Microglia M2A Polarization as Potential Link between Food Allergy and Autism Spectrum Disorders

{kind=link}
Abstract
:1. Introduction
2. Environmental Risk Factors for ASD
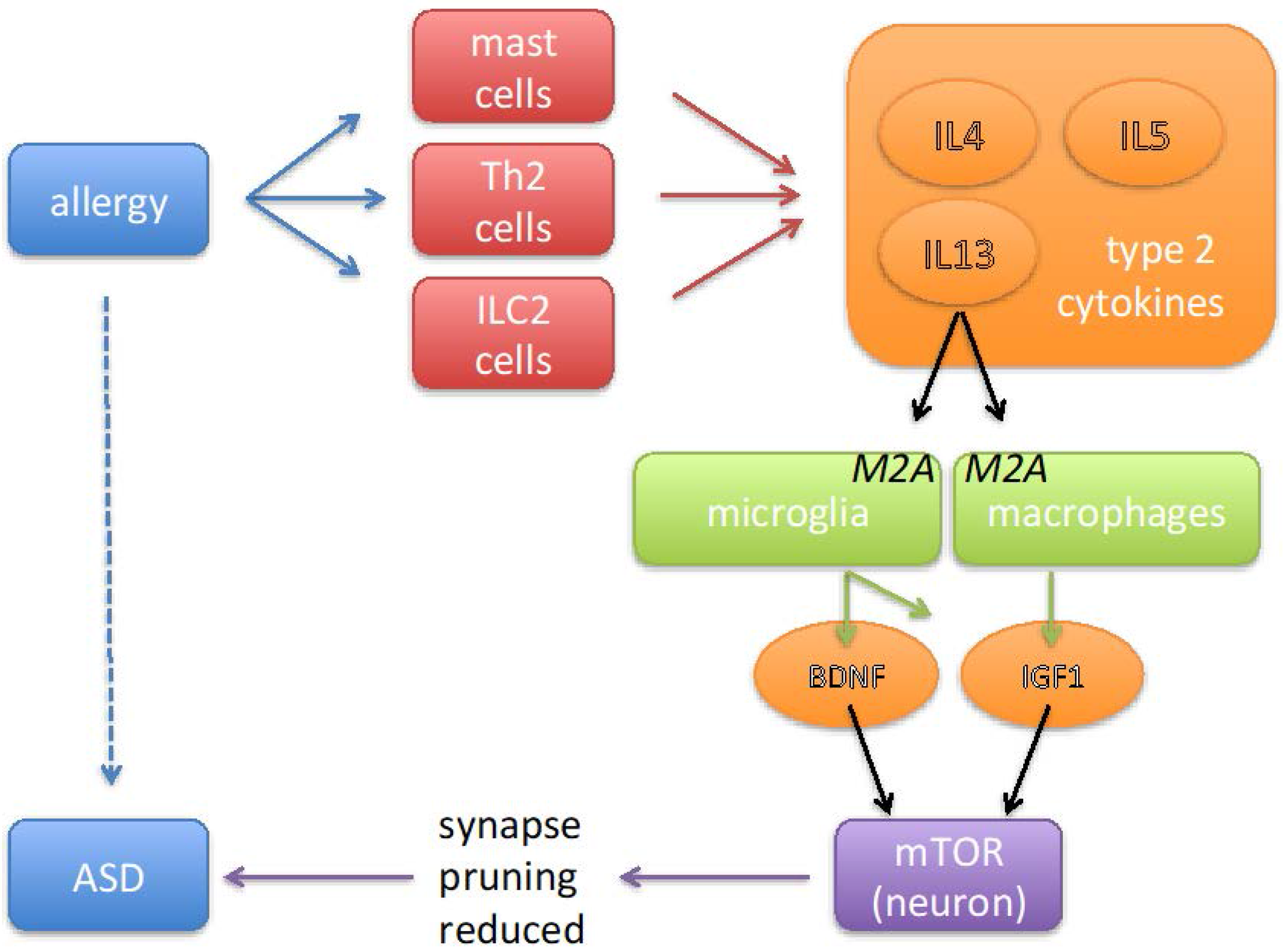
3. Mast Cells, Th2 Lymphocytes, and Cytokine Profiles in ASD
4. IL4 Induces M2A Polarization of Macrophages and Microglia
5. Changes in Post Mortem Brain of ASD
6. Growth Factors from M2A-Polarized Microglia Are Associated with ASD
7. Mechanisms by Which Growth Factors May Cause ASD Symptoms
8. Discussion
Acknowledgments
Conflicts of Interest
References
- Happe, F.; Ronald, A. The ‘fractionable autism triad’: A review of evidence from behavioural, genetic, cognitive and neural research. Neuropsychol. Rev. 2008, 18, 287–304. [Google Scholar] [PubMed]
- King, B.H.; Navot, N.; Bernier, R.; Webb, S.J. Update on diagnostic classification in autism. Curr. Opin. Psychiatry 2014, 27, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.K. The savant syndrome: Intellectual impairment and exceptional skill. Psychol. Bull. 1999, 125, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Young, A.M.; Chakrabarti, B.; Roberts, D.; Lai, M.C.; Suckling, J.; Baron-Cohen, S. From molecules to neural morphology: Understanding neuroinflammation in autism spectrum condition. Mol. Autism 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.R.; Iossifov, I.; Levy, D.; Ronemus, M.; Wigler, M.; Vitkup, D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 2011, 70, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Hussman, J.P.; Chung, R.H.; Griswold, A.J.; Jaworski, J.M.; Salyakina, D.; Ma, D.; Konidari, I.; Whitehead, P.L.; Vance, J.M.; Martin, E.R.; et al. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol. Autism 2011, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Toro, R.; Konyukh, M.; Delorme, R.; Leblond, C.; Chaste, P.; Fauchereau, F.; Coleman, M.; Leboyer, M.; Gillberg, C.; Bourgeron, T. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010, 26, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.P.; Wilkerson, J.R.; Guo, W.; Maksimova, M.A.; DeMartino, G.N.; Cowan, C.W.; Huber, K.M. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 2012, 151, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Eapen, V. Converging pathways in autism spectrum disorders: Interplay between synaptic dysfunction and immune responses. Front. Hum. Neurosci. 2013, 7, 738. [Google Scholar] [CrossRef] [PubMed]
- Krey, J.F.; Pasca, S.P.; Shcheglovitov, A.; Yazawa, M.; Schwemberger, R.; Rasmusson, R.; Dolmetsch, R.E. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 2013, 16, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Poultney, C.S.; Goldberg, A.P.; Drapeau, E.; Kou, Y.; Harony-Nicolas, H.; Kajiwara, Y.; De Rubeis, S.; Durand, S.; Stevens, C.; Rehnstrom, K.; et al. Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am. J. Hum. Genet. 2013, 93, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Klann, E.; Costa-Mattioli, M.; Zukin, R.S. Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J. Neurosci. 2015, 35, 13836–13842. [Google Scholar] [CrossRef] [PubMed]
- Sato, A. mTOR, a potential target to treat autism spectrum disorder. CNS Neurol. Disord. Drug Targets 2016, 15, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry 2015, 72, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tick, B.; Bolton, P.; Happe, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M. Environmental factors in autism. Front. Psychiatry 2012, 3, 118. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Asadi, S.; Patel, A.B. Focal brain inflammation and autism. J. Neuroinflamm. 2013, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; McAllister, A.K. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 469–486. [Google Scholar] [PubMed]
