Mitoxantrone is More Toxic than Doxorubicin in SH-SY5Y Human Cells: A ‘Chemobrain’ In Vitro Study


 ,
, 

Abstract
:1. Introduction
2. Results
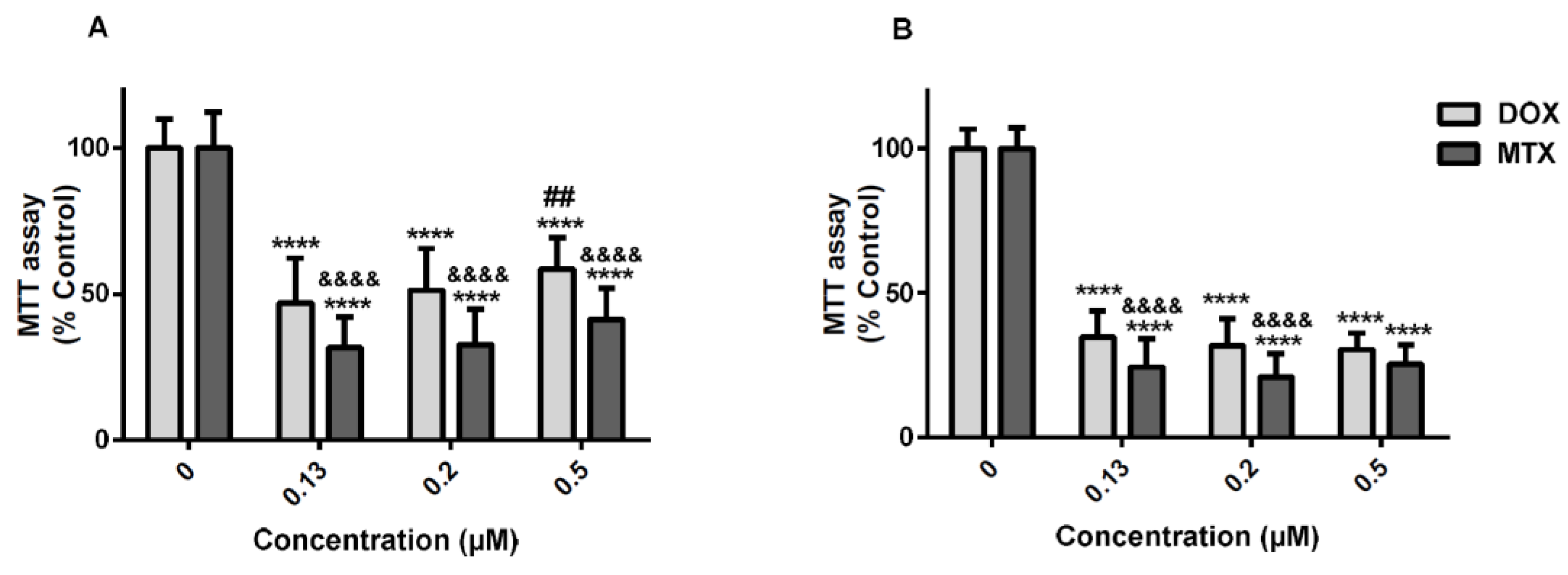
2.1. The Cytotoxicity of Mitoxantrone Was Significantly Higher Than That of Doxorubicin
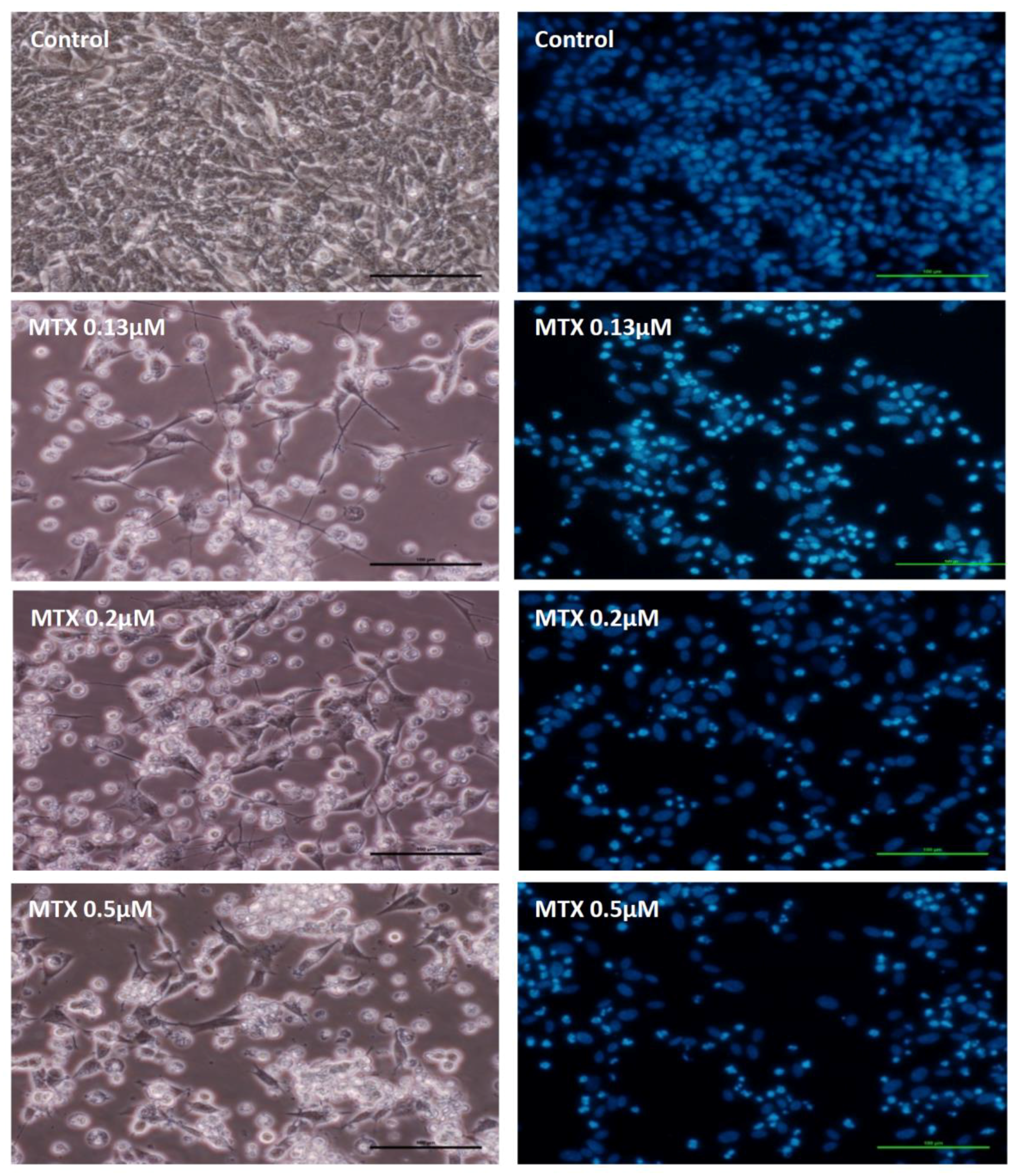
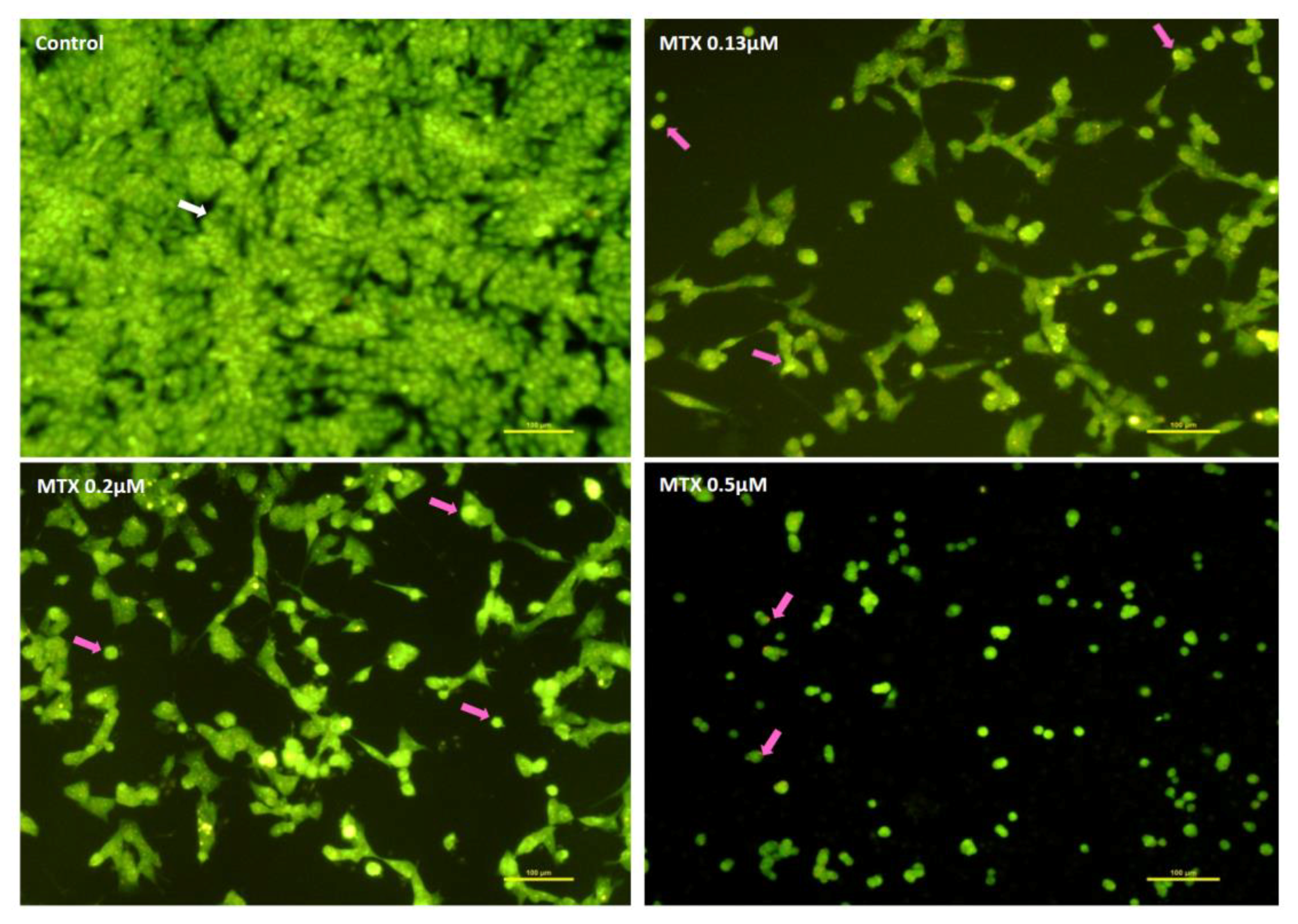
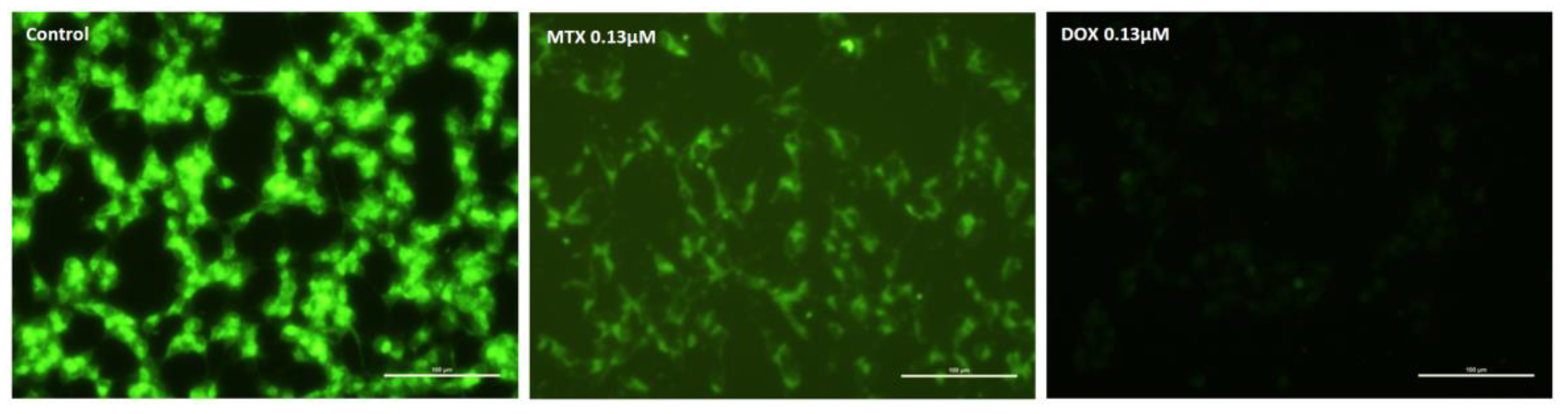
2.2. Mitoxantrone Led to Cellular Damage in SH-SY5Y Cells, with Signs of Apoptosis Most Evident at the Lowest Concentration after a 48-h Exposure
2.3. Mitoxantrone and Doxorubicin Caused Apoptosis in Undifferentiated SH-SY5Y Cells
2.4. Both Doxorubicin and Mitoxantrone Caused a Decrease in the Mitochondria Potential of Neuronal Cells at 0.13 μM
