
Microsatellites Reveal Genetic Homogeneity among Outbreak Populations of Crown-of-Thorns Starfish (Acanthaster cf. solaris) on Australia’s Great Barrier Reef

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
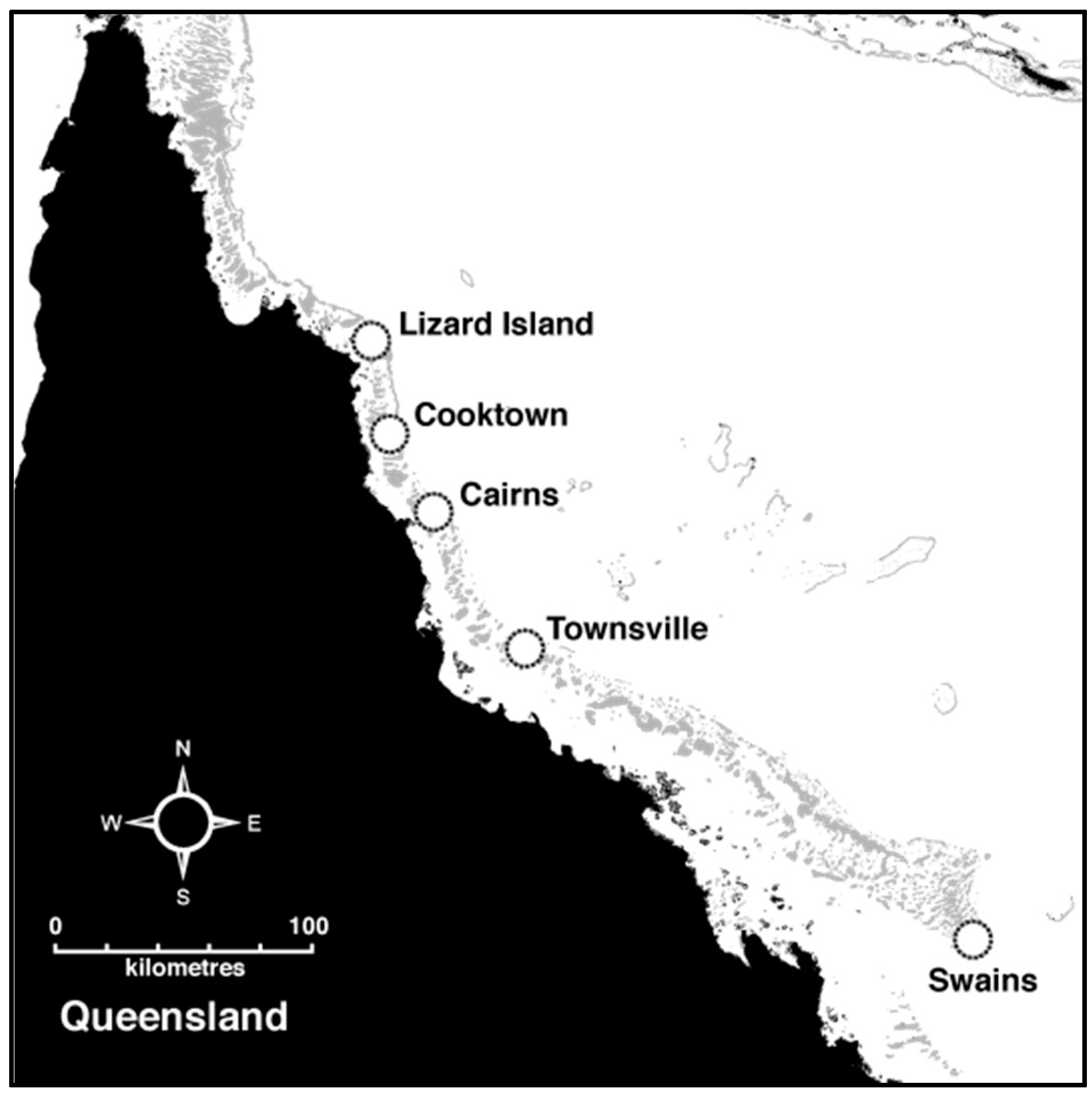
2.1. Sample Collection
2.2. Microsatellite Genotyping and Locus Characteristics
2.3. Diversity Analysis of Sampled Populations
2.4. Spatial Genetic Clustering
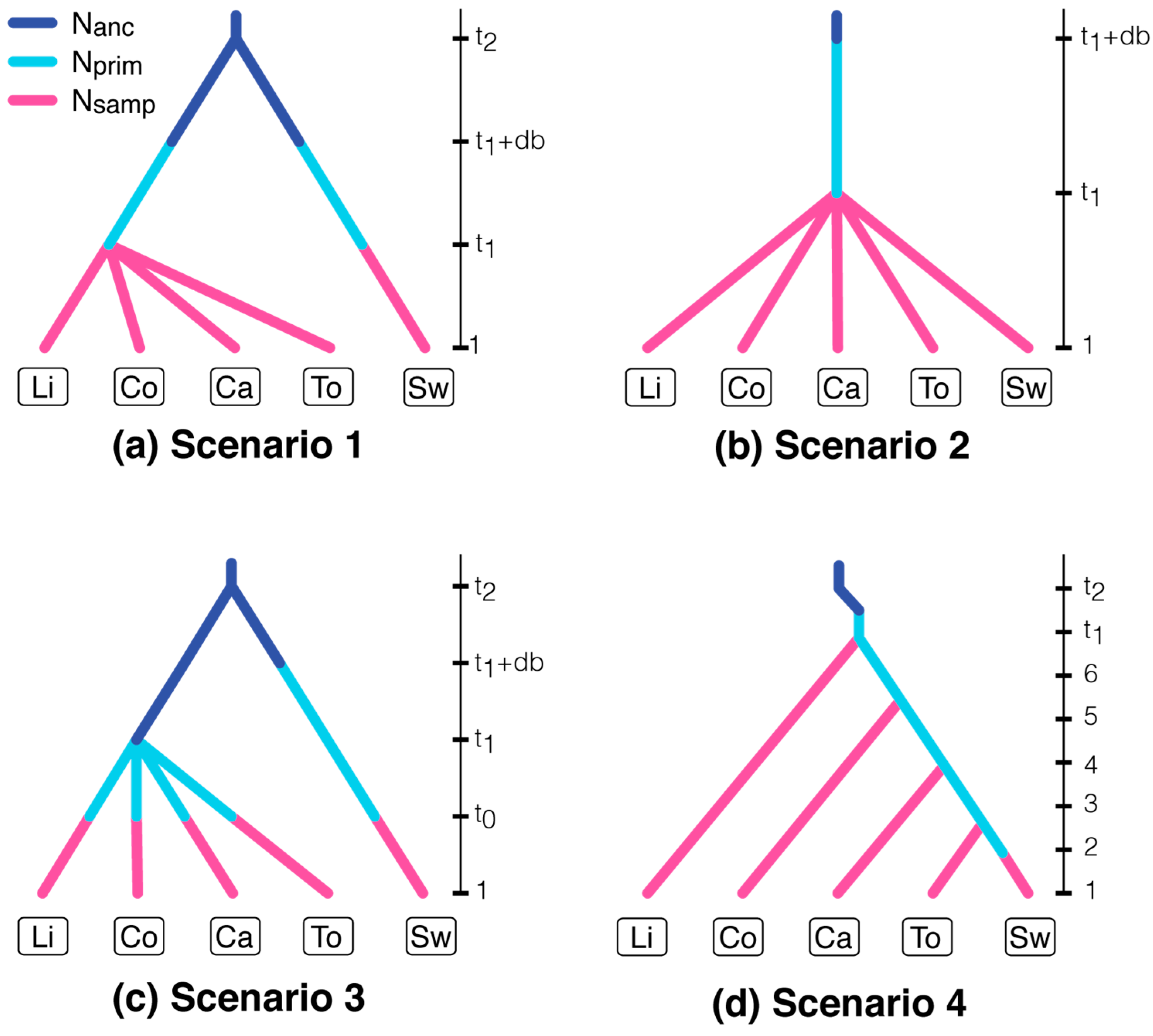
2.5. Approximate Bayesian Computation of Source Populations
2.6. Inbreeding
3. Results
3.1. Microsatellite Data
3.2. Spatial Patterns of Genetic Diversity
3.3. Inbreeding
3.4. ABC Framework
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Birkeland, C.E.; Lucas, J.S. Acanthaster. planci: Major Management Problem of Coral Reefs; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Pratchett, M.; Caballes, C.; Posada, J.R.; Sweatman, H. Limits to Understanding and Managing Outbreaks of Crown-of-Thorns Starfish (Acanthaster. spp.). Oceanogr. Mar. Biol. 2014, 52, 133–200. [Google Scholar]
- Moran, P.J.; De’ath, G. Estimates of the abundance of the crown-of-thorns starfish Acanthaster planci in outbreaking and non-outbreaking populations on reefs within the Great Barrier Reef. Mar. Biol. 1992, 113, 509–515. [Google Scholar] [CrossRef]
- Bruno, J.F.; Selig, E.R. Regional Decline of Coral Cover in the Indo-Pacific: Timing, Extent, and Subregional Comparisons. PLoS ONE 2007, 2, e711. [Google Scholar] [CrossRef] [PubMed]
- Trapon, M.L.; Pratchett, M.S.; Penin, L. Comparative Effects of Different Disturbances in Coral Reef Habitats in Moorea, French Polynesia. J. Mar. Biol. 2011, 1–11. [Google Scholar] [CrossRef]
- De’ath, G.; Fabricius, K.E.; Sweatman, H.; Puotinen, M. The 27-year decline of coral cover on the Great Barrier Reef and its causes. PNAS 2012, 104, 17995–17999. [Google Scholar] [CrossRef] [PubMed]
- Pisapia, C.; Burn, D.; Yoosuf, R.; Najeeb, A.; Anderson, K.D.; Pratchett, M.S. Coral recovery in the central Maldives archipelago since the last major mass-bleaching, in 1998. Sci. Rep. 2016, 6, 34720. [Google Scholar] [CrossRef] [PubMed]
- Moran, P.J. The Acanthaster phenomenon. Oceanogr. Mar. Biol. 1986, 24, 379–480. [Google Scholar]
- Babcock, R.C.; Milton, D.A.; Pratchett, M.S. Relationships between size and reproductive output in the crown-of-thorns starfish. Mar. Biol. 2016, 163, 234. [Google Scholar] [CrossRef]
- Uthicke, S.; Schaffelke, B.; Byrne, M. A boom–bust phylum? Ecological and evolutionary consequences of density variations in echinoderms. Ecol. Monogr. 2009, 79, 3–24. [Google Scholar] [CrossRef]
