The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention


Abstract
:1. Introduction
2. Structure and Regulation of Calcium/Calmodulin-Stimulated Protein Kinase (CAMK) Family Members
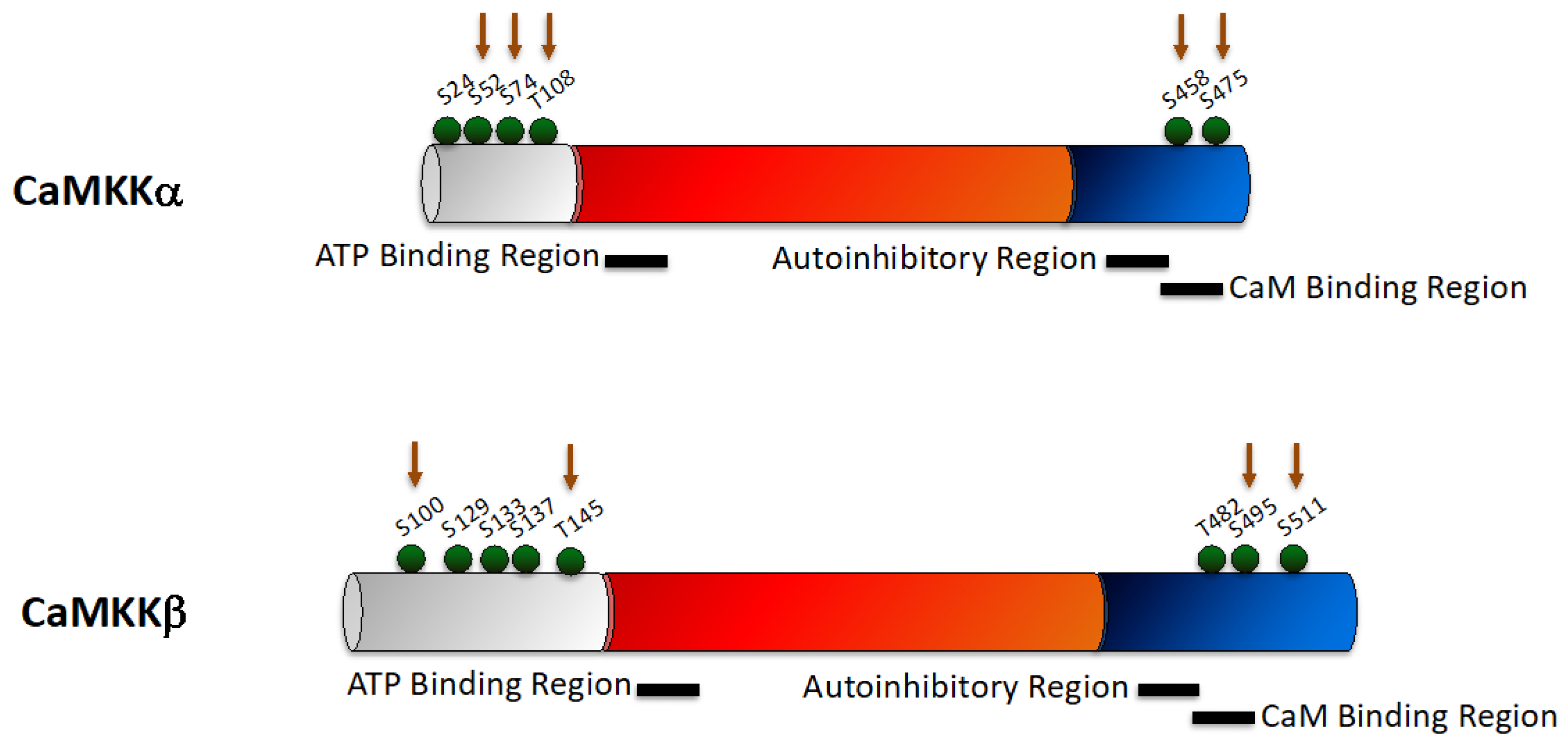
2.1. CaMKK
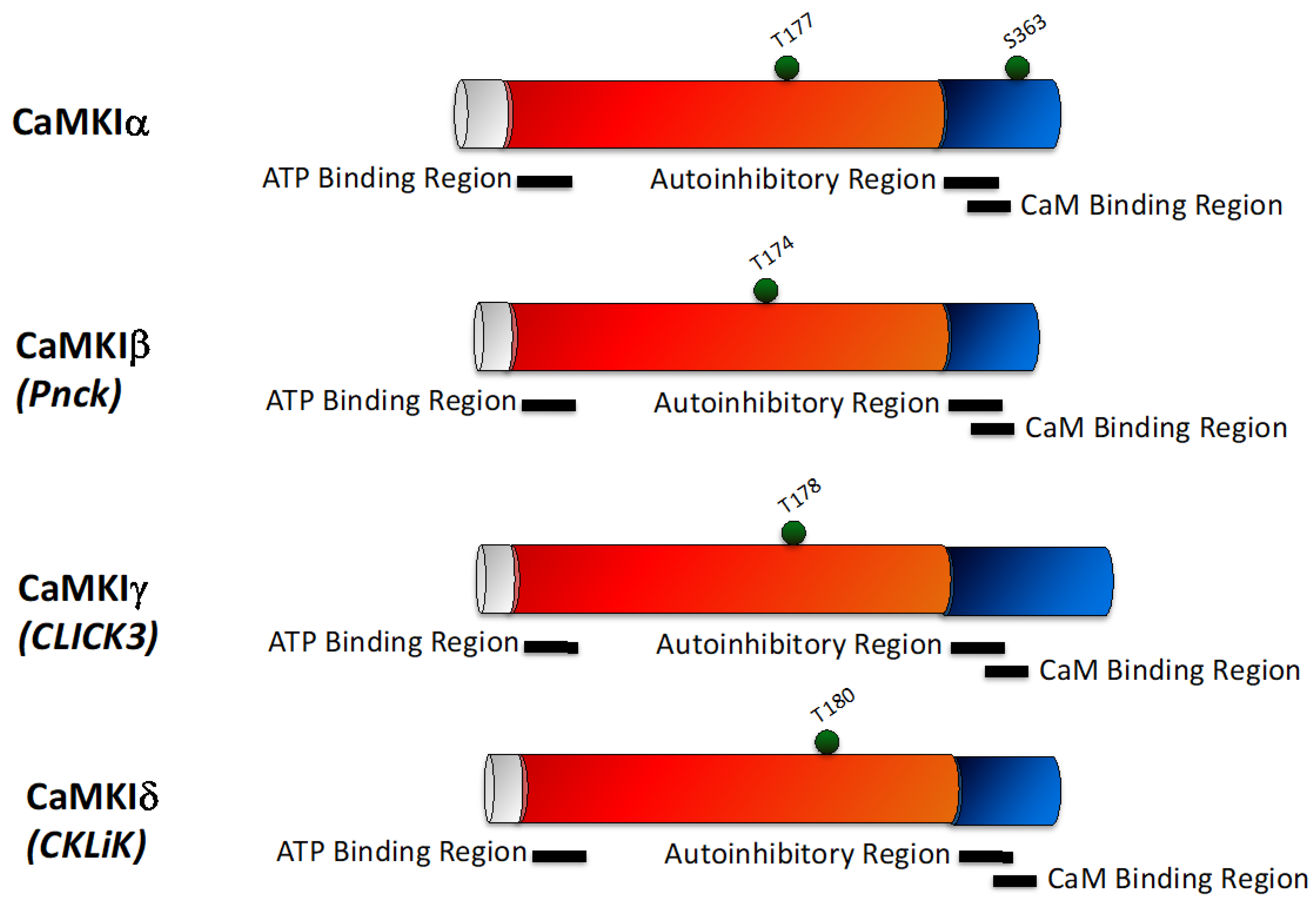
