Staphylococcus aureus Sequences from Osteomyelitic Specimens of a Pathological Bone Collection from Pre-Antibiotic Times

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
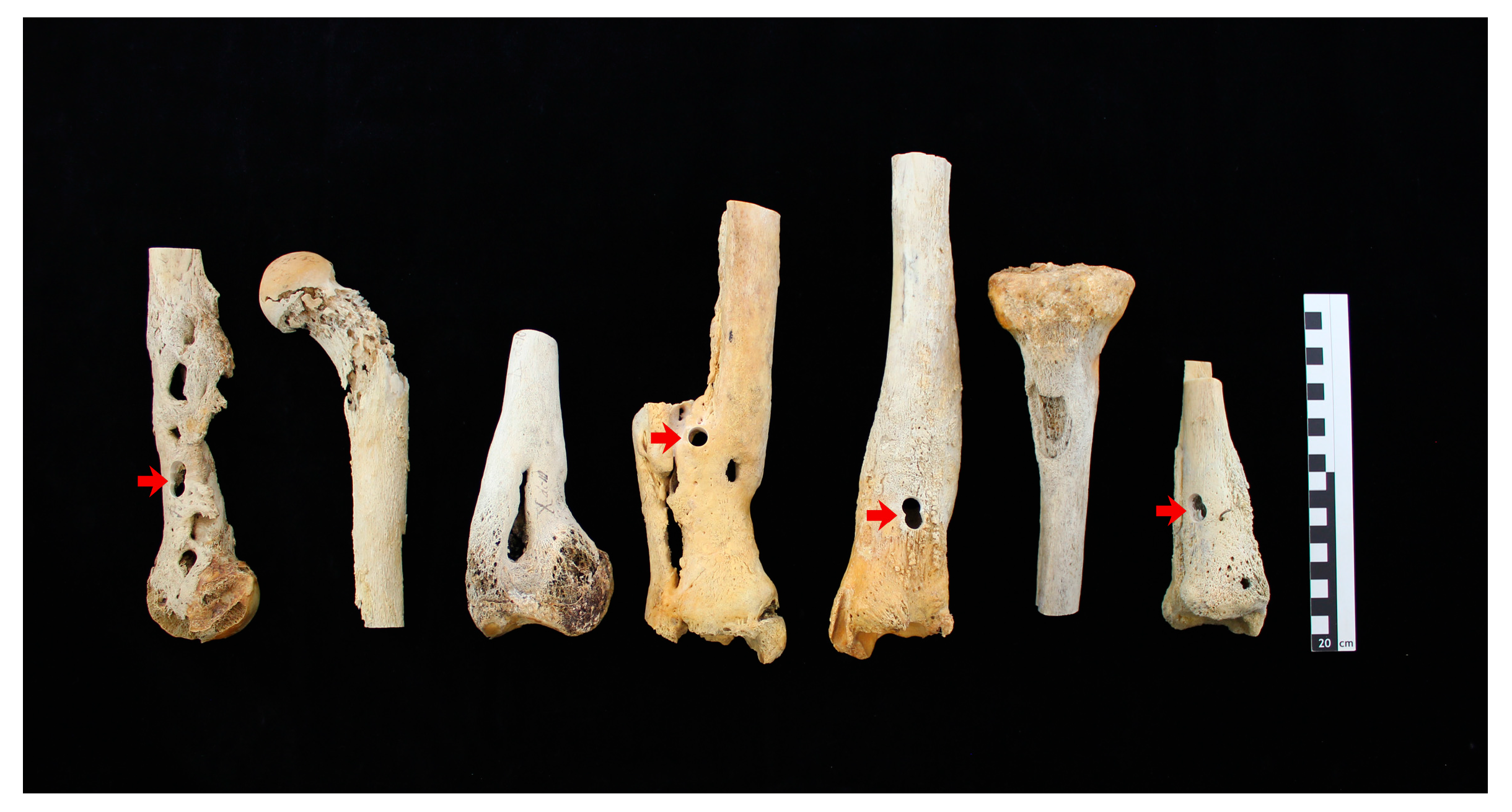
2.1.1. Osteomyelitic Bone Specimens
2.1.2. Staphylococcus aureus Genomic DNA
2.2. Methods
2.2.1. DNA Extraction
2.2.2. Primer Design
2.2.3. Amplification of S. aureus DNA
2.2.4. Sequence Analysis of PCR Products
2.2.5. Contamination Prevention
2.2.6. STR Typing
3. Results
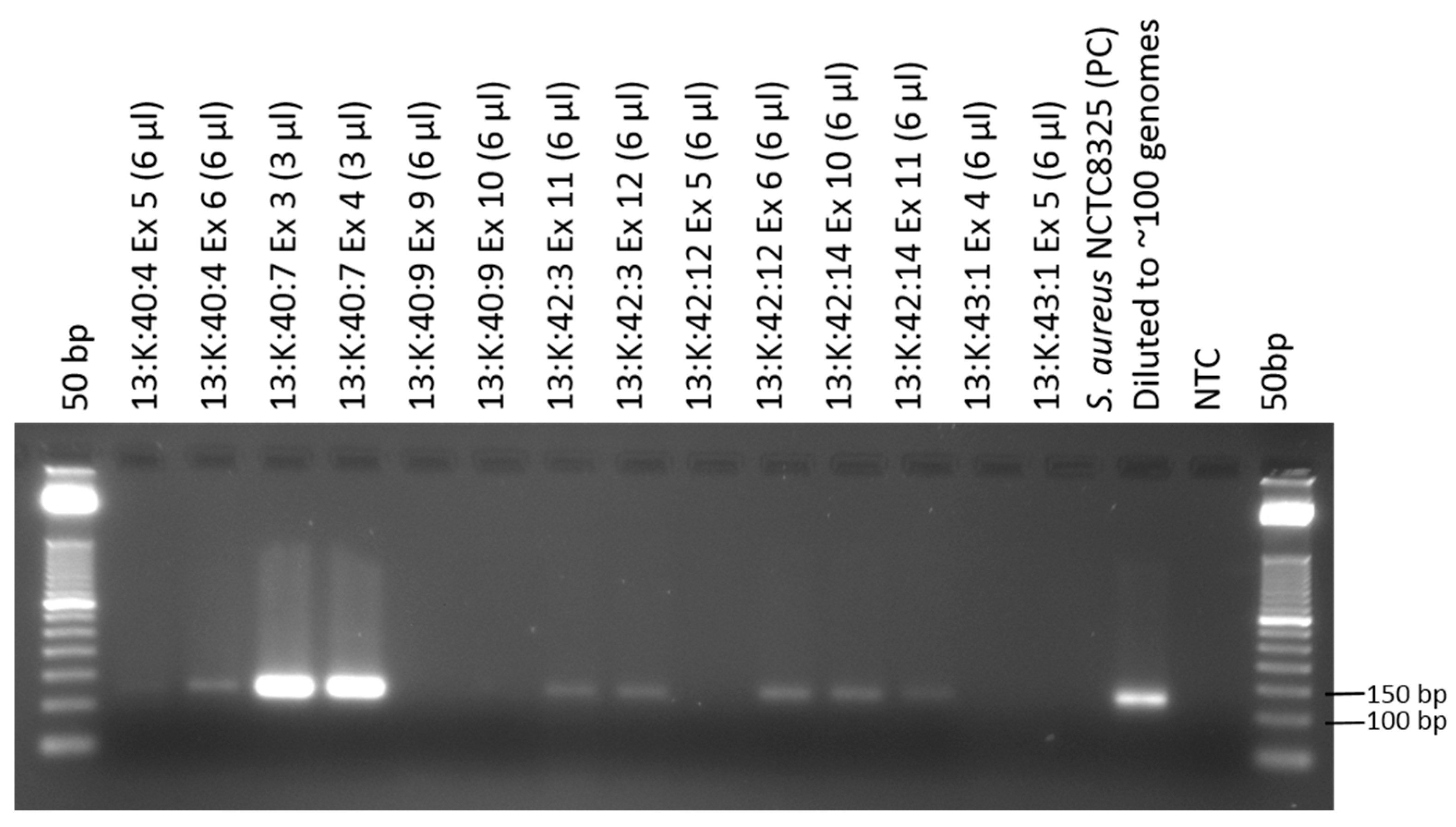
3.1. Amplification of Staphylococcus aureus Sequences from Osteomyelitic Bone Specimens
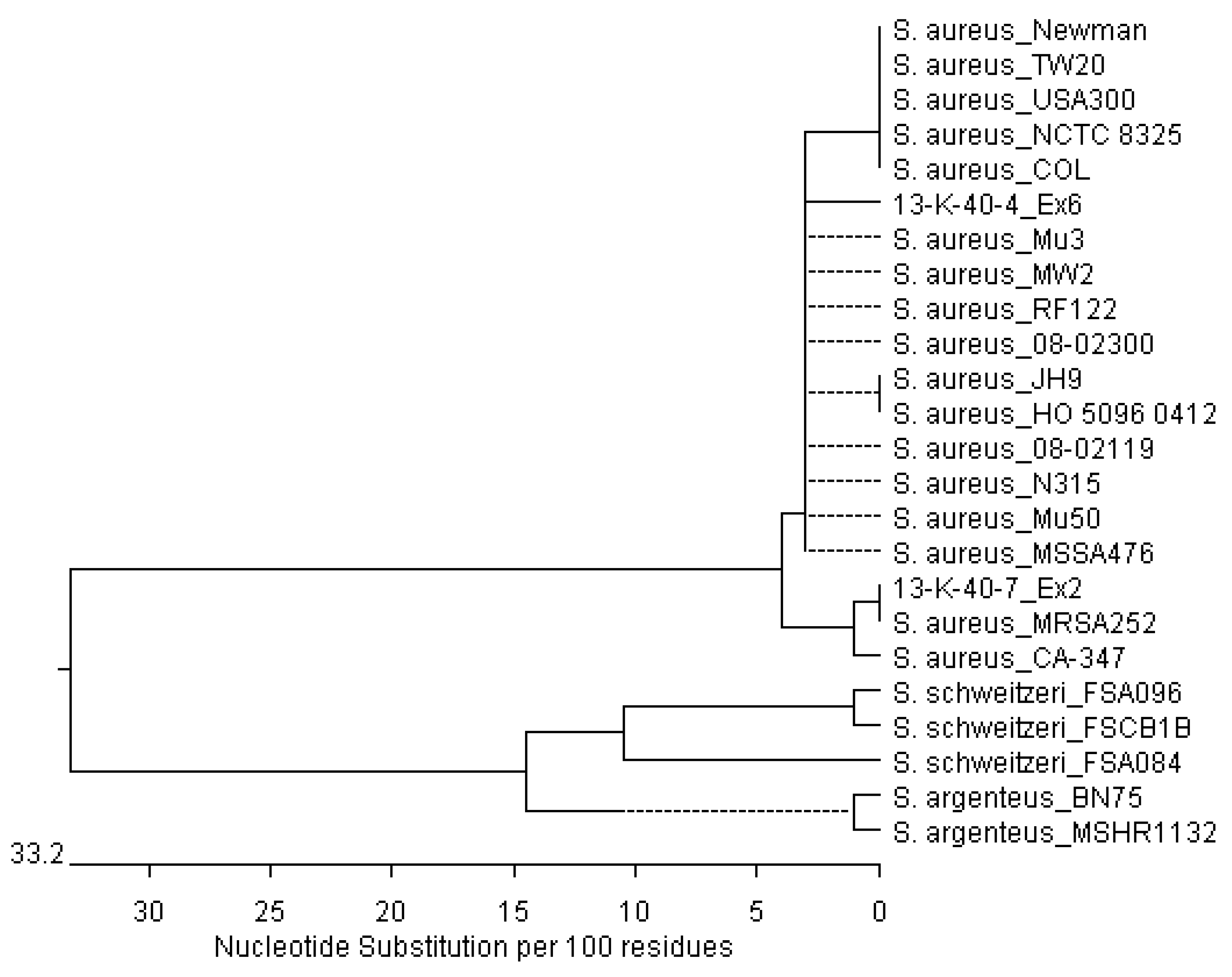
3.2. Sequencing of Staphylococcus aureus Sequences from Osteomyelitic Bone Specimens
3.3. STR Typing
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lew, D.P.; Waldvogel, F.A. Osteomyelitis. N. Engl. J. Med. 1997, 336, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Ortner, D.J. Infectious Diseases: Introduction, Biology, Osteomyelitis, Periostitis, Brucellosis, Glanders, and Septic Arthritis. In Identification of Pathological Conditions in Human Skeletal Remains; Ortner, D.J., Ed.; Academic Press: San Diego, CA, USA, 2003; ISBN 978-0-12-528628-2. [Google Scholar]
- Owen, W.B. The Diagnosis and Treatment of Osteomyelitis. Ann. Surg. 1936, 103, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.; Manchester, K. The Archaeology of Disease; The History Press: Stroud, Great Britain, 2010; ISBN 978-0-7524-9497-5. [Google Scholar]
- Skinner, D.; Keefer, C.S. Significance of Bacteremia Caused by Staphylococcus aureus: A Study of One Hundred and Twenty-Two Cases and a Review of the Literature Concerned with Experimental Infection in Animals. Arch. Intern. Med. 1941, 68, 851–875. [Google Scholar] [CrossRef]
- Fleming, A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to their Use in the Isolation of B. influenzæ. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- Chain, E.; Florey, H.W.; Adelaide, M.B.; Gardner, A.D.; Heatley, N.G.; Jennings, M.A.; Orr-Ewing, J.; Sanders, A.G. Penicillin as a chemotherapeutic agent. Lancet 1940, 236, 226–228. [Google Scholar] [CrossRef]
- Rammelkamp, C.H.; Maxon, T. Resistance of Staphylococcus aureus to the Action of Penicillin. Proc. Soc. Exp. Biol. Med. 1942, 51, 386–389. [Google Scholar] [CrossRef]
- Jevons, M.P. “Celbenin”—Resistant Staphylococci. Br. Med. J. 1961, 1, 124–125. [Google Scholar] [CrossRef]
- Parker, M.T.; Jevons, M.P. A Survey of Methicillin Resistance in Staphylococcus aureus. Postgrad. Med. J. 1964, 40, 170–178. [Google Scholar] [CrossRef]