- Lucas, J.S. Reproductive and larval biology of Acanthaster planci (L.) in Great Barrier Reef waters. Micronesica 1973, 9, 197–203. [Google Scholar]
- Birkeland, C. Terrestrial runoff as a cause of outbreaks of Acanthaster. planci (Echinodermata: Asteroidea). Mar. Biol. 1982, 69, 175–185. [Google Scholar] [CrossRef]
- Fabricius, K.E.; Okaji, K.; De’ath, G. Three lines of evidence to link outbreaks of the crown-of-thorns seastar Acanthaster planci to the release of larval food limitation. Coral Reefs 2010, 29, 593–605. [Google Scholar] [CrossRef]
- Pearson, R.G.; Endean, R. A Preliminary Study of the Coral Predator Acanthaster. planci (L.)(Asteroidea) on the Great Barrier Reef; Fisheries Notes; Queensland Department of Harbours and Marine: Brisbane, Australia, 1969; Volume 3, pp. 27–55. [Google Scholar]
- Vine, P.J. Crown of thorns (Acanthaster planci) plagues: The natural causes theory. Atoll Res. Bull. 1973, 166, 1–10. [Google Scholar] [CrossRef]
- Ganter, R. Oral History of Human Use and Experience of Crown of Thorns Starfish on the Great Barrier Reef; Report to the Great Barrier Reef Marine Park Authority; Great Barrier Reef Marine Park Authority: Townsville, Australia, 1987. [Google Scholar]
- Kenchington, R.A. Acanthaster planci on the Great Barrier Reef: Detailed surveys of four transects between 19° and 20°S. Biol. Conserv. 1976, 9, 165–179. [Google Scholar] [CrossRef]
- Kenchington, R.A. Growth and recruitment of Acanthaster planci (L.) on the Great Barrier Reef. Biol. Conserv. 1977, 11, 103–118. [Google Scholar] [CrossRef]
- Hock, K.; Wolff, N.H.; Condie, S.A.; Anthony, K.; Mumby, P.J. Connectivity networks reveal the risks of crown-of-thorns starfish outbreaks on the Great Barrier Reef. J. Appl. Ecol. 2014, 51, 1188–1196. [Google Scholar] [CrossRef]
- Sweatman, H.; Bass, D.K.; Cheal, A.J.; Coleman, G.; Miller, I.R.; Ninio, R.; Osborne, K.; Oxley, W.G.; Ryan, D.A.J.; Thompson, A.A.; et al. Long-Term Monitoring of the Great Barrier Reef. Status Report Number 3; Australian Institute of Marine Science: Townsville, Australia, 1998. [Google Scholar]
- Moore, R.J. Persistent and Transient Populations of the Crown-of-Thorns Starfish, Acanthaster Planci. In Acanthaster and the Coral Reef: A Theoretical Perspective; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar]
- Crowl, T.A.; Crist, T.O.; Parmenter, R.R.; Belovsky, G.; Lugo, A.E. The spread of invasive species and infectious disease as drivers of ecosystem change. Front. Ecol. Environ. 2008, 6, 238–246. [Google Scholar] [CrossRef]
