belterra, Estado do Pará; Boletim De Pesquisa E Desenvolvimento, Embrapa Amazônia Oriental: Belém, Brazil, 2001. [Google Scholar]
- Rodrigues, T.E.; Santos, P.L., dos; de Oliveira Júnior, R.C.; Valente, M.A.; Silva, J.M.L., da; Cardoso Júnior, E.Q. Caracterização dos Solos da Área do Planalto de Belterra, Município de Santarém, Estado do Pará; Embrapa Amazônia Oriental, Boletim De Pesquisa E Desenvolvimento: Belém, Brazil, 2001. [Google Scholar]
- Moyer, C.L.; Tiedje, J.M.; Dobbs, F.C.; Karl, D.M. Diversity of Deep-sea Hydrothermal Vent Archaea from Loihi Seamount, Hawaii. Deep-Sea Res. 1998, 45, 303–317. [Google Scholar] [CrossRef]
- Ewing, B.; Green, P. Base-Calling of Automated Sequencer Traces Using Phred. II Error Probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Gordon, D.; Abajian, C.; Green, P. Consed: A Geographical Tool for Sequence Finishing. Genome Res. 1998, 8, 195–202. [Google Scholar] [CrossRef]
- Schloss, P.D.; Handelsman, J. Introducing to DOTUR, a Computer Program for Defining Operacional Taxonomic Units and Estimating Species Richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]
- Singleton, D.R.; Furlong, M.A.; Rathbun, S.L.; Whitman, W.B. Quantitative Comparisons of 16S rRNA Gene Sequence Libraries from Environmental Samples. Appl. Environ. Microbiol. 2001, 67, 4374–4376. [Google Scholar] [CrossRef]
- Deevong, P.; Hattori, S.; Yamada, A.; Trakulnaleamsai, S.; Ohkuma, M.; Noparatnaraporn, N.; Kudo, T. Isolation and Detection of Methanogens from the Gut of Higher Térmites. Microbes Environ. 2004, 19, 221–226. [Google Scholar] [CrossRef]
- Edwards, T.; McBridge, B.C. New Method for the Isolation and Identification of Methanogenic Bacteria. Appl. Environ. Microbiol. 1975, 29, 540–545. [Google Scholar]
- Vazoller, R.F. Avaliação do Ecossistema Microbiano de um Biodigestor Anaeróbio de Fluxo Ascendente e Manta de Lodo, Operado com Vinhaça sob Condições Termofílicas; (Tese Doutorado em Hidráulica e Saneamento); Escola de Engenharia de São Carlos, Universidade de São Paulo: São Paulo, Brazil, 1995; p. 295. [Google Scholar]
- Schneider, R.P.; Ferreira, L.M.; Binder, P.; Bejarano, E.M.; Goes, K.P.; Slongo, E.; Machado, C.R.; Rosa, G.M.Z. Dynamics of Organic Carbon and of Bacterial Populations in a Conventional Pretreatment Train of a Reverse Osmosis Unit Experiencing Severe Biofouling. J. Membr. Sci. 2005, 266, 18–29. [Google Scholar] [CrossRef]
- Massana, R.; Murray, A.E.; Preston, C.M.; DeLong, E.F. Vertical Distribution and Phylogenetic Characterization of Marine Planktonic Archaea in the Santa Barbara Channel. Appl. Environ. Microbiol. 1997, 63, 50–56. [Google Scholar]
- Canhos, V.P.; Vazoller, R.F. Microrganismos & Vírus. In Biodiversidade do estado de São Paulo.; Fundacão de Amparo à Pesquisa do Estado de São Paulo: São Paulo, Brazil, 1999; pp. 3–23. [Google Scholar]
- Bintrin, S.B.; Donohue, T.J.; Handelsman, J.; Roberts, G.P.; Goodman, R.M. Molecular Phylogeny of Archaea from Soil. PNAS 1997, 94, 277–282. [Google Scholar] [CrossRef]
- Chaban, B.; Ng, S.Y.M.; Jarrel, K.F. Archaeal Habitats from the Extreme to the Ordinary. Can. J. Microbiol. 2006, 52, 73–116. [Google Scholar] [CrossRef]
- Ayton, J.; Aislabie, J.; Barker, G.M.; Saul, D.; Turner, S.S. Crenarchaeota Affiliated with Group 1.1b are Prevalent in Coastal Mineral Soils of the Ross Sea Region of Antartica. Environ. Microbiol. 2010, 12, 689–703. [Google Scholar] [CrossRef]