- Sutherland, R.; Rolinson, G.N. Characteristics of Methicillin-Resistant Staphylococci. J. Bacteriol. 1964, 87, 887–899. [Google Scholar] [PubMed]
- Peacock, S.J.; Paterson, G.K. Mechanisms of Methicillin Resistance in Staphylococcus aureus. Annu. Rev. Biochem. 2015, 84, 577–601. [Google Scholar] [CrossRef] [PubMed]
- Paul-Ehrlich-Gesellschaft für Chemotherapie, e.V. Studiengruppe Epidemiologie und Resistenzsituation bei Klinisch wichtigen Infektionserregern aus dem Ambulanten Versorgungsbereich bzw. aus dem dem Hospitalbereich Gegenüber Antibiotika. Available online: http://www.p-e-g.org/econtext/Berichte%20der%20Studien/ (accessed on 18 May 2017).
- European Antimicrobial Resistance Surveillance Network (EARS-Net). Annual Surveillance Reports on Antimicrobial Resistance. Available online: http://ecdc.europa.eu/en/antimicrobial-resistance/surveillance-and-disease-data/report (accessed on 18 May 2017).
- Deurenberg, R.H.; Stobberingh, E.E. The evolution of Staphylococcus aureus. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2008, 8, 747–763. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.; Waglechner, N.; Pawlowski, A.; Koteva, K.; Banks, E.D.; Johnston, M.D.; Barton, H.A.; Wright, G.D. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS ONE 2012, 7, e34953. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the antibiotic resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Drancourt, M.; Raoult, D. Palaeomicrobiology: Current issues and perspectives. Nat. Rev. Microbiol. 2005, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Zink, A.R.; Reischl, U.; Wolf, H.; Nerlich, A.G. Molecular analysis of ancient microbial infections. FEMS Microbiol. Lett. 2002, 213, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Salo, W.L.; Aufderheide, A.C.; Buikstra, J.; Holcomb, T.A. Identification of Mycobacterium tuberculosis DNA in a pre-Columbian Peruvian mummy. Proc. Natl. Acad. Sci. USA 1994, 91, 2091–2094. [Google Scholar] [CrossRef] [PubMed]
- Spigelman, M.; Lemma, E. The use of the polymerase chain reaction (PCR) to detect Mycobacterium tuberculosis in ancient skeletons. Int. J. Osteoarchaeol. 1993, 3, 137–143. [Google Scholar] [CrossRef]
- Baron, H.; Hummel, S.; Herrmann, B. Mycobacterium tuberculosis Complex DNA in Ancient Human Bones. J. Archaeol. Sci. 1996, 23, 667–671. [Google Scholar] [CrossRef]