- Suarez, A.V.; Holway, D.A.; Case, T.J. Patterns of spread in biological invasions dominated by long-distance jump dispersal: Insights from Argentine ants. PNAS 2001, 98, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Pratchett, M.S. Dynamics of an outbreak population of Acanthaster planci at Lizard Island, northern Great Barrier Reef (1995-1999). Coral Reefs 2005, 24, 453–462. [Google Scholar] [CrossRef]
- Guillemaud, T.; Ciosi, M.; Lombaert, E.; Estoup, A. Biological invasions in agricultural settings: Insights from evolutionary biology and population genetics. Comptes Rendus Biol. 2011, 334, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Handley, L.J.L.; Estoup, A.; Evans, D.M.; Thomas, C.E.; Lombaert, E.; Facon, B.; Aebi, A.; Roy, H.E. Ecological genetics of invasive alien species. BioControl 2011, 56, 409–428. [Google Scholar] [CrossRef]
- Lowe, W.H.; Allendorf, F.W. What can genetics tell us about population connectivity? Mol. Ecol. 2010, 19, 3038–3051. [Google Scholar] [CrossRef] [PubMed]
- Lombaert, E.; Guillemaud, T.; Thomas, C.E.; Lawson Handley, L.J.; Li, J.; Wang, S.; Pang, H.; Goryacheva, I.; Zakharov, I.A.; Jousselin, E.; et al. Inferring the origin of populations introduced from a genetically structured native range by approximate Bayesian computation: Case study of the invasive ladybird Harmonia axyridis. Mol. Ecol. 2011, 20, 4654–4670. [Google Scholar] [CrossRef] [PubMed]
- Estoup, A.; Lombaert, E.; Marin, J.-M.; Guillemaud, T.; Pudlo, P.; Robert, C.P.; Cornuet, J.-M. Estimation of demo-genetic model probabilities with Approximate Bayesian Computation using linear discriminant analysis on summary statistics. Mol. Ecol. Res. 2012, 12, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.A.; Zhang, W.; Balding, D.J. Approximate Bayesian Computation in Population Genetics. Genetics 2002, 162, 2025–2035. [Google Scholar] [PubMed]
- Yalcindag, E.; Elguero, E.; Arnathau, C.; Durand, P.; Akiana, J.; Anderson, T.J.; Aubouy, A.; Balloux, F.; Besnard, P.; Bogreau, H.; et al. Multiple independent introductions of Plasmodium falciparum in South America. PNAS 2012, 109, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, M.C.; Austerlitz, F.; Giraud, T.; Labbé, F.; Papura, D.; Richard Cervera, S.; Delmotte, F. Genetic signature of a range expansion and leap-frog event after the recent invasion of Europe by the grapevine downy mildew pathogen Plasmopara. viticola. Mol. Ecol. 2013, 22, 2771–2786. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, N.; Nagai, S.; Hamaguchi, M.; Lian, C.L.; Nadaoka, K. Development of microsatellite markers for the crown-of-thorns starfish Acanthaster planci. Mol. Ecol. Notes 2006, 6, 141–143. [Google Scholar] [CrossRef]
