A Novel Interaction Between the TLR7 and a Colchicine Derivative Revealed Through a Computational and Experimental Study

 , ,
, , 

Abstract
:1. Introduction
2. Results and discussion
2.1. Homology Modeling
2.2. Molecular Docking
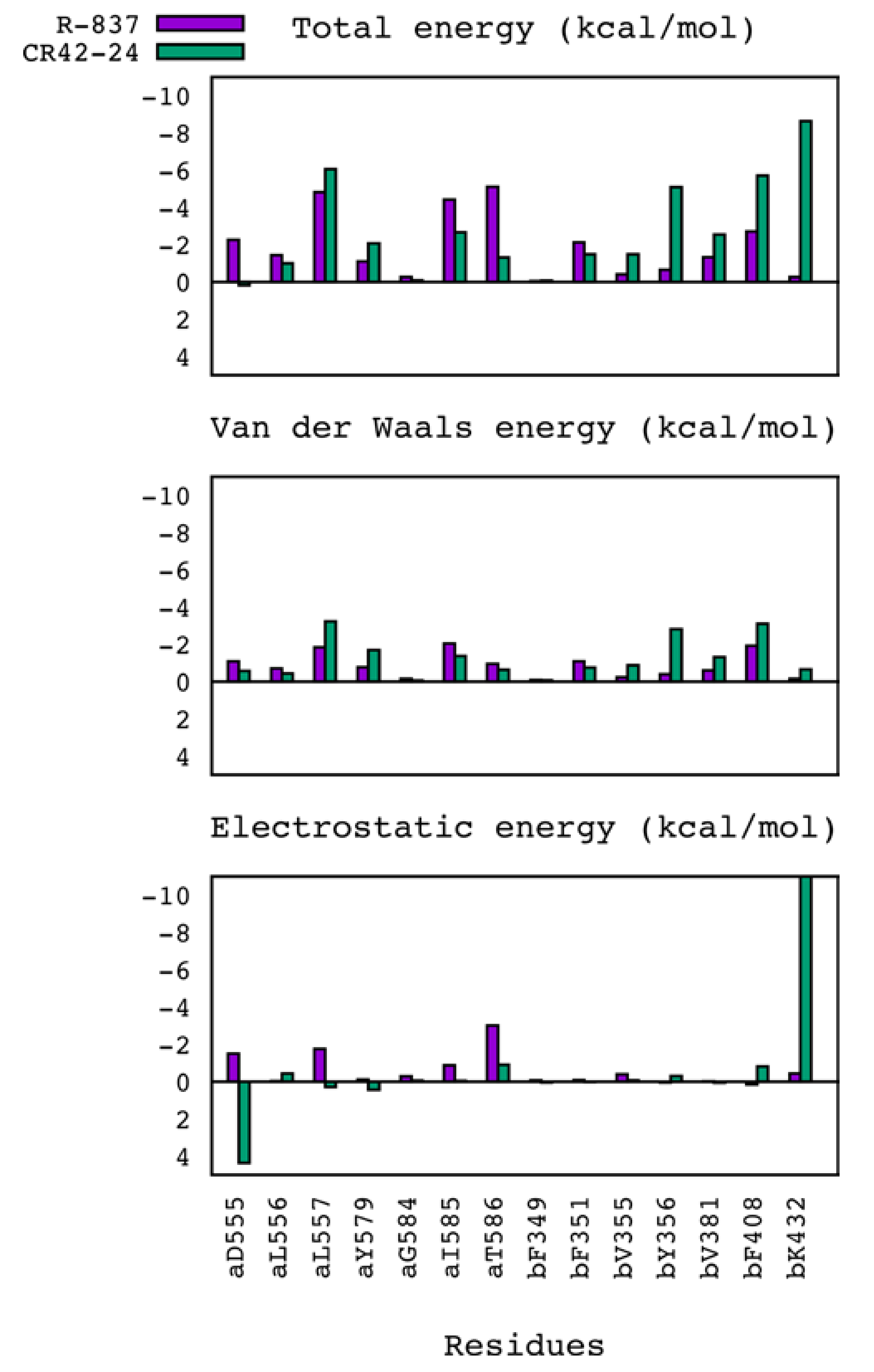
2.3. Molecular Dynamics Simulations and MM/GBSA Calculations
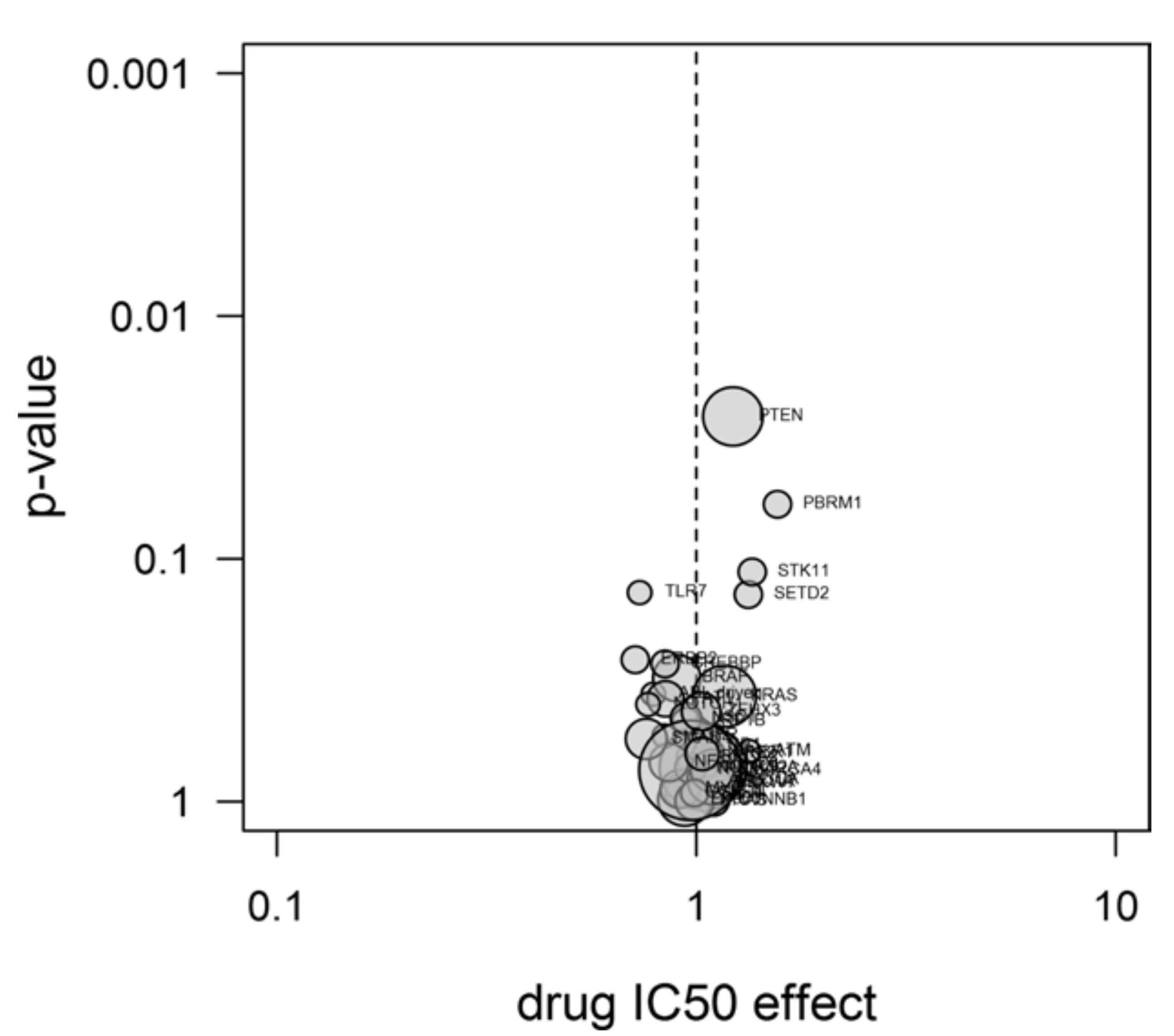
2.4. Experiments
3. Materials and Methods
3.1. Model Building and Validation
3.2. Ligand Preparation and Molecular Docking
3.3. Molecular Dynamics Simulations
3.4. MM/GBSA Binding Energy Calculations
3.5. Cell Preparation
3.6. Compound Preparation
3.7. Cell Proliferation Assay
3.8. Data Analysis
3.9. Curve Fitting
3.10. Cell Genetics
3.11. Drug Sensitivity
4. Conclusions
Supplementary materials
Acknowledgments
Author contributions
Conflict of interests
References
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Smits, E.L.J.M.; Ponsaerts, P.; Berneman, Z.N.; Van Tendeloo, V.F.I. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist 2008, 13, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Forsbach, A.; Müller, C.; Montino, C.; Kritzler, A.; Nguyen, T.; Weeratna, R.; Jurk, M.; Vollmer, J. Negative regulation of the type I interferon signaling pathway by synthetic Toll-like receptor 7 ligands. J. Interferon Cytokine Res. 2012, 32, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Chuang, T.-H.; Redecke, V.; She, L.; Pitha, P.M.; Carson, D.A.; Raz, E.; Cottam, H.B. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: Activation of Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2003, 100, 6646–6651. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.P.; Schön, M. TLR7 and TLR8 as targets in cancer therapy. Oncogene 2008, 27, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Petricevic, B.; Wessner, B.; Sachet, M.; Vrbanec, D.; Spittler, A.; Bergmann, M. CL097, a TLR7/8 ligand, inhibits TLR-4--dependent