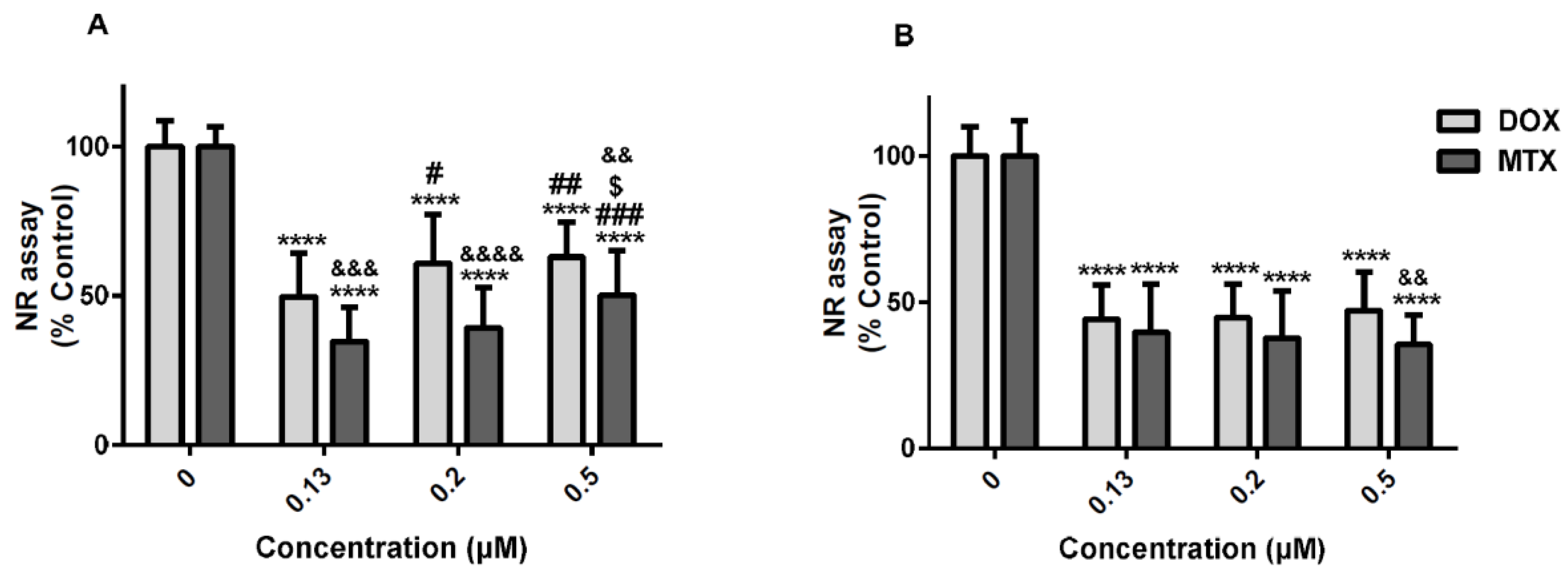
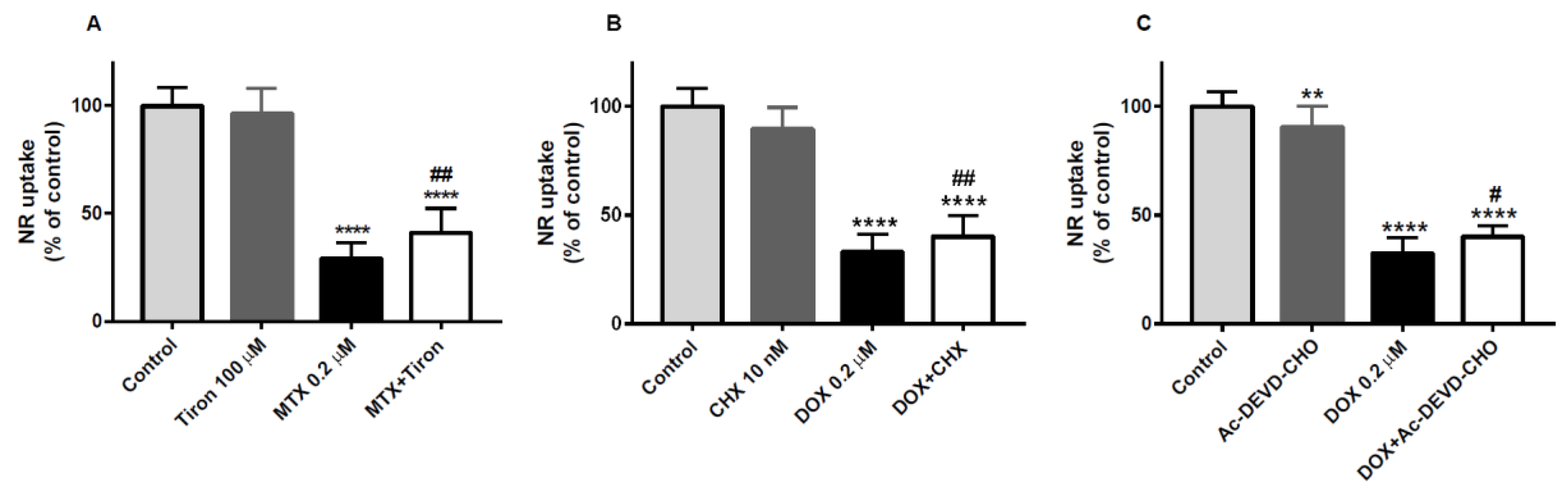
2.5. Tiron, an Antioxidant, was the Only Drug That Partially Prevented the Cytotoxicity of Mitoxantrone in the Neutral Red Uptake Assay
2.6. Cycloheximide, a Protein Synthesis Inhibitor, and Ac-DEVD-CHO, an Inhibitor of Caspase-3, Partially Counteracted the Doxorubicin-Induced Toxicity
2.7. Doxorubicin Caused Greater Cytotoxicity in Undifferentiated SH-SY5Y Cells than in Differentiated Cells According to the MTT Reduction Assay
2.8. Mitoxantrone is More Neurotoxic than Doxorubicin in Differentiated SH-SY5Y Cells in the MTT Reduction Assay
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Undifferentiated SH-SY5Y Cells
4.4. Differentiated SH-SY5Y Cells
4.5. Cytotoxicity Evaluation