- Yasuda, N.; Nagai, S.; Hamaguchi, M.; Nadaoka, K. Seven new microsatellite markers for crown-of-thorns starfish Acanthaster planci. Plank 2007, 2, 103–106. [Google Scholar] [CrossRef]
- Wainwright, B.J.; Arlyza, I.S.; Karl, S.A. Eighteen microsatellite loci for the crown-of-thorns starfish, Acanthaster planci. Conserv. Genet. Res. 2012, 4, 861–863. [Google Scholar] [CrossRef]
- Harrison, H.B.; Saenz-Agudelo, P.; Al-Salamah, M. Microsatellite multiplex assay for the coral-eating crown-of-thorns starfish, Acanthaster. cf. planci. Conserv. Genet. 2015, 7, 627–630. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Alberto, F. MsatAllele_1.0: An R package to visualize the binning of microsatellite alleles. J. Heredity 2009, 100, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. An exact test for population differentiation. Evolution 1995, 49, 1280–1283. [Google Scholar] [CrossRef]
- Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Res. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.; Cockerham, C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.; Wills, D.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Brookfield, J.F. A simple new method for estimating null allele frequency from heterozygote deficiency. Mol. Ecol. 1996, 5, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G.; van Tienderen, P.H. genotype and genodive: Two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 2004, 4, 792–794. [Google Scholar] [CrossRef]
- Hedrick, P. Large variance in reproductive success and the Ne/N ratio. Evolution 2005, 59, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Ryman, N.; Palm, S. POWSIM: A computer program for assessing statistical power when testing for genetic differentiation. Mol. Ecol. Notes 2006, 6, 600–602. [Google Scholar] [CrossRef]
- Cornuet, J.-M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Leblois, R.; Marin, J.-M.; Estoup, A. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.-M.; Ravigné, V.; Estoup, A. Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1.0). BMC Bioinform. 2010, 11, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S. Isolation by Distance. Genetics 1943, 28, 114–138. [Google Scholar] [PubMed]