activation of IRAK-M and BCL-3 expression. Shock 2009, 32, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Surber, C.; French, L.E.; Stockfleth, E. Resiquimod, a topical drug for viral skin lesions and skin cancer. Expert Opin. Investig. Drugs 2013, 22, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.J.; O’Neill, L.A.J. New developments in Toll-like receptor targeted therapeutics. Curr. Opin. Pharmacol. 2012, 12, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Koski, G.K.; Czerniecki, B.J. Combining Innate Immunity with Radiation Therapy for Cancer Treatment. Clin. Cancer Res. 2005, 11, 7–11. [Google Scholar] [PubMed]
- Hotz, C.; Bourquin, C. Systemic cancer immunotherapy with Toll-like receptor 7 agonists: Timing is everything. Oncoimmunology 2012, 1, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, E.C.; Liu, H.; Schwartz, M.J.; Scherr, D.S. Toll-like receptor 7 agonist therapy with imidazoquinoline enhances cancer cell death and increases lymphocytic infiltration and proinflammatory cytokine production in established tumors of a renal cell carcinoma mouse model. J. Oncol. 2012, 2012, 103298. [Google Scholar] [CrossRef] [PubMed]
- Ochi, A.; Graffeo, C.S.; Zambirinis, C.P.; Rehman, A.; Hackman, M.; Fallon, N.; Barilla, R.M.; Henning, J.R.; Jamal, M.; Rao, R.; et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J. Clin. Invest. 2012, 122, 4118–4129. [Google Scholar] [CrossRef] [PubMed]
- Eigenbrod, T.; Dalpke, A.H. TLR7 inhibition: A novel strategy for pancreatic cancer treatment? JAKSTAT 2013, 2, e23011. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Liu, F.; Zhou, H.; Zhai, S.; Yan, B. Novel Natural Product- and Privileged Scaffold-Based Tubulin Inhibitors Targeting the Colchicine Binding Site. Molecules 2016, 21, 1375. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Goping, I.S.; Rieger, A.; Mane, J.; Huzil, J.T.; Banerjee, A.; Luduena, R.F.; Hossani, B.; Winter, P.; Tuszynski, J.A. Novel Colchicine Derivatives and Their Anti-Cancer Activity. Curr. Top. Med. Chem. 2016, 17, 2538–2558. [Google Scholar] [CrossRef] [PubMed]