- Donoghue, H.D.; Spigelman, M.; Zias, J.; Gernaey-Child, A.M.; Minnikin, D.E. Mycobacterium tuberculosis complex DNA in calcified pleura from remains 1400 years old. Lett. Appl. Microbiol. 1998, 27, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.M.; Goyal, M.; Legge, A.J.; Shaw, R.J.; Young, D. Genotypic analysis of Mycobacterium tuberculosis from medieval human remains. Microbiology 1999, 145, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Rafi, A.; Spigelman, M.; Stanford, J.; Lemma, E.; Donoghue, H.; Zias, J. Mycobacterium leprae DNA from ancient bone detected by PCR. Lancet Lond. Engl. 1994, 343, 1360–1361. [Google Scholar] [CrossRef]
- Haas, C.J.; Zink, A.; Pálfi, G.; Szeimies, U.; Nerlich, A.G. Detection of leprosy in ancient human skeletal remains by molecular identification of Mycobacterium leprae. Am. J. Clin. Pathol. 2000, 114, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, H.D.; Holton, J.; Spigelman, M. PCR primers that can detect low levels of Mycobacterium leprae DNA. J. Med. Microbiol. 2001, 50, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Drancourt, M.; Aboudharam, G.; Signoli, M.; Dutour, O.; Raoult, D. Detection of 400-year-old Yersinia pestis DNA in human dental pulp: An approach to the diagnosis of ancient septicemia. Proc. Natl. Acad. Sci. USA 1998, 95, 12637–12640. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Aboudharam, G.; Crubézy, E.; Larrouy, G.; Ludes, B.; Drancourt, M. Molecular identification by “suicide PCR” of Yersinia pestis as the agent of Medieval Black Death. Proc. Natl. Acad. Sci. USA 2000, 97, 12800–12803. [Google Scholar] [CrossRef] [PubMed]
- Drancourt, M.; Tran-Hung, L.; Courtin, J.; de Lumley, H.; Raoult, D. Bartonella quintana in a 4000-year-old human tooth. J. Infect. Dis. 2005, 191, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Grumbkow, P.V.; Zipp, A.; Seidenberg, V.; Fehren-Schmitz, L.; Kempf, V.A.J.; Gross, U.; Hummel, S. Brief communication: Evidence of Bartonella quintana infections in skeletons of a historical mass grave in Kassel, Germany. Am. J. Phys. Anthropol. 2011, 146, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, B.; Grupe, G.; Hummel, S.; Piepenbrink, H.; Schutkowski, H. Prähistorische Anthropologie: Leitfaden der Feld- und Labormethoden; Springer: Berlin Heidelberg, Germany, 1990; ISBN 0-387-52541-6. [Google Scholar]
- Ancient DNA: Recovery and Analysis of Genetic Material from Paleontological, Archaeological, Museum, Medical, and Forensic Specimens; Herrmann, B.; Hummel, S. (Eds.) Springer: New York, NY, USA, 1994; ISBN 13: 9780387943084. [Google Scholar]
- Gillaspy, A.F.; Worrell, V.; Orvis, J.; Roe, B.A.; Dyer, D.W.; Iandolo, J.J. The Staphylococcus aureus NCTC 8325 Genome. In Gram-Positive Pathogens, 2nd ed.; American Society of Microbiology: Washington, DC, USA, 2006; pp. 381–412. [Google Scholar] [CrossRef]