4.6. MTT Reduction Assay
4.7. Neutral Red Lysosomal Uptake Assay
4.8. Microscopic Evaluation of the Cells
4.8.1. Phase Contrast Microscopy
4.8.2. Hoechst Staining
4.8.3. Ethidium Bromide and Acridine Orange Staining
4.8.4. Evaluation of the Mitochondrial Membrane Potential
5. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| BBB | Blood-brain barrier |
| CHX | Cycloheximide |
| CNS | Central nervous system |
| DMEM | Dulbecco’s modified Eagle medium |
| DOX | Doxorubicin |
| EMA | European Medicines Agency |
| HBSS | Hanks’ balanced salt solution |
| MS | Multiple sclerosis |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| MTX | Mitoxantrone |
| NAC | N-Acetyl-l-cysteine |
| NR | Neutral Red |
| PBS | Phosphate buffered saline |
| RA | Retinoic acid |
| TPA | 12-O-tetradecanoylphorbol 13-acetate |
References
- GBD. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar]
- Hrynchak, I.; Sousa, E.; Pinto, M.; Costa, V.M. The importance of drug metabolites synthesis: The case-study of cardiotoxic anticancer drugs. Drug Metab. Rev. 2017, 49, 158–196. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-C.; Smieliauskas, F.; Geynisman, D.; Kelly, R.; Smith, T. Trends in the cost and use of targeted cancer therapies for the privately insured noneldery: 2001 to 2011. J. Clin. Oncol. 2015, 33, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Novantrone and Associated Names; EMA: London, UK, 2016; Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Novantrone_30/WC500209683.pdf (accessed on 29 April 2018).