- Nicol, G.W.; Tscherko, D.; Chang, L.; Hammesfahr, U.; Prosser, J.I. Crenarchaeal Community Assembly and Microdiversity in Developing Soils at Two Sites Associated with Deglaciation. Environ. Microbiol. 2006, 8, 1382–1393. [Google Scholar] [CrossRef]
- Könneke, M.; Bernhard, A.E.; de la Torre, J.R.; Walker, C.B.; Waterbury, J.B.; Stahl, D.A. Isolation of an Autotrophic Ammonia–Oxidizing Marine Archaeon. Nature 2005, 437, 543–546. [Google Scholar] [CrossRef]
- Preston, C.M.; Wu, K.Y.; Molinski, T.F.; DeLong, E.F. A Psychrophilic Crenarchaeon Inhabits a Marine Sponge: Cenarchaeum Symbiosum gen. nov., sp. nov. PNAS 1996, 93, 6241–6246. [Google Scholar] [CrossRef]
- Jurgens, G.; Lindstrom, K.; Saano, A. Novel Group within the Kingdom Crenarchaeota from Boreal Forest Soil. Appl. Environ. Microbiol. 1997, 63, 803–805. [Google Scholar]
- Pesaro, M.; Widmer, F. Identification of Novel Crenarchaeota and Euryarchaeota Clusters Associated with Different Depth Layers of a Forest Soil. FEMS Microbiol. Ecol. 2002, 42, 89–98. [Google Scholar] [CrossRef]
- Kemnitz, D.; Kolb, S.; Conrad, R. High Abundance of Crenarchaeota in a Temperate Acidic Forest Soil. FEMS Microbiol. Ecol. 2007, 60, 447–448. [Google Scholar]
- Kim, B.S.; Oh, H.M.; Kang, H.; Chun, J. Archaeal Diversity in Tidal Flat Sediment as Revealed by 16S rDNA Analysis. J. Microbiol. 2005, 43, 144–151. [Google Scholar]
- Meuer, J.; Kuettner, H.C.; Zhang, J.K.; Hedderich, R.; Metcalf, W.W. Genetic Analysis of the Archaeon Methanosarcina Barkeri FUSARO Reveals a Central Role for Ech Hydrogenase and Ferredoxin in Methanogenesis and Carbon Fixation. PNAS 2002, 99, 5632–5637. [Google Scholar] [CrossRef]
- Castro, H.; Ogram, A.; Reddy, K.R. Phylogenetic Characterization of Methanogenic Assemblages in Eutrophic and Oligotrophic Areas of the Florida Everglades. Appl. Environ. Microbiol. 2004, 70, 6559–6568. [Google Scholar] [CrossRef]
- Conrad, R. Contribution of Hydrogen to Methane Production and Control of Hydrogen Concentrations in Methanogenic Soils and Sediments. FEMS Microbiol. Ecol. 1999, 28, 193–202. [Google Scholar] [CrossRef]
- Marick, T.; Fischer, H.; Conen, F.; Smith, K. Seasonal Variations in Stable Carbon and Hydrogen Isotope Ratio in Methane from Rice Fields. Global Biogeochem. Cycles 2002, 16, 41–52. [Google Scholar]
- Moura, J.M.S. Variação na Composição Isotópica do Metano (CH4) em Sedimentos de Igarapés na Amazônia Oriental.; (Dissertação Mestrado em Ecologia de Agroecossistemas); Escola Superior de Agricultura “Luiz de Queiroz”; Universidade de São Paulo: Piracicaba, Brazil, 2005; p. 82. [Google Scholar]
- Whiticar, M.J. Carbon and Hydrogen Isotope Systematic of Bacterial Formation and Oxidation of Methane. Chem. Geol. 1999, 161, 291–314. [Google Scholar] [CrossRef]
- Kemnitz, D.; Kolb, S.; Conrad, R. Phenotypic Characterization of Rice Cluster III Archaea Without Prior Isolation by Applying Quantitative Polymerase Chain Reaction to an Enrichment Culture. Environ. Microbiol. 2005, 7, 553–565. [Google Scholar] [CrossRef]
- Schloss, P.D.; Handelsman, J. Introducing SONS, a Tool for Operational Taxonomic Unit–based Comparisons of Microbial Community Memberships and Structures. Appl. Environ. Microbiol. 2006, 72, 6773–6779. [Google Scholar] [CrossRef]
- Torsvik, V.; Goksoyr, J.; Daae, F.L. High Diversity in DNA of Soil Bacteria. Appl. Environ. Microbiol. 1990, 56, 782. [Google Scholar]