- Courbet, A.; Bec, N.; Constant, C.; Larroque, C.; Pugniere, M.; El Messaoudi, S.; Zghaib, Z.; Khier, S.; Deleuze-Masquefa, C.; Gattacceca, F. Imidazoquinoxaline anticancer derivatives and imiquimod interact with tubulin: Characterization of molecular microtubule inhibiting mechanisms in correlation with cytotoxicity. PLoS ONE 2017, 12, e0182022. [Google Scholar] [CrossRef] [PubMed]
- De Martino, G.; Edler, M.C.; La Regina, G.; Coluccia, A.; Barbera, M.C.; Barrow, D.; Nicholson, R.I.; Chiosis, G.; Brancale, A.; Hamel, E.; et al. New arylthioindoles: Potent inhibitors of tubulin polymerization. 2. Structure-activity relationships and molecular modeling studies. J. Med. Chem. 2006, 49, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Deriu, M.A.; Licandro, G.; Prunotto, A.; Danani, A.; Tuszynski, J.A. Structure Based Modeling of Small Molecules Binding to the TLR7 by Atomistic Level Simulations. Molecules 2015, 20, 8316–8340. [Google Scholar] [CrossRef] [PubMed]
- Maschalidi, S.; Hässler, S.; Blanc, F.; Sepulveda, F.E.; Tohme, M.; Chignard, M.; van Endert, P.; Si-Tahar, M.; Descamps, D.; Manoury, B. Asparagine endopeptidase controls anti-influenza virus immune responses through TLR7 activation. PLoS Pathog. 2012, 8, e1002841. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Gong, J.; Jamitzky, F.; Heckl, W.M.; Stark, R.W.; Rössle, S.C. Homology modeling of human Toll-like receptors TLR7, 8, and 9 ligand-binding domains. Protein Sci. 2009, 18, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Gong, J.; Rössle, S.C.; Jamitzky, F.; Heckl, W.M.; Stark, R.W. A leucine-rich repeat assembly approach for homology modeling of the human TLR5-10 and mouse TLR11-13 ectodomains. J. Mol. Model. 2011, 17, 27–36. [Google Scholar] [CrossRef] [PubMed]