- Kemp, B.M.; Smith, D.G. Use of bleach to eliminate contaminating DNA from the surface of bones and teeth. Forensic Sci. Int. 2005, 154, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Barta, J.L.; Monroe, C.; Kemp, B.M. Further evaluation of the efficacy of contamination removal from bone surfaces. Forensic Sci. Int. 2013, 231, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Frischalowski, M.; Seidenberg, V.; Grosskopf, B.; Wulf, F.-W.; Hummel, S. Molekulargenetische Untersuchung des Verwandtschaftsverhältnisses von möglichen Mutter-Kind-Bestattungen aus dem frühneuzeitlichen Eldagsen. Nachrichten aus Niedersachsens Urgeschichte 2015, 84, 193–206. [Google Scholar]
- Hummel, S. Ancient DNA Typing: Methods, Strategies and Applications; Springer Science & Business Media: New York, NY, USA, 2003; ISBN 978-3-540-43037-7. [Google Scholar]
- Shortle, D. A genetic system for analysis of staphylococcal nuclease. Gene 1983, 22, 181–189. [Google Scholar] [CrossRef]
- Bodén, M.K.; Flock, J.I. Cloning and characterization of a gene for a 19 kDa fibrinogen-binding protein from Staphylococcus aureus. Mol. Microbiol. 1994, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinform. Oxf. Engl. 2008, 24, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Frénay, H.M.; Theelen, J.P.; Schouls, L.M.; Vandenbroucke-Grauls, C.M.; Verhoef, J.; van Leeuwen, W.J.; Mooi, F.R. Discrimination of epidemic and nonepidemic methicillin-resistant Staphylococcus aureus strains on the basis of protein A gene polymorphism. J. Clin. Microbiol. 1994, 32, 846–847. [Google Scholar] [PubMed]
- Frénay, H.M.; Bunschoten, A.E.; Schouls, L.M.; van Leeuwen, W.J.; Vandenbroucke-Grauls, C.M.; Verhoef, J.; Mooi, F.R. Molecular typing of methicillin-resistant Staphylococcus aureus on the basis of protein A gene polymorphism. Eur. J. Clin. Microbiol. Infect. Dis. 1996, 15, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Strommenger, B.; Kettlitz, C.; Weniger, T.; Harmsen, D.; Friedrich, A.W.; Witte, W. Assignment of Staphylococcus Isolates to Groups by spa Typing, SmaI Macrorestriction Analysis, and Multilocus Sequence Typing. J. Clin. Microbiol. 2006, 44, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Llamas, B.; Valverde, G.; Fehren-Schmitz, L.; Weyrich, L.S.; Cooper, A.; Haak, W. From the field to the laboratory: Controlling DNA contamination in human ancient DNA research in the high-throughput sequencing era. STAR Sci. Technol. Archaeol. Res. 2017, 3, 1–14. [Google Scholar] [CrossRef]
- Seidenberg, V.; Schilz, F.; Pfister, D.; Georges, L.; Fehren-Schmitz, L.; Hummel, S. A new miniSTR heptaplex system for genetic fingerprinting of ancient DNA from archaeological human bone. J. Archaeol. Sci. 2012, 39, 3224–3229. [Google Scholar] [CrossRef]