- Becker, K.G. Autism, asthma, inflammation, and the hygiene hypothesis. Med. Hypotheses 2007, 69, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Gurney, J.G.; McPheeters, M.L.; Davis, M.M. Parental report of health conditions and health care use among children with and without autism: National survey of children’s health. Arch. Pediatr. Adolesc. Med. 2006, 160, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Lyall, K.; Van de Water, J.; Ashwood, P.; Hertz-Picciotto, I. Asthma and allergies in children with autism spectrum disorders: Results from the CHARGE study. Autism Res. 2015, 8, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Croen, L.A.; Grether, J.K.; Yoshida, C.K.; Odouli, R.; Van de Water, J. Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: A case-control study. Arch. Pediatr. Adolesc. Med. 2005, 159, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Entringer, S.; Kumsta, R.; Nelson, E.L.; Hellhammer, D.H.; Wadhwa, P.D.; Wust, S. Influence of prenatal psychosocial stress on cytokine production in adult women. Dev. Psychobiol. 2008, 50, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.; Fedulov, A.V.; Kobzik, L. Maternal stress during pregnancy increases neonatal allergy susceptibility: Role of glucocorticoids. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L141–L148. [Google Scholar] [CrossRef] [PubMed]
- von Hertzen, L.C. Maternal stress and T-cell differentiation of the developing immune system: Possible implications for the development of asthma and atopy. J. Allergy Clin. Immunol. 2002, 109, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Kinney, D.K.; Munir, K.M.; Crowley, D.J.; Miller, A.M. Prenatal stress and risk for autism. Neurosci. Biobehav. Rev. 2008, 32, 1519–1532. [Google Scholar] [CrossRef] [PubMed]
- Ronald, A.; Pennell, C.E.; Whitehouse, A.J. Prenatal maternal stress associated with ADHD and autistic traits in early childhood. Front. Psychol. 2010, 1, 223. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, E.S.; Pinto-Mariz, F.; Bastos-Pinto, S.; Pontes, A.T.; Prado, E.A.; deAzevedo, L.C. Immune allergic response in Asperger syndrome. J. Neuroimmunol. 2009, 216, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Farmer, C.A.; Thurm, A.; Shebl, F.M.; Ilieva, J.; Kalra, S.; Swedo, S. Serum and cerebrospinal fluid immune mediators in children with autistic disorder: A longitudinal study. Mol. Autism 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Angelidou, A.; Alysandratos, K.D.; Asadi, S.; Zhang, B.; Francis, K.; Vasiadi, M.; Kalogeromitros, D.; Theoharides, T.C. Brief report: “Allergic symptoms” in children with autism spectrum disorders. More than meets the eye? J. Autism Dev. Disord. 2011, 41, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Chawarska, K.; Campbell, D.; Chen, L.; Shic, F.; Klin, A.; Chang, J. Early generalized overgrowth in boys with autism. Arch. Gen. Psychiatry 2011, 68, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Carper, R.; Akshoomoff, N. Evidence of brain overgrowth in the first year of life in autism. JAMA 2003, 290, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Nordahl, C.W.; Lange, N.; Li, D.D.; Barnett, L.A.; Lee, A.; Buonocore, M.H.; Simon, T.J.; Rogers, S.; Ozonoff, S.; Amaral, D.G. Brain enlargement is associated with regression in preschool-age boys with autism spectrum disorders. Proc. Natl. Acad. Sci. USA 2011, 108, 20195–20200. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.; Militerni, R.; Frolli, A.; Bravaccio, C.; Gritti, A.; Elia, M.; Curatolo, P.; Manzi, B.; Trillo, S.; Lenti, C.; et al. Clinical, morphological, and biochemical correlates of head circumference in autism. Biol. Psychiatry 2007, 62, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.; Doull, I.; Pearce, N.; Cheng, S.; Leadbitter, P.; Holgate, S.; Beasley, R. The relationship between anthropometric measurements at birth: Asthma and atopy in childhood. Clin. Exp. Allergy 1999, 29, 330–333. [Google Scholar] [CrossRef]