- Coltman, D.W.; Pilkington, J.G.; Smith, J.A.; Pemberton, J.M. Parasite-Mediated Selection against Inbred Soay Sheep in a Free-Living, Island Population. Evolution 1999, 53, 1259. [Google Scholar] [CrossRef]
- Amos, W.; Worthington Wilmer, J.; Fullard, K.; Burg, T.M.; Croxall, J.P.; Bloch, D.; Coulson, T. The influence of parental relatedness on reproductive success. Proc. R. Soc. Lond. Biol. 2001, 268, 2021–2027. [Google Scholar] [CrossRef] [PubMed]
- Alho, J.S.; Välimäki, K.; Merilä, J. Rhh: An R extension for estimating multilocus heterozygosity and heterozygosity-heterozygosity correlation. Mol. Ecol. Res. 2010, 10, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Dyer, R.J.; Nason, J.D. Population Graphs: The graph theoretic shape of genetic structure. Mol. Ecol. 2004, 13, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Clobert, J.; Danchin, E.; Dhondt, A.A.; Nichols, J.D. Dispersal; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Sax, D.F.; Stachowicz, J.J.; Gaines, S.D. Species Invasions: Insight into Ecology, Evolution and Biogeography; Sinauer Associates Incorporated: Sunderland, Massachusetts, 2005. [Google Scholar]
- Pearson, R.G. Recovery and Recolonization of Coral Reefs. Mar. Ecol. Prog. Ser. 1981, 4, 105–122. [Google Scholar] [CrossRef]
- Francois, O.; Durand, E. Spatially explicit Bayesian clustering models in population genetics. Mol. Ecol. Res. 2010, 10, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Benzie, J.A.H.; Wakeford, M. Genetic structure of crown-of-thorns starfish (Acanthaster. planci) on the Great Barrier Reef, Australia: Comparison of two sets of outbreak populations occurring ten years apart. Mar. Biol. 1997, 129, 149–157. [Google Scholar] [CrossRef]
- Muirhead, J.R.; Gray, D.K.; Kelly, D.W.; Ellis, S.M.; Heath, D.D.; Macisaac, H.J. Identifying the source of species invasions: Sampling intensity vs. genetic diversity. Mol. Ecol. 2008, 17, 1020–1035. [Google Scholar] [CrossRef] [PubMed]
- Geller, J.B.; Darling, J.A.; Carlton, J.T. Genetic perspectives on marine biological invasions. Ann. Rev. Mar. Sci. 2010, 2, 367–393. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, M.A.; Chong-Seng, K.M.; Pratchett, D.J.; Thompson, C.A.; Messmer, V.; Pratchett, M.S. Age and Growth of An Outbreaking Acanthaster cf. solaris Population within the Great Barrier Reef. Diversity 2017. accepted. [Google Scholar]
- Yasuda, N.; Hamaguchi, M.; Sasaki, M.; Nagai, S.; Saba, M.; Nadaoka, K. Complete mitochondrial genome sequences for Crown-of-thorns starfish Acanthaster planci and Acanthaster brevispinus. BMC Genom. 2006, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Vanhatalo, J.; Hosack, G.R.; Sweatman, H. Spatiotemporal modelling of crown-of-thorns starfish outbreaks on the Great Barrier Reef to inform control strategies. J. Appl. Ecol. 2016, 1–10. [Google Scholar] [CrossRef]