- Goodin, D.S.; Arnason, B.G.; Coyle, P.K.; Frohman, E.M.; Paty, D.W. The use of mitoxantrone (Novantrone) for the treatment of multiple sclerosis: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2003, 61, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef] [PubMed]
- Marriott, J.J.; Miyasaki, J.M.; Gronseth, G.; O’Connor, P.W. Evidence Report: The efficacy and safety of mitoxantrone (Novantrone) in the treatment of multiple sclerosis: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology(CME). Neurology 2010, 74, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Recommendations on the Use of Caelyx (Doxorubicin Hydrochloride); EMA: London, UK, 2011; Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2011/11/WC500117926.pdf (accessed on 29 April 2018).
- Frei, E.I.; Eder, J.P. Combination chemotherapy. In Holland-Frei Cancer Medicine., 6th ed.; Kufe, D.W., Pollock, R.E., Bast, R.C., Weichselbaum, R.R., Eds.; BC Decker: Hamilton, ON, USA, 2003. Available online: https://www.ncbi.nlm.nih.gov/books/NBK13955/ (accessed on 29 April 2018).
- Costa, V.M.; Carvalho, F.; Duarte, J.A.; Bastos M., L.; Remiâo, F. The heart as a target for xenobiotic toxicity: The cardiac susceptibility to oxidative stress. Chem. Res. Toxicol. 2013, 26, 1285–1311. [Google Scholar] [CrossRef] [PubMed]
- Mladenka, P.; Applova, L.; Patocka, J.; Costa, V.M.; Remiao, F.; Pourova, J.; Mladenka, A.; Karlickova, J.; Jahodar, L.; Voprsalova, M.; et al. Comprehensive review of cardiovascular toxicity of drugs and related agents. Med. Res. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Reis-Mendes, A.F.; Sousa, E.; de Lourdes Bastos, M.; Costa, V.M. The role of the metabolism of anticancer drugs in their induced-cardiotoxicity. Curr. Drug Metab. 2015, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Ahles, T.A.; Saykin, A.J. Candidate mechanisms for chemotherapy-induced cognitive changes. Nat. Rev. Cancer 2007, 7, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jungsuwadee, P.; Vore, M.; Butterfield, D.A.; St Clair, D.K. Collateral damage in cancer chemotherapy: Oxidative stress in nontargeted tissues. Mol. Interv. 2007, 7, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.E.; Dodd, M.J.; Miaskowski, C.A.; Dowling, G.A.; Kramer, J. Preliminary results of a longitudinal study of changes in cognitive function in breast cancer patients undergoing chemotherapy with doxorubicin and cyclophosphamide. Psychooncology 2008, 17, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.E.; Cooper, B.A.; Dodd, M.J.; Miaskowski, C.A. A prospective longitudinal study of chemotherapy-induced cognitive changes in breast cancer patients. Support. Care Cancer 2011, 19, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Ahles, T.A.; Saykin, A.J.; Furstenberg, C.T.; Cole, B.; Mott, L.A.; Skalla, K.; Whedon, M.B.; Bivens, S.; Mitchell, T.; Greenberg, E.R.; et al. Neuropsychologic impact of standard-dose systemic chemotherapy in long-term survivors of breast cancer and lymphoma. J. Clin. Oncol. 2002, 20, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.A.; Meisel, A.; Carvalho, F.D.; Bastos, M.L. Neuronal nitric oxide synthase is a key factor in doxorubicin-induced toxicity to rat-isolated cortical neurons. Neurotox. Res. 2011, 19, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.A.; Meisel, A.; Dirnagl, U.; Carvalho, F.D.; Bastos, M.L. Doxorubicin induces biphasic neurotoxicity to rat cortical neurons. Neurotoxicology 2008, 29, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Tangpong, J.; Cole, M.P.; Sultana, R.; Estus, S.; Vore, M.; St Clair, W.; Ratanachaiyavong, S.; St Clair, D.K.; Butterfield, D.A. Adriamycin-mediated nitration of manganese superoxide dismutase in the central nervous system: Insight into the mechanism of chemobrain. J. Neurochem. 2007, 100, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Taube, F.; Stölzel, F.; Thiede, C.; Ehninger, G.; Laniado, M.; Schaich, M. Increased incidence of central nervous system hemorrhages in patients with secondary acute promyelocytic leukemia after treatment of multiple sclerosis with mitoxantrone? Haematologica 2011, 96, e31–e32. [Google Scholar] [CrossRef] [PubMed]