- Katz, K.A.; Pocock, S.J.; Strachan, D.P. Neonatal head circumference, neonatal weight, and risk of hayfever, asthma and eczema in a large cohort of adolescents from Sheffield, England. Clin. Exp. Allergy 2003, 33, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Oryszczyn, M.P.; Annesi-Maesano, I.; Campagna, D.; Sahuquillo, J.; Huel, G.; Kauffmann, F. Head circumference at birth and maternal factors related to cord blood total IgE. Clin. Exp. Allergy 1999, 29, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Eviston, D.P.; Minasyan, A.; Mann, K.P.; Campbell, D.E.; Nanan, R.K. In utero head circumference is associated with childhood allergy. Front. Pediatr. 2015, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, L.; Zhu, T.; Huang, J.; Qu, Y.; Mu, D. Peripheral brain-derived neurotrophic factor in autism spectrum disorder: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 31241. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Geier, D.A.; Sykes, L.K.; Geier, M.R. Relevance of neuroinflammation and encephalitis in autism. Front. Cell. Neurosci. 2015, 9, 519. [Google Scholar] [CrossRef] [PubMed]
- Onore, C.; Careaga, M.; Ashwood, P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun. 2012, 26, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ishizuka, T.; Okayama, Y. Human mast cells and basophils as sources of cytokines. Clin. Exp. Allergy 2000, 30, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Tsilioni, I.; Patel, A.B.; Doyle, R. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl. Psychiatry 2016, 6, e844. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.E.; Croen, L.A.; Braunschweig, D.; Yoshida, C.K.; Grether, J.; Hansen, R.; Kharrazi, M.; Ashwood, P.; Van de Water, J. Increased midgestational IFN-gamma, IL-4 and IL-5 in women bearing a child with autism: A case-control study. Mol. Autism 2011, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.W.; Larsen, N.; Grove, J.; Norgaard-Pedersen, B.; Thorsen, P.; Mortensen, E.L.; Hougaard, D.M. Amniotic fluid inflammatory cytokines: Potential markers of immunologic dysfunction in autism spectrum disorders. World J. Biol. Psychiatry 2013, 14, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, P.; Goines, P.E.; Tancredi, D.J.; Ashwood, P.; Hansen, R.L.; Hertz-Picciotto, I.; Van de Water, J. Neonatal cytokine profiles associated with autism spectrum disorder. Biol. Psychiatry 2017, 81, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Guest, P.C.; Rahmoune, H.; Wang, L.; Levin, Y.; Ingudomnukul, E.; Ruta, L.; Kent, L.; Spain, M.; Baron-Cohen, S.; Bahn, S. Sex-specific serum biomarker patterns in adults with Asperger’s syndrome. Mol. Psychiatry 2011, 16, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Aggarwal, S.; Rashanravan, B.; Lee, T. Th1- and Th2-like cytokines in CD4+ and CD8+ T cells in autism. J. Neuroimmunol. 1998, 85, 106–109. [Google Scholar] [CrossRef]