- TSeng, C.-Y.; Gajewski, M.; Danani, A.; Tuszynski, J.A. Homology and Molecular Dynamics Models of Toll-Like Receptor 7 Protein and Its Dimerization. Chem. Biol. Drug Des. 2014. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Tanji, H.; Ohto, U.; Shibata, T.; Miyake, K.; Shimizu, T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 2013, 339, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); Chemical Computing Group, Inc.: Montreal, QC, Canada, 2017. [Google Scholar]
- Kelly, K. Multiple Sequence and Structure Alignment in MOE. Available online: http://www.chemcomp.com/journal/align.htm (accessed on 10 June 2014).
- AmberTools 12 Reference Manual. Available online: http://ambermd.org/doc12/AmberTools12.pdf (accessed on 13 February 2018).
- Hoffmann, R. An Extended Hückel Theory. I. Hydrocarbons. J. Chem. Phys. 1963, 39, 1397. [Google Scholar] [CrossRef]
- Labute, P. Protonate 3D: Assignment of Macromolecular Protonation State and Geometry. Available online: http://www.ccl.net/cca/documents/proton/ (accessed on 31 March 2014).
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollmann, P.; Case, D. Antechamber, An Accessory Software Package For Molecular Mechanical Calculation. J. Comput. Chem. 2005, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Schrödinger LigPrep. Available online: http://www.schrodinger.com/productpage/14/10/ (accessed on 24 January 2014).
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Wolfson, H.J. Efficient detection of three-dimensional structural motifs in biological macromolecules by computer vision techniques. Proc. Natl. Acad. Sci. USA 1991, 88, 10495–10499. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A. The Generalized Born Model: Its Foundation, Applications, and Limitations. Available online: http://people.cs.vt.edu/~onufriev/PUBLICATIONS/gbreview.pdf (accessed on 9 June 2014).