- Boiardi, A.; Eoli, M.; Salmaggi, A.; Lamperti, E.; Botturi, A.; Broggi, G.; Bissola, L.; Finocchiaro, G.; Silvani, A. Systemic temozolomide combined with loco-regional mitoxantrone in treating recurrent glioblastoma. J. Neurooncol. 2005, 75, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Alexander, J.S. Blood-brain barrier disruption in multiple sclerosis. Mult. Scler. 2003, 9, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Honer, M.; Menke-Moellers, I.; Berthold, F. Antiproliferative potential of cytostatic drugs on neuroblastoma cells in vitro. Eur. J. Cancer 1995, 31, 616–621. [Google Scholar] [CrossRef]
- Jantas, D.; Greda, A.; Leskiewicz, M.; Grygier, B.; Pilc, A.; Lason, W. Neuroprotective effects of mGluR II and III activators against staurosporine- and doxorubicin-induced cellular injury in SH-SY5Y cells: New evidence for a mechanism involving inhibition of AIF translocation. Neurochem. Int. 2015, 88, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Jantas, D.; Lason, W. Protective effect of memantine against doxorubicin toxicity in primary neuronal cell cultures: Influence a development stage. Neurotox. Res. 2009, 15, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Jantas, D.; Pytel, M.; Mozrzymas, J.W.; Leskiewicz, M.; Regulska, M.; Antkiewicz-Michaluk, L.; Lason, W. The attenuating effect of memantine on staurosporine-, salsolinol- and doxorubicin-induced apoptosis in human neuroblastoma SH-SY5Y cells. Neurochem. Int. 2008, 52, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Gaman, A.M.; Uzoni, A.; Popa-Wagner, A.; Andrei, A.; Petcu, E.B. The role of oxidative stress in etiopathogenesis of chemotherapy induced cognitive impairment (CICI)-”Chemobrain”. Aging Dis. 2016, 7, 307–317. [Google Scholar] [PubMed]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remiao, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Homolya, L.; Orban, T.I.; Csanady, L.; Sarkadi, B. Mitoxantrone is expelled by the ABCG2 multidrug transporter directly from the plasma membrane. Biochim. Biophys. Acta 2011, 1808, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Grewaal, D.; Green, R.M.; Mikhael, N.; Goel, R.; Montpetit, V.A.; Redmond, M.D. Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer Res. 1993, 13, 1945–1952. [Google Scholar] [PubMed]
- Green, R.M.; Stewart, D.J.; Hugenholtz, H.; Richard, M.T.; Thibault, M.; Montpetit, V. Human central nervous system and plasma pharmacology of mitoxantrone. J. Neurooncol. 1988, 6, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Geiger, S.; Baur, D.; Hausmann, A.; Tischer, J.; Horster, S.; Stemmler, H.J. Cardiotoxicity after anthracycline treatment in survivors of adult cancers: Monitoring by USCOM, Echocardiography and Serum Biomarkers. World J. Oncol. 2013, 4, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.M.; Eksborg, S.; Björk, O.; Abrahamsson, J.; Behrendtz, M.; Castor, A.; Forestier, E.; Lönnerholm, G. Pharmacokinetics of doxorubicin in children with acute lymphoblastic leukemia: Multi-institutional collaborative study. Med. Pediatr. Oncol. 2002, 38, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Palle, J.; Frost, B.-M.; Peterson, C.; Gustafsson, G.; Hellebostad, M.; Kanerva, J.; Schmiegelow, K. Nordic Society for Pediatric Hematology and Oncology. Doxorubicin pharmacokinetics is correlated to the effect of induction therapy in children with acute myeloid leukemia. Anticancer Drugs 2006, 17, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Barpe, D.R.; Rosa, D.D.; Froehlich, P.E. Pharmacokinetic evaluation of doxorubicin plasma levels in normal and overweight patients with breast cancer and simulation of dose adjustment by different indexes of body mass. Eur. J. Pharm. Sci. 2010, 41, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Voon, P.J.; Yap, H.L.; Ma, C.Y.; Lu, F.; Wong, A.L.; Sapari, N.S.; Soong, R.; Soh, T.I.; Goh, B.C.; Lee, H.S.; et al. Correlation of aldo-ketoreductase (AKR) 1C3 genetic variant with doxorubicin pharmacodynamics in Asian breast cancer patients. Br. J. Clin. Pharmacol. 2013, 75, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Alberts, D.S.; Peng, Y.M.; Bowden, G.T.; Dalton, W.S.; Mackel, C. Pharmacology of mitoxantrone: Mode of action and pharmacokinetics. Investig. New Drugs 1985, 3, 101–107. [Google Scholar] [CrossRef]