- Molloy, C.A.; Morrow, A.L.; Meinzen-Derr, J.; Schleifer, K.; Dienger, K.; Manning-Courtney, P.; Altaye, M.; Wills-Karp, M. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol. 2006, 172, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Matsuzaki, H.; Iwata, K.; Kameno, Y.; Shimmura, C.; Kawai, S.; Yoshihara, Y.; Wakuda, T.; Takebayashi, K.; Takagai, S.; et al. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PLoS ONE 2011, 6, e20470. [Google Scholar] [CrossRef] [PubMed]
- Careaga, M.; Murai, T.; Bauman, M.D. Maternal immune activation and autism spectrum disorder: From rodents to nonhuman and human primates. Biol. Psychiatry 2017, 81, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W.; Frank, B.C.; Heine, S.; Lee, N.H.; Quackenbush, J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genom. 2006, 7, 118. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Licona-Limon, P.; Kim, L.K.; Palm, N.W.; Flavell, R.A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Georas, S.N.; Guo, J.; De Fanis, U.; Casolaro, V. T-helper cell type-2 regulation in allergic disease. Eur. Respir. J. 2005, 26, 1119–1137. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.E.; Ashwood, P. Cytokine dysregulation in autism spectrum disorders (ASD): Possible role of the environment. Neurotoxicol. Teratol. 2013, 36, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.A.; Bennett, B.L.; Graham, N.M.; Pirozzi, G.; Stahl, N.; Yancopoulos, G.D. Targeting key proximal drivers of type 2 inflammation in disease. Nat. Rev. Drug Discov. 2016, 15, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflamm. 2016, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Talpalar, A.E.; Ben-Yaakov, K.; Schwartz, M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol. Cell. Neurosci. 2005, 29, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, C.A.; Ziats, M.N.; Rennert, O.M. A Non-inflammatory role for microglia in autism spectrum disorders. Front. Neurol. 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Vargas, D.L.; Zimmerman, A.W. Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry 2005, 17, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.M.; Charych, E.; Lee, A.W.; Moller, T. Kynurenines in CNS disease: Regulation by inflammatory cytokines. Front. Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Kita, T.; Morrison, P.F.; Heyes, M.P.; Markey, S.P. Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the L-kynurenine and quinolinic acid pools in brain. J. Neurochem. 2002, 82, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [PubMed]
- Neurauter, G.; Schrocksnadel, K.; Scholl-Burgi, S.; Sperner-Unterweger, B.; Schubert, C.; Ledochowski, M.; Fuchs, D. Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr. Drug Metab. 2008, 9, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.W.; Jyonouchi, H.; Comi, A.M.; Connors, S.L.; Milstien, S.; Varsou, A.; Heyes, M.P. Cerebrospinal fluid and serum markers of inflammation in autism. Pediatr. Neurol. 2005, 33, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ellis, S.E.; Ashar, F.N.; Moes, A.; Bader, J.S.; Zhan, J.; West, A.B.; Arking, D.E. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 2014, 5, 5748. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.S.; Zhao, M.L.; Derico, L.; Choi, N.; Lee, S.C. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: Differential regulation by inflammatory mediators. J. Neuroinflamm. 2013, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Wynes, M.W.; Riches, D.W. Induction of macrophage insulin-like growth factor-I expression by the Th2 cytokines IL-4 and IL-13. J. Immunol. 2003, 171, 3550–3559. [Google Scholar] [CrossRef] [PubMed]