- Bornerman, J.; Triplett, E.W. Molecular Diversity in Soils from Eastern Amazonia: Evidence for Unusual Microorganisms and Microbial Population Shifts Associated with Deforestation. Appl. Environ. Microbiol. 1997, 63, 2647–2653. [Google Scholar]
- Fierer, N.; Jackson, R.B. The Diversity and Biogeography of Soil Bacterial Communities. PNAS 2006, 103, 626–631. [Google Scholar] [CrossRef]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor–Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Buckley, D.H.; Graber, J.R.; Schmidt, T.M. Phylogenetic Analysis of Nonthermophilic Members of the Kingdom Crenarchaeota and their Diversity and Abundance in Soils. Appl. Environ. Microbiol. 1998, 64, 4333–4339. [Google Scholar]
- Clementino, M.M.; Fernandes, C.C.; Vieira, R.P.; Cardoso, A.M.; Polycarpo, C.R.; Martins, O.B. Archaeal Diversity in Naturally Occurring and Impacted Environments from a Tropical Region. J. Appl. Microbiol. 2006, 103, 141–151. [Google Scholar]
- Chauhan, A.; Reddy, K.R.; Ogram, A.V. Syntrophic-Archaeal Associations in a Nutrient–Impacted Freshwater Marsh. J. Appl. Microbiol. 2006, 100, 73–84. [Google Scholar] [CrossRef]
- Stein, L.Y.; La Duc, M.T.; Grundl, T.J.; Nealson, K.H. Bacterial and Archaeal Populations Associated with Freshwater Ferromanganous Micronodules and Sediments. Environ. Microbiol. 2001, 3, 10–18. [Google Scholar] [CrossRef]
- Erkel, C.; Kube, M.; Reinhardt, R.; Liesack, W. Genome of Rice Cluster I Archaea—The Key Methane Producers in the Rice Rhizosphere. Science 2006, 313, 370–372. [Google Scholar] [CrossRef]
- Lueders, T.; Friedrich, M.W. Archaeal Population Dynamics during Sequential Reduction Processes in Rice Field Soil. Appl. Environ. Microbiol. 2000, 66, 2732–2742. [Google Scholar] [CrossRef]
- Chelius, M.K.; Triplett, E.W. The Diversity of Archaea and Bacteria in Association with the Roots of Zea Mays L. Microb. Ecol. 2001, 41, 252–263. [Google Scholar]
- Yan, B.; Hong, K.; Yu, Z.N. Archaeal Communities in Mangrove Soil Characterized by 16S rRNA Gene Clones. J. Microbiol. 2006, 44, 566–571. [Google Scholar]
- Kasai, Y.; Takahata, Y.; Hoaki, T.; Watanabe, K. Physiological and Molecular Characterization of a Microbial Community Established in Unsaturated, Petroleum-Contaminated Soil. Environ. Microbiol. 2005, 7, 806–818. [Google Scholar] [CrossRef]
- Donovan, S.E.; Purdy, K.J.; Kane, M.D.; Eggleton, P. Comparison of Euryarchaea Strains in the Guts and Food-soil of the Soil–feeding Termite Cubitermes Fungifaber across Different Soil Types. Appl. Environ. Microbiol. 2004, 70, 3884–3892. [Google Scholar] [CrossRef]
- Winter, C.; Smit, A.; Herndl, G.L.; Weinbauer, M.G. Impact of Virioplankton on Archaeal and Bacterial Community Richness as Assessed in Seawater Batch Cultures. Appl. Environ. Microbiol. 2004, 70, 804–813. [Google Scholar] [CrossRef]
- Cury, J.C. Diversidade de Bacteria e Archaea em Solos de Mangue e Marisma.; (Tese Doutorado em Solos e Nutrição de Plantas); Escola Superior de Agricultura “Luiz de Queiroz”; Universidade de São Paulo: Piracicaba, Brazil, 2006; p. 147. [Google Scholar]
- Garcia, J.L.; Patel, B.K.C.; Ollivier, B. Taxonomic Phylogentic and Ecological Diversity of Methanogenic Archaea. Anaerobe 2000, 6, 205–226. [Google Scholar] [CrossRef]
- Madigan, M.T.; Martinko, J.M.; Parker, J. Microbiologia de Brock, 10th ed.; Editora Pearson Education do Brasil: São Paulo, Brasil, 2004. [Google Scholar]