- Harmsen, D.; Claus, H.; Witte, W.; Rothgänger, J.; Claus, H.; Turnwald, D.; Vogel, U. Typing of Methicillin-Resistant Staphylococcus aureus in a University Hospital Setting by Using Novel Software for spa Repeat Determination and Database Management. J. Clin. Microbiol. 2003, 41, 5442–5448. [Google Scholar] [CrossRef] [PubMed]
- Schaumburg, F.; Pauly, M.; Schubert, G.; Shittu, A.; Tong, S.; Leendertz, F.; Peters, G.; Becker, K. Characterization of a Novel Thermostable Nuclease Homolog (NucM) in a Highly Divergent Staphylococcus aureus Clade. J. Clin. Microbiol. 2014, 52, 4036–4038. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.C.; Holden, M.T.G.; Tong, S.Y.C.; Castillo-Ramirez, S.; Clarke, L.; Quail, M.A.; Currie, B.J.; Parkhill, J.; Bentley, S.D.; Feil, E.J.; Giffard, P.M. A very early-branching Staphylococcus aureus lineage lacking the carotenoid pigment staphyloxanthin. Genome Biol. Evol. 2011, 3, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.; Rickmeyer, J.; Gajdiss, M.; Thye, T.; Lorenzen, S.; Reif, M.; Josten, M.; Szekat, C.; Melo, L.D.R.; Schmithausen, R.M.; et al. Differentiation of Staphylococcus argenteus (formerly: Staphylococcus aureus clonal complex 75) by mass spectrometry from S. aureus using the first strain isolated from a wild African great ape. Int. J. Med. Microbiol. IJMM 2017, 307, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Holden, M.T.G.; Feil, E.J.; Lindsay, J.A.; Peacock, S.J.; Day, N.P.J.; Enright, M.C.; Foster, T.J.; Moore, C.E.; Hurst, L.; Atkin, R.; et al. Complete genomes of two clinical Staphylococcus aureus strains: Evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. USA 2004, 101, 9786–9791. [Google Scholar] [CrossRef] [PubMed]
- Stegger, M.; Driebe, E.M.; Roe, C.; Lemmer, D.; Bowers, J.R.; Engelthaler, D.M.; Keim, P.; Andersen, P.S. Genome Sequence of Staphylococcus aureus Strain CA-347, a USA600 Methicillin-Resistant Isolate. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Neoh, H.; Cui, L.; Yuzawa, H.; Takeuchi, F.; Matsuo, M.; Hiramatsu, K. Mutated Response Regulator graR Is Responsible for Phenotypic Conversion of Staphylococcus aureus from Heterogeneous Vancomycin-Intermediate Resistance to Vancomycin-Intermediate Resistance. Antimicrob. Agents Chemother. 2008, 52, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Takeuchi, F.; Kuroda, M.; Yuzawa, H.; Aoki, K.; Oguchi, A.; Nagai, Y.; Iwama, N.; Asano, K.; Naimi, T.; et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet Lond. Engl. 2002, 359, 1819–1827. [Google Scholar] [CrossRef]