- O’Kusky, J.; Ye, P. Neurodevelopmental effects of insulin-like growth factor signaling. Front. Neuroendocrinol. 2012, 33, 230–251. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Popken, G.J.; Kemper, A.; McCarthy, K.; Popko, B.; D’Ercole, A.J. Astrocyte-specific overexpression of insulin-like growth factor-I promotes brain overgrowth and glial fibrillary acidic protein expression. J. Neurosci. Res. 2004, 78, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.L.; Hediger, M.L.; Molloy, C.A.; Chrousos, G.P.; Manning-Courtney, P.; Yu, K.F.; Brasington, M.; England, L.J. Elevated levels of growth-related hormones in autism and autism spectrum disorder. Clin. Endocrinol. 2007, 67, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Gosset, P.; Cormier-Daire, V.; Odent, S.; Amiel, J.; Giurgea, I.; Nassogne, M.C.; Pasquier, L.; Munnich, A.; Romana, S.; et al. Overgrowth and trisomy 15q26.1-qter including the IGF1 receptor gene: Report of two families and review of the Literature. Eur. J. Hum. Genet. 2002, 10, 699–706. [Google Scholar] [PubMed]
- Chakrabarti, B.; Dudbridge, F.; Kent, L.; Wheelwright, S.; Hill-Cawthorne, G.; Allison, C.; Banerjee-Basu, S.; Baron-Cohen, S. Genes related to sex steroids, neural growth, and social-emotional behavior are associated with autistic traits, empathy, and Asperger syndrome. Autism Res. 2009, 2, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, L.; Zhu, T.; Huang, J.; Qu, Y.; Mu, D. Association between asthma and autism spectrum disorder: A meta-analysis. PLoS ONE 2016, 11, e0156662. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Lee, M.L.; Martin-Ruiz, C.M.; Court, J.A.; Volsen, S.G.; Merrit, J.; Folly, E.; Iversen, P.E.; Bauman, M.L.; Perry, R.H.; et al. Cholinergic activity in autism: Abnormalities in the cerebral cortex and basal forebrain. Am. J. Psychiatry 2001, 158, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Raznahan, A.; Toro, R.; Proitsi, P.; Powell, J.; Paus, T.; Bolton, F.P.; Murphy, D.G. A functional polymorphism of the brain derived neurotrophic factor gene and cortical anatomy in autism spectrum disorder. J. Neurodev. Disord. 2009, 1, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.M.; Paquin, A.; Nutt, S.L.; Kaplan, D.R.; Miller, F.D. Endogenous microglia regulate development of embryonic cortical precursor cells. J. Neurosci. Res. 2011, 89, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Gross, C.T. Microglia in development: Linking brain wiring to brain environment. Neuron Glia Biol. 2011, 7, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Pucci, L.; Bezzi, P. Astrocytes and microglia and their potential link with autism spectrum disorders. Front. Cell. Neurosci. 2016, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, J.S.; Castren, E. Mice with altered BDNF signaling as models for mood disorders and antidepressant effects. Front. Behav. Neurosci. 2014, 8, 143. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, F.; Silverman, J.L.; Aney, J.; Tian, Q.; Barkan, C.L.; Chadman, K.K.; Crawley, J.N. Working memory deficits, increased anxiety-like traits, and seizure susceptibility in BDNF overexpressing mice. Learn. Mem. 2011, 18, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Ajo, R.; Cacicedo, L.; Navarro, C.; Sanchez-Franco, F. Growth hormone action on proliferation and differentiation of cerebral cortical cells from fetal rat. Endocrinology 2003, 144, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Pathania, M.; Yan, L.D.; Bordey, A. A symphony of signals conducts early and late stages of adult neurogenesis. Neuropharmacology 2010, 58, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the brain: A cytokine to remember. J. Immunol. 2012, 189, 4213–4219. [Google Scholar] [CrossRef] [PubMed]