- Batra, V.K.; Morrison, J.A.; Woodward, D.L.; Siverd, N.S.; Yacobi, A. Pharmacokinetics of mitoxantrone in man and laboratory animals. Drug Metab. Rev. 1986, 17, 311–329. [Google Scholar] [CrossRef] [PubMed]
- Andersson, B.S.; Eksborg, S.; Vidal, R.F.; Sundberg, M.; Carlberg, M. Anthraquinone-induced cell injury: Acute toxicity of carminomycin, epirubicin, idarubicin and mitoxantrone in isolated cardiomyocytes. Toxicology 1999, 135, 11–20. [Google Scholar] [CrossRef]
- Koceva-Chyla, A.; Jedrzejczak, M.; Skierski, J.; Kania, K.; Jozwiak, Z. Mechanisms of induction of apoptosis by anthraquinone anticancer drugs aclarubicin and mitoxantrone in comparison with doxorubicin: Relation to drug cytotoxicity and caspase-3 activation. Apoptosis 2005, 10, 1497–1514. [Google Scholar] [CrossRef] [PubMed]
- Kluza, J.; Marchetti, P.; Gallego, M.A.; Lancel, S.; Fournier, C.; Loyens, A.; Beauvillain, J.C.; Bailly, C. Mitochondrial proliferation during apoptosis induced by anticancer agents: Effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene 2004, 23, 7018–7030. [Google Scholar] [CrossRef] [PubMed]
- Reis-Mendes, A.; Gomes, A.S.; Carvalho, R.A.; Carvalho, F.; Remião, F.; Pinto, M.; Bastos, M.L.; Sousa, E.; Costa, V.M. Naphthoquinoxaline metabolite of mitoxantrone is less cardiotoxic than the parent compound and it can be a more cardiosafe drug in anticancer therapy. Arch. Toxicol. 2017, 91, 1871–1890. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.S.; Costa, V.M.; Diniz, C.; Fresco, P. Potentiation of cytotoxicity of paclitaxel in combination with Cl-IB-MECA in human C32 metastatic melanoma cells: A new possible therapeutic strategy for melanoma. Biomed. Pharmacother. 2013, 67, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.K.; Lee, J.H.; Shin, H.J.; Kim, J.H.; Choi, S. Structural and functional analysis of cell adhesion and nuclear envelope nano-topography in cell death. Sci. Rep. 2015, 5, 15623. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Ahir, M.; Sil, P.C. Doxorubicin-induced neurotoxicity is attenuated by a 43-kD protein from the leaves of Cajanus indicus L. via NF-kappaB and mitochondria dependent pathways. Free Radic. Res. 2012, 46, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Santos, R.X.; Carvalho, C.; Correia, S.; Pereira, G.C.; Pereira, S.S.; Oliveira, P.J.; Santos, M.S.; Proenca, T.; Moreira, P.I. Doxorubicin increases the susceptibility of brain mitochondria to Ca(2+)-induced permeability transition and oxidative damage. Free Radic. Biol. Med. 2008, 45, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.S.; Nogueira, T.B.; Costa, V.M.; Branco, P.S.; Ferreira, L.M.; Fernandes, E.; Bastos, M.L.; Meisel, A.; Carvalho, F.; Capela, J.P. Neurotoxicity of “ecstasy” and its metabolites in human dopaminergic differentiated SH-SY5Y cells. Toxicol. Lett. 2013, 216, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Feio-Azevedo, R.; Costa, V.M.; Ferreira, L.M.; Branco, P.S.; Pereira, F.C.; Bastos, M.L.; Carvalho, F.; Capela, J.P. Toxicity of the amphetamine metabolites 4-hydroxyamphetamine and 4-hydroxynorephedrine in human dopaminergic differentiated SH-SY5Y cells. Toxicol. Lett. 2017, 269, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Kim, S.D.; Lee, M.H.; Lee, H.S.; Lee, I.S.; Sung, J.K.; Yoon, Y.S. Protective effects of N-acetylcysteine and selenium against doxorubicin toxicity in rats. J. Vet. Sci. 2003, 4, 129–136. [Google Scholar] [PubMed]
- Chwastek, J.; Jantas, D.; Lason, W. The ATM kinase inhibitor KU-55933 provides neuroprotection against hydrogen peroxide-induced cell damage via a gammaH2AX/p-p53/caspase-3-independent mechanism: Inhibition of calpain and cathepsin D. Int. J. Biochem. Cell Biol. 2017, 87, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Rossato, L.G.; Costa, V.M.; Vilas-Boas, V.; de Lourdes Bastos, M.; Rolo, A.; Palmeira, C.; Remiao, F. Therapeutic concentrations of mitoxantrone elicit energetic imbalance in H9c2 cells as an earlier event. Cardiovasc. Toxicol. 2013, 13, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossato, L.G.; Costa, V.M.; Dallegrave, E.; Arbo, M.; Silva, R.; Ferreira, R.; Amado, F.; Dinis-Oliveira, R.J.; Duarte, J.A.; de Lourdes Bastos, M.; et al. Mitochondrial cumulative damage induced by mitoxantrone: Late onset cardiac energetic impairment. Cardiovasc. Toxicol. 2014, 14, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomkins, C.E.; Edwards, S.N.; Tolkovsky, A.M. Apoptosis is induced in post-mitotic rat sympathetic neurons by arabinosides and topoisomerase II inhibitors in the presence of NGF. J. Cell. Sci. 1994, 107 Pt 6, 1499–1507. [Google Scholar] [PubMed]