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uitdehaag, J.C.M.; de Roos, J.A.D.M.; van Doornmalen, A.M.; Prinsen, M.B.W.; de Man, J.; Tanizawa, Y.; Kawase, Y.; Yoshino, K.; Buijsman, R.C.; Zaman, G.J.R. Comparison of the Cancer Gene Targeting and Biochemical Selectivities of All Targeted Kinase Inhibitors Approved for Clinical Use. PLoS ONE 2014, 9, e92146. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Li, C.; Zhao, F. S.; Zhang, L.; Ng, T. B.; Jin, G.; Sha, O. Anti-tumor Activity of Toll-Like Receptor 7 Agonists. Front. Pharmacol. 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]







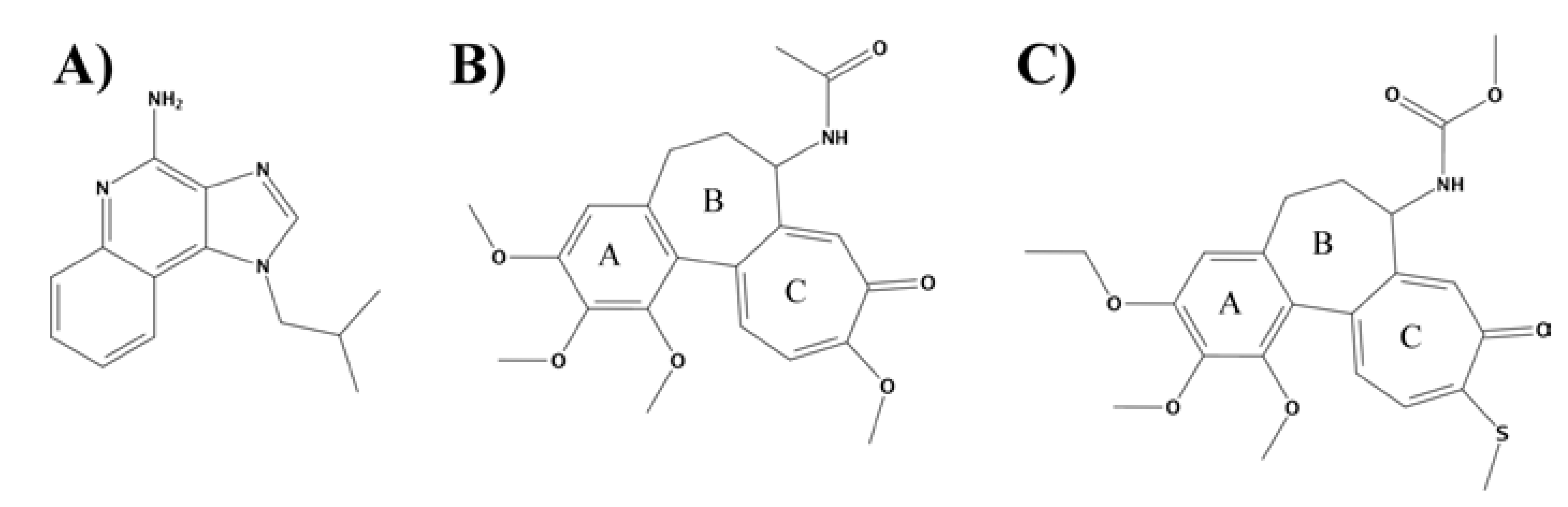{kind=link}
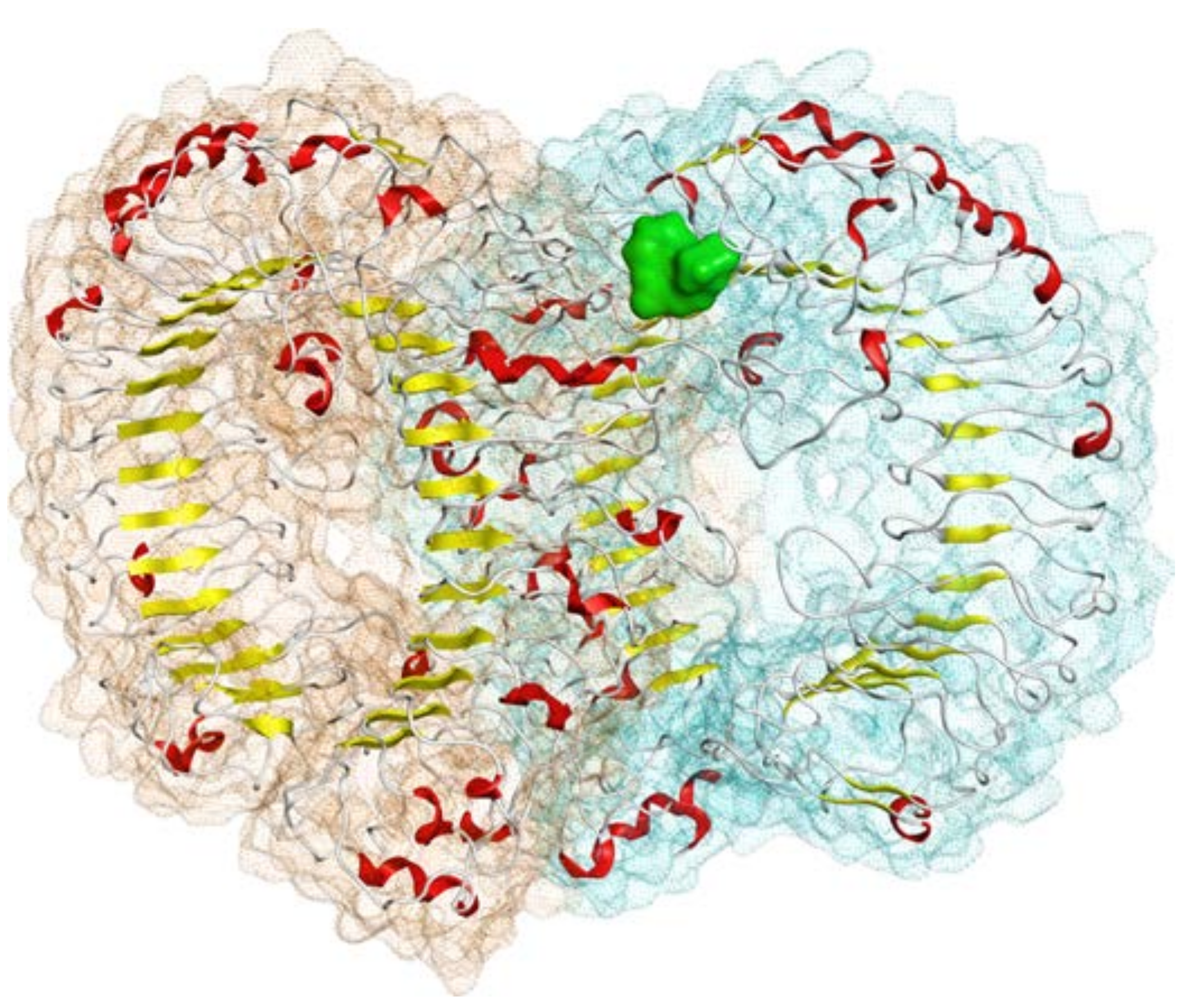{kind=link}
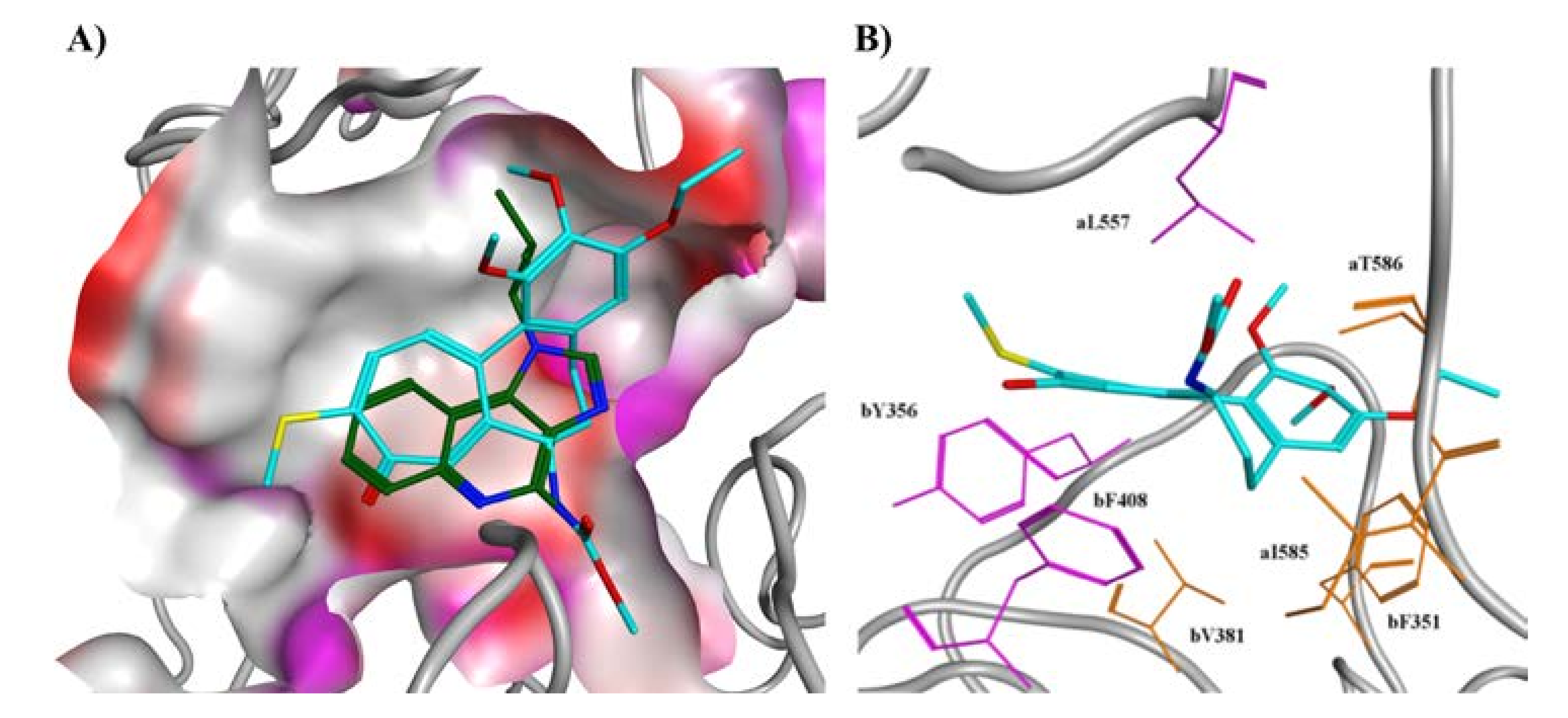{kind=link}
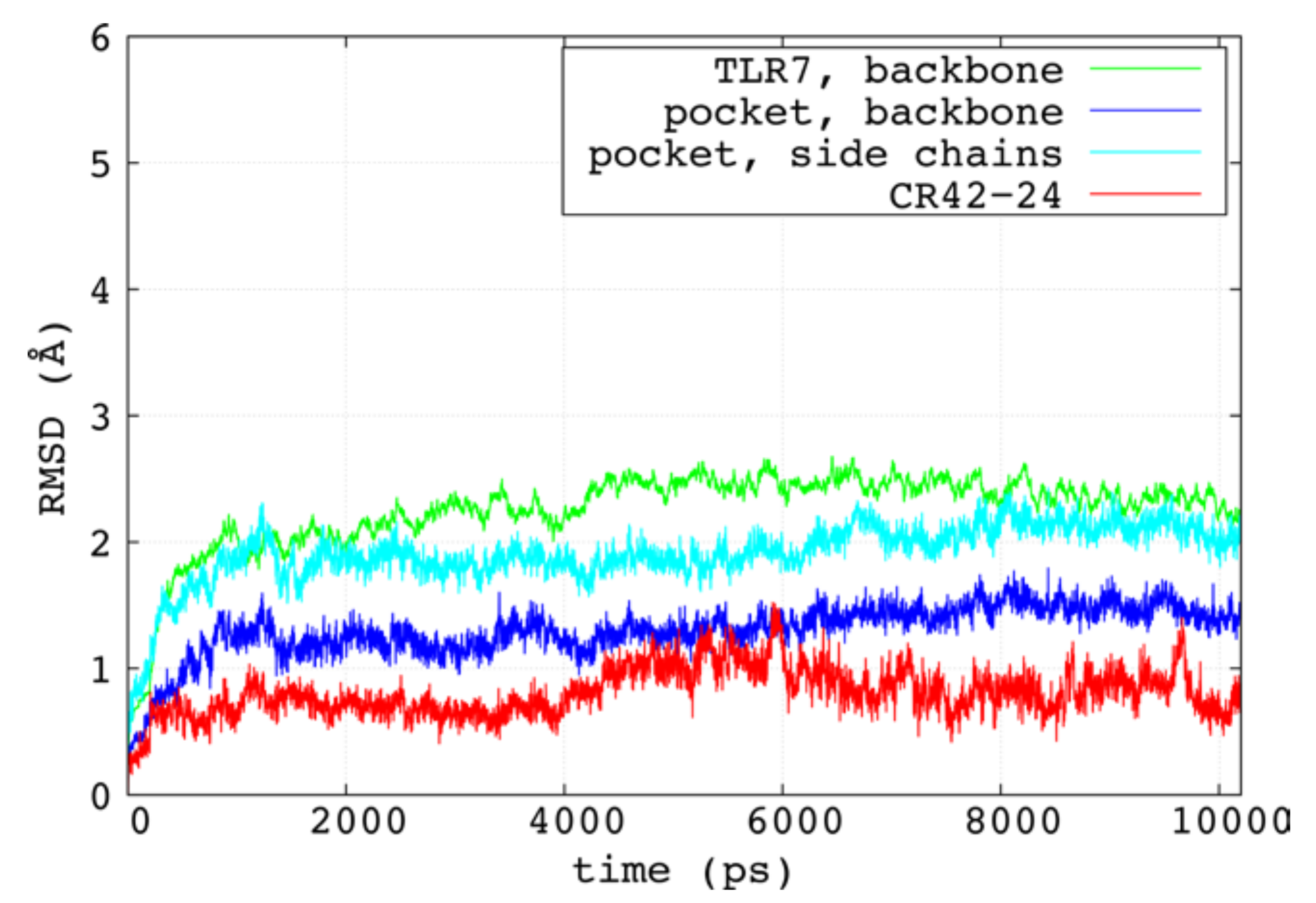{kind=link}
{kind=link}
{kind=link}
| MM/GBSA ΔG (kcal/mol) | |||||
|---|---|---|---|---|---|
| Compound | Total | Van der Waals | Electrostatic | Polar solvation | Non-polar solvation |
| R-837 | −31.081 (2.531) | −35.028 (2.600) | −18.251 (2.912) | 27.331 (2.653) | −5.133 (0.216) |