- Reeve, J.N.; Nölling, J.; Morgan, R.M.; Smith, D.R. Methanogenesis: Genes, Genomes, and Who’s on First? J. Bacteriol. 1997, 179, 5975–5986. [Google Scholar]
- Balch, W.E.; Fox, G.E.; Magrum, L.J.; Woese, C.R.; Wolfe, R.S. Methanogens: Reevaluation of a Unique Biological Group. Microbiol. Rev. 1979, 43, 260–296. [Google Scholar]
- Oremland, R.S. Biogeochemistry of Methanogenic Bacteria. In Biology of Anaerobic Microorganisms; Zehnder, A.J.B., Ed.; John Wiley & Sons: Wageningen, Netherlands, 1988. [Google Scholar]
- Hungate, R.E. A Roll-Tube Method for Cultivation of Strict Anaerobes. Methods Microbiol. 1969, 3, 117–132. [Google Scholar] [CrossRef]
- Thórsson, A.E.; Sverrisson, H.; Anamthawat-Jónsson, K. Genotyping Icelandic Isolates of Rhizobia Based on rDNA-RFLP. Icel. Agric. Sci. 2000, 13, 17–25. [Google Scholar]
- Wright, A.D.G.; Pimm, C. Improved Strategy for Presumptive Identification of Methanogens Using 16S Riboprinting. J. Microbiol. Methods 2003, 55, 337–349. [Google Scholar] [CrossRef]
- Whitby, C.; Earl, J.; Lanyon, C.; Gray, S.; Robinson, J.; Meadows, J.; Edwards, C. The Molecular Diversity of the Methanogenic Community in a Hypereutrophic Freshwater Lake Determined by PCR-RFLP. J. Appl. Microbiol. 2004, 97, 973–984. [Google Scholar] [CrossRef]
- Joulian, C.; Ollivier, B.; Patel, B.K.C.; Roger, P.A. Phenotypic and Phylogenetic Characterization of Dominant Culturable Methanogens Isolated from Ricefield Soils. FEMS Microbiol. Ecol. 1998, 25, 135–145. [Google Scholar] [CrossRef]
- Simankova, M.V.; Kotsyurbenko, O.R.; Lueders, T.; Nozhevnikova, A.N.; Wagner, B.; Conrad, R.; Friedrich, M.W. Isolation and Characterization of New Strains of Methanogens from Cold Terrestrial Habitats. Syst. Appl. Microbiol. 2003, 26, 312–318. [Google Scholar] [CrossRef]
- Garrity, G.M.; Holt, J.H. A Road Map to the Manual. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Boone, D.R., Castenholz, G.M., Garrity, G.M., Eds.; Springer Verlag: Berlin, Germany, 2001. [Google Scholar]
- Le Mer, J.; Roger, P. Production, Oxidation, Emission and Consumption of Methane by Soils: A review. Eur. J. Soil Sci. 2001, 37, 25–50. [Google Scholar]
- Adashi, K. Isolation of Hydrogenotrophic Methanogenic Archaea from a Subtropical Paddy Field. FEMS Microbiol. Ecol. 1999, 30, 77–85. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pazinato, J.M.; Paulo, E.N.; Mendes, L.W.; Vazoller, R.F.; Tsai, S.M. Molecular Characterization of the Archaeal Community in an Amazonian Wetland Soil and Culture-Dependent Isolation of Methanogenic Archaea. Diversity 2010, 2, 1026-1047. https://doi.org/10.3390/d2071026
Pazinato JM, Paulo EN, Mendes LW, Vazoller RF, Tsai SM. Molecular Characterization of the Archaeal Community in an Amazonian Wetland Soil and Culture-Dependent Isolation of Methanogenic Archaea. Diversity. 2010; 2(7):1026-1047. https://doi.org/10.3390/d2071026
Chicago/Turabian StylePazinato, Jeanedy M., Ezio N. Paulo, Lucas W. Mendes, Rosana F. Vazoller, and Siu M. Tsai. 2010. "Molecular Characterization of the Archaeal Community in an Amazonian Wetland Soil and Culture-Dependent Isolation of Methanogenic Archaea" Diversity 2, no. 7: 1026-1047. https://doi.org/10.3390/d2071026
APA StylePazinato, J. M., Paulo, E. N., Mendes, L. W., Vazoller, R. F., & Tsai, S. M. (2010). Molecular Characterization of the Archaeal Community in an Amazonian Wetland Soil and Culture-Dependent Isolation of Methanogenic Archaea. Diversity, 2(7), 1026-1047. https://doi.org/10.3390/d2071026