- Barbosa, D.J.; Capela, J.P.; de Lourdes Bastos, M.; Carvalho, F. In vitro models for neurotoxicology research. Toxicol. Res. 2015, 4, 801–842. [Google Scholar] [CrossRef]
- Wenker, S.D.; Chamorro, M.E.; Vota, D.M.; Callero, M.A.; Vittori, D.C.; Nesse, A.B. Differential antiapoptotic effect of erythropoietin on undifferentiated and retinoic acid-differentiated SH-SY5Y cells. J. Cell. Biochem. 2010, 110, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Lombet, A.; Zujovic, V.; Kandouz, M.; Billardon, C.; Carvajal-Gonzalez, S.; Gompel, A.; Rostene, W. Resistance to induced apoptosis in the human neuroblastoma cell line SK-N-SH in relation to neuronal differentiation. Role of Bcl-2 protein family. Eur. J. Biochem. 2001, 268, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Giordano, S.; Zelickson, B.R.; S Johnson, M.; A Benavides, G.; Ouyang, X.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic. Biol. Med. 2011, 51, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Xun, Z.; Lee, D.-Y.; Lim, J.; Canaria, C.A.; Barnebey, A.; Yanonne, S.M.; McMurray, C.T. Retinoic acid-induced differentiation increases the rate of oxygen consumption and enhances the spare respiratory capacity of mitochondria in SH-SY5Y cells. Mech. Ageing Dev. 2012, 133, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Strigun, A.; Wahrheit, J.; Niklas, J.; Heinzle, E.; Noor, F. Doxorubicin increases oxidative metabolism in HL-1 cardiomyocytes as shown by 13C metabolic flux analysis. Toxicol. Sci. 2012, 125, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, D.J.; Capela, J.P.; Feio-Azevedo, R.; Teixeira-Gomes, A.; Bastos, M.L.; Carvalho, F. Mitochondria: Key players in the neurotoxic effects of amphetamines. Arch. Toxicol. 2015, 89, 1695–1725. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | ||||
|---|---|---|---|---|
| MTX | Control | 0.13 μM | 0.2 μM | 0.5 μM |
| Condensed nuclei | 4 ± 4 | 205 ± 111 | 130 ± 29 | 117 ± 29 |
| Number of cells | 439 ± 102 | 357 ± 95 | 259 ± 19 | 212 ± 15 |
| Ratio of condensed nuclei/number of cells | 0.89 ± 0.76 | 57.04 ± 24.98 * | 50.09 ± 9.30 | 54.87 ± 9.54 |
| DOX | Control | 0.13 μM | 0.2 μM | 0.5 μM |
| Condensed nuclei | 2 ± 1 | 43 ± 8 | 26 ± 12 | 84 ± 14 |
| Number of cells | 436 ± 98 | 263 ± 29 | 186 ± 59 | 170 ± 18 |
| Ratio of condensed nuclei/number of cells | 0.39 ± 0.27 | 16.20 ± 2.05 | 13.53 ± 2.57 | 49.00 ± 4.00 ** |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, D.; Pinho, R.; Correia, V.; Soares, J.; Bastos, M.D.L.; Carvalho, F.; Capela, J.P.; Costa, V.M. Mitoxantrone is More Toxic than Doxorubicin in SH-SY5Y Human Cells: A ‘Chemobrain’ In Vitro Study. Pharmaceuticals 2018, 11, 41. https://doi.org/10.3390/ph11020041
Almeida D, Pinho R, Correia V, Soares J, Bastos MDL, Carvalho F, Capela JP, Costa VM. Mitoxantrone is More Toxic than Doxorubicin in SH-SY5Y Human Cells: A ‘Chemobrain’ In Vitro Study. Pharmaceuticals. 2018; 11(2):41. https://doi.org/10.3390/ph11020041
Chicago/Turabian StyleAlmeida, Daniela, Rita Pinho, Verónica Correia, Jorge Soares, Maria De Lourdes Bastos, Félix Carvalho, João Paulo Capela, and Vera Marisa Costa. 2018. "Mitoxantrone is More Toxic than Doxorubicin in SH-SY5Y Human Cells: A ‘Chemobrain’ In Vitro Study" Pharmaceuticals 11, no. 2: 41. https://doi.org/10.3390/ph11020041
APA StyleAlmeida, D., Pinho, R., Correia, V., Soares, J., Bastos, M. D. L., Carvalho, F., Capela, J. P., & Costa, V. M. (2018). Mitoxantrone is More Toxic than Doxorubicin in SH-SY5Y Human Cells: A ‘Chemobrain’ In Vitro Study. Pharmaceuticals, 11(2), 41. https://doi.org/10.3390/ph11020041