| CR42-24 | −36.055 (3.090) | −46.841 (2.719) | −24.050 (5.273) | 41.089 (5.105) | −6.252 (0.239) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, F.; Deriu, M.A.; Barakat, K.; Danani, A.; Tuszynski, J. A Novel Interaction Between the TLR7 and a Colchicine Derivative Revealed Through a Computational and Experimental Study. Pharmaceuticals 2018, 11, 22. https://doi.org/10.3390/ph11010022
Gentile F, Deriu MA, Barakat K, Danani A, Tuszynski J. A Novel Interaction Between the TLR7 and a Colchicine Derivative Revealed Through a Computational and Experimental Study. Pharmaceuticals. 2018; 11(1):22. https://doi.org/10.3390/ph11010022
Chicago/Turabian StyleGentile, Francesco, Marco A. Deriu, Khaled Barakat, Andrea Danani, and Jack Tuszynski. 2018. "A Novel Interaction Between the TLR7 and a Colchicine Derivative Revealed Through a Computational and Experimental Study" Pharmaceuticals 11, no. 1: 22. https://doi.org/10.3390/ph11010022
APA StyleGentile, F., Deriu, M. A., Barakat, K., Danani, A., & Tuszynski, J. (2018). A Novel Interaction Between the TLR7 and a Colchicine Derivative Revealed Through a Computational and Experimental Study. Pharmaceuticals, 11(1), 22. https://doi.org/10.3390/ph11010022