- Peltier, J.; O’Neill, A.; Schaffer, D.V. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev. Neurobiol. 2007, 67, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Bosnjak, B.; Stelzmueller, B.; Erb, K.J.; Epstein, M.M. Treatment of allergic asthma: Modulation of Th2 cells and their responses. Respir. Res. 2011, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J. Glucocorticoids and the Th1/Th2 balance. Ann. N. Y. Acad. Sci. 2004, 1024, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Kalkman, H.O.; Feuerbach, D. Antidepressant therapies inhibit inflammation and microglial M1-polarization. Pharmacol. Ther. 2016, 163, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Derecki, N.C.; Quinnies, K.M.; Kipnis, J. Alternatively activated myeloid (M2) cells enhance cognitive function in immune compromised mice. Brain Behav. Immun. 2011, 25, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 2010, 70, 304–322. [Google Scholar] [PubMed]
- Branum, A.M.; Lukacs, S.L. Food allergy among children in the United States. Pediatrics 2009, 124, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Sheikh, A.; Strachan, D.P.; Anderson, H.R. Time trends in allergic disorders in the UK. Thorax 2007, 62, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Charpin, D.; Gouitaa, M. Why is the prevalence of allergic diseases increasing? A critical assessment of some classical risk factors. Mediat. Inflamm. 2001, 10, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Hadley, C. Food allergies on the rise? Determining the prevalence of food allergies, and how quickly it is increasing, is the first step in tackling the problem. EMBO Rep. 2006, 7, 1080–1083. [Google Scholar] [CrossRef] [PubMed]
- Pennesi, C.M.; Klein, L.C. Effectiveness of the gluten-free, casein-free diet for children diagnosed with autism spectrum disorder: Based on parental report. Nutr. Neurosci. 2012, 15, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Ou, J.J.; Li, Y.M.; Xiang, D.X. Dietary supplement for core symptoms of autism spectrum disorder: Where are we now and where should we go? Front. Psychiatry 2017, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Todoroki, M.; Nishida, Y.; Tanabe, M.; Misawa, M. A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am. J. Respir. Cell Mol. Biol. 2009, 41, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Tsilioni, I.; Leeman, S.E.; Theoharides, T.C. Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef]
- Wu, J.; de Theije, C.G.; da Silva, S.L.; Abbring, S.; van der Horst, H.; Broersen, L.M.; Willemsen, L.; Kas, M.; Garssen, J.; Kraneveld, A.D. Dietary interventions that reduce mTOR activity rescue autistic-like behavioral deficits in mice. Brain Behav. Immun. 2017, 59, 273–287. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalkman, H.O.; Feuerbach, D. Microglia M2A Polarization as Potential Link between Food Allergy and Autism Spectrum Disorders. Pharmaceuticals 2017, 10, 95. https://doi.org/10.3390/ph10040095
Kalkman HO, Feuerbach D. Microglia M2A Polarization as Potential Link between Food Allergy and Autism Spectrum Disorders. Pharmaceuticals. 2017; 10(4):95. https://doi.org/10.3390/ph10040095
Chicago/Turabian StyleKalkman, Hans O., and Dominik Feuerbach. 2017. "Microglia M2A Polarization as Potential Link between Food Allergy and Autism Spectrum Disorders" Pharmaceuticals 10, no. 4: 95. https://doi.org/10.3390/ph10040095
APA StyleKalkman, H. O., & Feuerbach, D. (2017). Microglia M2A Polarization as Potential Link between Food Allergy and Autism Spectrum Disorders. Pharmaceuticals, 10(4), 95. https://doi.org/10.3390/ph10040095