- Herron-Olson, L.; Fitzgerald, J.R.; Musser, J.M.; Kapur, V. Molecular Correlates of Host Specialization in Staphylococcus aureus. PLoS ONE 2007, 2, e1120. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.E.; Layer, F.; Fuchs, S.; Bender, J.K.; Fiedler, S.; Werner, G.; Strommenger, B. Complete Genome Sequences of Two Methicillin-Sensitive Staphylococcus aureus Isolates Representing a Population Subset Highly Prevalent in Human Colonization. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, M.M.; Wu, S.W.; Zhou, Y.; Sieradzki, K.; de Lencastre, H.; Richardson, P.; Bruce, D.; Rubin, E.; Myers, E.; Siggia, E.D.; et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2007, 104, 9451–9456. [Google Scholar] [CrossRef] [PubMed]
- Holden, M.T.G.; Hsu, L.-Y.; Kurt, K.; Weinert, L.A.; Mather, A.E.; Harris, S.R.; Strommenger, B.; Layer, F.; Witte, W.; de Lencastre, H.; et al. A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res. 2013, 23, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.; Oguchi, A.; Aoki, K.; Nagai, Y.; et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet Lond. Engl. 2001, 357, 1225–1240. [Google Scholar] [CrossRef]
- Gill, P.; Whitaker, J.; Flaxman, C.; Brown, N.; Buckleton, J. An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci. Int. 2000, 112, 17–40. [Google Scholar] [CrossRef]
- Advanced Topics in Forensic DNA Typing: Interpretation; Butler, J.M. (Ed.) Academic Press: San Diego, CA, USA, 2015; ISBN 978-0-12-405213-0. [Google Scholar]
- Kluytmans, J.; van Belkum, A.; Verbrugh, H. Nasal carriage of Staphylococcus aureus: Epidemiology, underlying mechanisms, and associated risks. Clin. Microbiol. Rev. 1997, 10, 505–520. [Google Scholar] [PubMed]
- Cespedes, C.; Saïd-Salim, B.; Miller, M.; Lo, S.-H.; Kreiswirth, B.N.; Gordon, R.J.; Vavagiakis, P.; Klein, R.S.; Lowy, F.D. The Clonality of Staphylococcus aureus Nasal Carriage. J. Infect. Dis. 2005, 191, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Votintseva, A.A.; Miller, R.R.; Fung, R.; Knox, K.; Godwin, H.; Peto, T.E.A.; Crook, D.W.; Bowden, R.; Walker, A.S. Multiple-Strain Colonization in Nasal Carriers of Staphylococcus aureus. J. Clin. Microbiol. 2014, 52, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, F.R.; Kennedy, A.D.; Chen, L.; Wardenburg, J.B.; Kobayashi, S.D.; Mathema, B.; Braughton, K.R.; Whitney, A.R.; Villaruz, A.E.; Martens, C.A.; et al. Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2011, 108, 18091–18096. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Takigawa, W.; Tanigawa, K.; Nakamura, K.; Ishido, Y.; Kawashima, A.; Wu, H.; Akama, T.; Sue, M.; Yoshihara, A.; et al. Detection of Mycobacterium leprae DNA from Archaeological Skeletal Remains in Japan Using Whole Genome Amplification and Polymerase Chain Reaction. PLoS ONE 2010, 5, e12422. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, A.S.; Kennedy, S.L.; Müller, R.; Stephens, R.H.; Holst, M.; Caffell, A.C.; Roberts, C.A.; Brown, T.A. Genotype of a historic strain of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2012, 109, 18511–18516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, M.; Seifert, L.; Hänsch, S.; Wagner, D.M.; Birdsell, D.; Parise, K.L.; Wiechmann, I.; Grupe, G.; Thomas, A.; Keim, P.; et al. Yersinia pestis DNA from Skeletal Remains from the 6th Century AD Reveals Insights into Justinianic Plague. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuenemann, V.J.; Singh, P.; Mendum, T.A.; Krause-Kyora, B.; Jäger, G.; Bos, K.I.; Herbig, A.; Economou, C.; Benjak, A.; Busso, P.; et al. Genome-wide comparison of medieval and modern Mycobacterium leprae. Science 2013, 341, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.M.; Tucker, K.; Butler, R.; Pike, A.W.G.; Lewis, J.; Roffey, S.; Marter, P.; Lee, O.Y.-C.; Wu, H.H.T.; Minnikin, D.E.; et al. Detection and strain typing of ancient Mycobacterium leprae from a medieval leprosy hospital. PLoS ONE 2013, 8, e62406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendum, T.A.; Schuenemann, V.J.; Roffey, S.; Taylor, G.M.; Wu, H.; Singh, P.; Tucker, K.; Hinds, J.; Cole, S.T.; Kierzek, A.M.; et al. Mycobacterium leprae genomes from a British medieval leprosy hospital: Towards understanding an ancient epidemic. BMC Genomics 2014, 15, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, R.; Roberts, C.A.; Brown, T.A. Genotyping of ancient Mycobacterium tuberculosis strains reveals historic genetic diversity. Proc. R Soc. B 2014, 281, 20133236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, K.I.; Herbig, A.; Sahl, J.; Waglechner, N.; Fourment, M.; Forrest, S.A.; Klunk, J.; Schuenemann, V.J.; Poinar, D.; Kuch, M.; et al. Eighteenth century Yersinia pestis genomes reveal the long-term persistence of an historical plague focus. eLife 2016, 5, e12994. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.; Harbeck, M.; Keller, M.; Spyrou, M.A.; Rott, A.; Trautmann, B.; Scholz, H.C.; Päffgen, B.; Peters, J.; McCormick, M.; et al. A High-Coverage Yersinia pestis Genome from a Sixth-Century Justinianic Plague Victim. Mol. Biol. Evol. 2016, 33, 2911–2923. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Krafft, A.E.; Bijwaard, K.E.; Fanning, T.G. Initial genetic characterization of the 1918 “Spanish” influenza virus. Science 1997, 275, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Devault, A.M.; Golding, G.B.; Waglechner, N.; Enk, J.M.; Kuch, M.; Tien, J.H.; Shi, M.; Fisman, D.N.; Dhody, A.N.; Forrest, S.; et al. Second-Pandemic Strain of Vibrio cholerae from the Philadelphia Cholera Outbreak of 1849. N. Engl. J. Med. 2014, 370, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Maixner, F.; Krause-Kyora, B.; Turaev, D.; Herbig, A.; Hoopmann, M.R.; Hallows, J.L.; Kusebauch, U.; Vigl, E.E.; Malfertheiner, P.; Megraud, F.; et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science 2016, 351, 162–165. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Name | Primer Length | Sequence | Position in S. aureus Reference Sequence (NCTC 8325, Accession CP000253.1) | Product Length |
|---|---|---|---|---|---|
| Forward | S.aur nuc up 2 | 26-mer | 5’-GGCAATTGTTTCAATATTACTTATAG-3’ | 800069…800094 | 122 bp |
| Reverse | S.aur nuc low | 28-mer | 5’-TTGAAACTACAACTAAAGTTAACACTAA-3’ | 800163…800190 | |
| Forward | S.aur fib up | 20-mer | 5’-GAAGGATACGGTCCAAGAGA-3’ | 1073163…1073128 | 111 bp |
| Reverse | S.aur fib low | 23-mer | 5’-AGGTGTTGAGTTAAATTTTGGTC-3’ | 1073251…1073273 |
| Bone Specimen | Sampling Position (Figure 1) | nuc | fib |
|---|---|---|---|
| 13:K:40:4 | diaphysis | + | + |
| metaphysis | + | + | |
| 13:K:40:7 | diaphysis | + | + |
| 13:K:40:9 | diaphysis | + | + |
| 13:K:42:3 | diaphysis | + | + |
| metaphysis | − | + | |
| 13:K:42:12 | diaphysis | + | + |
| metaphysis | + | + | |
| 13:K:42:14 | diaphysis | + | + |
| 13:K:43:1 | diaphysis | − | − |
| metaphysis | − | + |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flux, A.L.; Mazanec, J.; Strommenger, B.; Hummel, S. Staphylococcus aureus Sequences from Osteomyelitic Specimens of a Pathological Bone Collection from Pre-Antibiotic Times. Diversity 2017, 9, 43. https://doi.org/10.3390/d9040043
Flux AL, Mazanec J, Strommenger B, Hummel S. Staphylococcus aureus Sequences from Osteomyelitic Specimens of a Pathological Bone Collection from Pre-Antibiotic Times. Diversity. 2017; 9(4):43. https://doi.org/10.3390/d9040043
Chicago/Turabian StyleFlux, Anna Lena, Janine Mazanec, Birgit Strommenger, and Susanne Hummel. 2017. "Staphylococcus aureus Sequences from Osteomyelitic Specimens of a Pathological Bone Collection from Pre-Antibiotic Times" Diversity 9, no. 4: 43. https://doi.org/10.3390/d9040043
APA StyleFlux, A. L., Mazanec, J., Strommenger, B., & Hummel, S. (2017). Staphylococcus aureus Sequences from Osteomyelitic Specimens of a Pathological Bone Collection from Pre-Antibiotic Times. Diversity, 9(4), 43. https://doi.org/10.3390/d9040043