- Uthicke, S.; Doyle, J.; Duggan, S.; Yasuda, N.; McKinnon, A.D. Outbreak of coral-eating Crown-of-Thorns creates continuous cloud of larvae over 320 km of the Great Barrier Reef. Sci. Rep. 2015, 5, 16885. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, J.; Matthews, S.; Schultz, D.; Messmer, V.; Hoey, A.; Pratchett, M. Modelling Growth of Juvenile Crown-of-Thorns Starfish on the Northern Great Barrier Reef. Diversity 2016, 9, 1. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Region | Sampling Site | Latitude | Longitude | Date of Collection | N | AR | Ho | Hs |
|---|---|---|---|---|---|---|---|---|
| Lizard Is | Lizard Island reefs | −14.6916 | 145.4479 | Oct 2013–Feb 2015 | 385 | 5.6 | 0.673 | 0.685 |
| Lizard Is | MacGillivray Reef | −14.6524 | 145.4892 | Oct 2013 | 192 | 5.4 | 0.675 | 0.685 |
| Lizard Is | Nth Direction | −14.7445 | 145.5399 | Oct 2013 | 247 | 5.6 | 0.674 | 0.690 |
| Lizard Is | Sth Direction | −14.8562 | 145.4825 | Oct 2013 | 182 | 5.3 | 0.672 | 0.684 |
| Cooktown | Emily Reef | −15.6325 | 145.6511 | Feb 2014 | 278 | 5.5 | 0.687 | 0.688 |
| Cooktown | Endeavour Reef | −15.7823 | 145.5847 | Feb 2014 | 208 | 5.6 | 0.683 | 0.692 |
| Cooktown | Pickersgill Reef | −15.8838 | 145.5640 | Feb 2014 | 151 | 5.6 | 0.670 | 0.693 |
| Cooktown | Spitfire Reef | −16.1148 | 145.6424 | Oct 2013 | 155 | 5.4 | 0.673 | 0.688 |
| Cairns | Arlington Reef | −16.7749 | 145.9767 | Sept 2014 | 262 | 5.4 | 0.670 | 0.685 |
| Cairns | Hedley Reef | −17.2474 | 146.4637 | Sept 2014 | 275 | 5.4 | 0.666 | 0.684 |
| Cairns | McCulloch Reef | −17.2996 | 146.4257 | Sept 2014 | 265 | 5.4 | 0.673 | 0.686 |
| Townsville | Townsville reefs | −18.4303 | 146.8209 | Nov 2014 | 137 | 5.5 | 0.651 | 0.682 |
| Swains | Swains reefs | −22.3112 | 152.6720 | May 2015 | 68 | 5.5 | 0.685 | 0.694 |
| Locus name | N | Na | Ho | He | Missing Data (%) | HWE-p | HWE-h | F |
|---|---|---|---|---|---|---|---|---|
| AP1 | 2705 | 4 | 0.648 | 0.657 | 0.0 | 0.399 | 0.159 | 0.0091 |
| AP12QS | 2703 | 6 | 0.490 | 0.514 | 0.1 | 0.002 | 0.001 | 0.0321 |
| AP654 | 2689 | 11 | 0.685 | 0.702 | 0.6 | 0.389 | 0.047 | 0.0089 |
| AP9 | 2702 | 23 | 0.845 | 0.843 | 0.1 | 0.471 | 0.360 | 0.0009 |
| Hisayo01 | 2688 | 32 | 0.804 | 0.829 | 0.6 | 0.010 | 0.003 | 0.0179 |
| Maki03 | 2701 | 28 | 0.775 | 0.792 | 0.1 | 0.001 | 0.005 | 0.0123 |
| Sayo03 | 2659 | 9 | 0.492 | 0.519 | 1.7 | 0.010 | 0.006 | 0.0273 |
| AP11QS | 2703 | 11 | 0.751 | 0.762 | 0.1 | 0.171 | 0.119 | 0.0056 |
| AP30QS | 2705 | 9 | 0.617 | 0.625 | 0.0 | 0.010 | 0.001 | 0.0635 |
| AyU03 | 2704 | 29 | 0.885 | 0.876 | 0.0 | 0.276 | 0.917 | −0.0041 |
| Yukina06 | 2681 | 32 | 0.878 | 0.917 | 0.9 | 0.000 | 0.000 | 0.0287 |
| AP5QS | 2705 | 7 | 0.590 | 0.600 | 0.0 | 0.000 | 0.014 | 0.0211 |
| Apl07 | 2440 | 7 | 0.390 | 0.409 | 9.8 | 0.040 | 0.009 | 0.0269 |
| Apl19 | 2474 | 62 | 0.949 | 0.965 | 8.5 | 0.644 | 0.010 | 0.0066 |
| Etsuko01 | 2657 | 10 | 0.772 | 0.777 | 1.8 | 0.001 | 0.183 | 0.0045 |
| Sayo01 | 2704 | 7 | 0.214 | 0.220 | 0.0 | 0.095 | 0.035 | 0.0148 |
| Apl02 | 2577 | 10 | 0.653 | 0.669 | 4.7 | 0.335 | 0.032 | 0.0108 |
| Source of Variation | Nested in | % var 1 | F-stat | F-value | SD 2 | CI 2.5% | CI 97.5% | p-Value 3 |
|---|---|---|---|---|---|---|---|---|
| Within Individual | 0.98 | Fit | 0.020 | 0.004 | 0.012 | 0.028 | - | |
| Among Individual | Population | 0.02 | Fis | 0.020 | 0.004 | 0.012 | 0.028 | 0.000 |
| Among Reefs | Region | 0 | Fsc | 0.000 | 0.000 | 0.000 | 0.000 | 0.903 |
| Among Region | 0 | Fct | 0.000 | 0.000 | 0.000 | 0.000 | 0.830 |
| Lizard Island | MacGillivray | North Direction | South Direction | Emily | Endeavour | Pickersgill | Spitfire | Arlington | Hedley | McCulloch | Townsville | Swains | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lizard Island | 0.210 | 0.958 | 0.391 | 0.721 | 0.990 | 0.707 | 0.302 | 0.752 | 0.333 | 0.826 | 0.730 | 0.952 | |
| MacGillivray | 0.000 | 0.380 | 0.749 | 0.380 | 0.657 | 0.988 | 0.407 | 0.358 | 0.210 | 0.764 | 0.585 | 0.634 | |
| North Direction | 0.000 | 0.000 | 0.316 | 0.662 | 0.992 | 0.784 | 0.158 | 0.331 | 0.480 | 0.700 | 0.954 | 0.949 | |
| Soutth Direction | 0.000 | 0.000 | 0.000 | 0.827 | 0.764 | 0.248 | 0.380 | 0.622 | 0.960 | 0.821 | 0.980 | 0.857 | |
| Emily | 0.000 | 0.000 | 0.000 | 0.000 | 0.912 | 0.678 | 0.570 | 0.672 | 0.425 | 0.845 | 0.948 | 0.854 | |
| Endeavour | −0.001 | 0.000 | −0.001 | 0.000 | −0.001 | 0.876 | 0.551 | 0.637 | 0.850 | 0.889 | 0.991 | 1.000 | |
| Pickersgill | 0.000 | −0.001 | 0.000 | 0.000 | 0.000 | −0.001 | 0.349 | 0.553 | 0.096 | 0.484 | 0.601 | 0.745 | |
| Spitfire | 0.000 | 0.000 | 0.001 | 0.000 | 0.000 | 0.000 | 0.000 | 0.060 | 0.202 | 0.712 | 0.703 | 0.874 | |
| Arlington | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.001 | 0.717 | 0.865 | 0.854 | 0.739 | |
| Hedley | 0.000 | 0.000 | 0.000 | −0.001 | 0.000 | 0.000 | 0.001 | 0.000 | 0.000 | 0.892 | 0.948 | 0.952 | |
| McCulloch | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.822 | 0.895 | |
| Townsville | 0.000 | 0.000 | −0.001 | −0.001 | −0.001 | −0.001 | 0.000 | 0.000 | −0.001 | −0.001 | 0.000 | 0.986 | |
| Swains | −0.001 | 0.000 | −0.001 | −0.001 | −0.001 | −0.002 | −0.001 | −0.001 | −0.001 | −0.001 | −0.001 | −0.002 |
| Parameter | Definition | Distribution (Interval, Mean, SD) |
|---|---|---|
| Nanc | Effective population size of an ancestral population | Normal (103 to 104, 5 × 103, 103) |
| Nprim | Effective population size of a primary outbreak | Normal (103 to 104, 4 × 103, 8 × 102) |
| Nsamp | Effective population size of a sampled population | Normal (5 × 104 to 2 × 105, 8 × 104, 104) |
| t1 | Divergence time of independent outbreaks | Uniform (1 to 10) |
| db | Foundation time of a primary outbreak | Uniform (1 to 10) |
| t2 | Divergence time of northern and southern populations | Uniform (102 to 1.5 × 103) |
| Mean μ | Mean mutation rate | Gamma (10−4 to 10−3, 10−4, 2) |
| Mean P | Mean of the geometric distribution of the number of repeats | Gamma (10−2 to 10−0, 5.5 × 10−1, 3) |
| Mean μSNI | Mean single nucleotide insertion/deletion mutation rate | Gamma (10−8 to 5 × 10−5, 1.5 × 10−5, 3) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison, H.B.; Pratchett, M.S.; Messmer, V.; Saenz-Agudelo, P.; Berumen, M.L. Microsatellites Reveal Genetic Homogeneity among Outbreak Populations of Crown-of-Thorns Starfish (Acanthaster cf. solaris) on Australia’s Great Barrier Reef. Diversity 2017, 9, 16. https://doi.org/10.3390/d9010016
Harrison HB, Pratchett MS, Messmer V, Saenz-Agudelo P, Berumen ML. Microsatellites Reveal Genetic Homogeneity among Outbreak Populations of Crown-of-Thorns Starfish (Acanthaster cf. solaris) on Australia’s Great Barrier Reef. Diversity. 2017; 9(1):16. https://doi.org/10.3390/d9010016
Chicago/Turabian StyleHarrison, Hugo B., Morgan S. Pratchett, Vanessa Messmer, Pablo Saenz-Agudelo, and Michael L. Berumen. 2017. "Microsatellites Reveal Genetic Homogeneity among Outbreak Populations of Crown-of-Thorns Starfish (Acanthaster cf. solaris) on Australia’s Great Barrier Reef" Diversity 9, no. 1: 16. https://doi.org/10.3390/d9010016
APA StyleHarrison, H. B., Pratchett, M. S., Messmer, V., Saenz-Agudelo, P., & Berumen, M. L. (2017). Microsatellites Reveal Genetic Homogeneity among Outbreak Populations of Crown-of-Thorns Starfish (Acanthaster cf. solaris) on Australia’s Great Barrier Reef. Diversity, 9(1), 16. https://doi.org/10.3390/d9010016