2.2. CaMKI
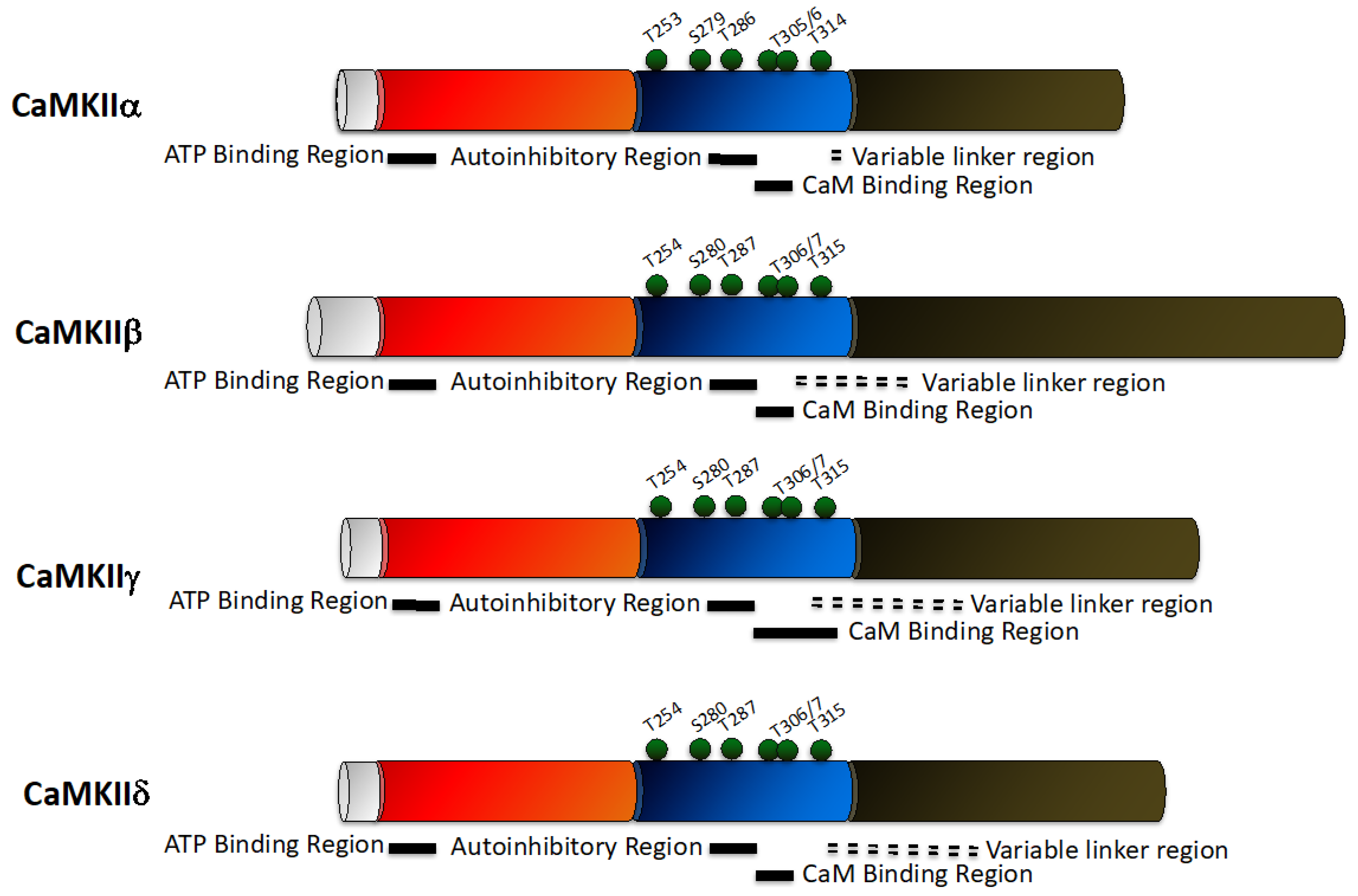
2.3. CaMKII
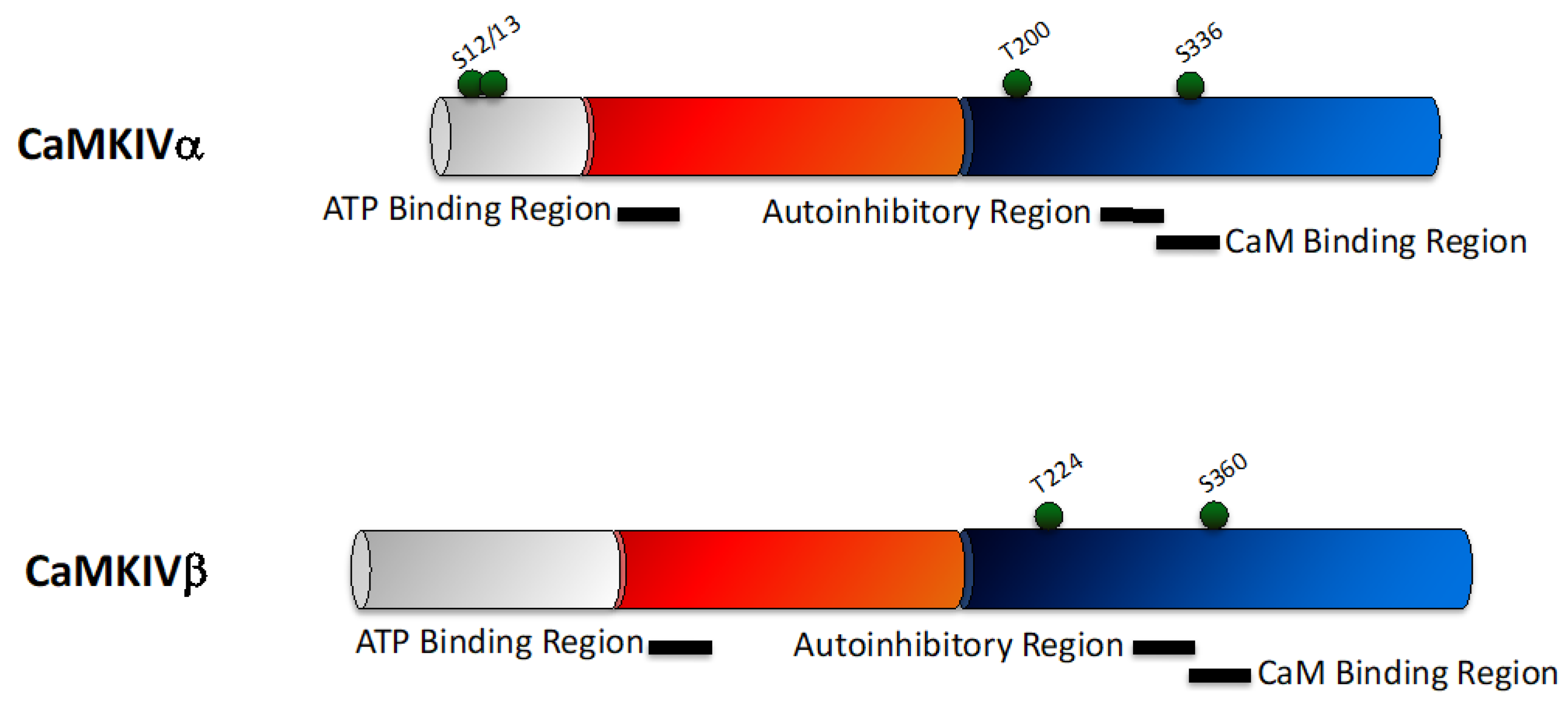
2.4. CaMKIV
3. The Role of CaMK Family Members in Cancer
3.1. CaMKK
3.2. CaMKI
3.3. CaMKII
3.4. CaMKIV
4. The CaMK Family Are Potential Anti-Cancer Therapeutic Targets
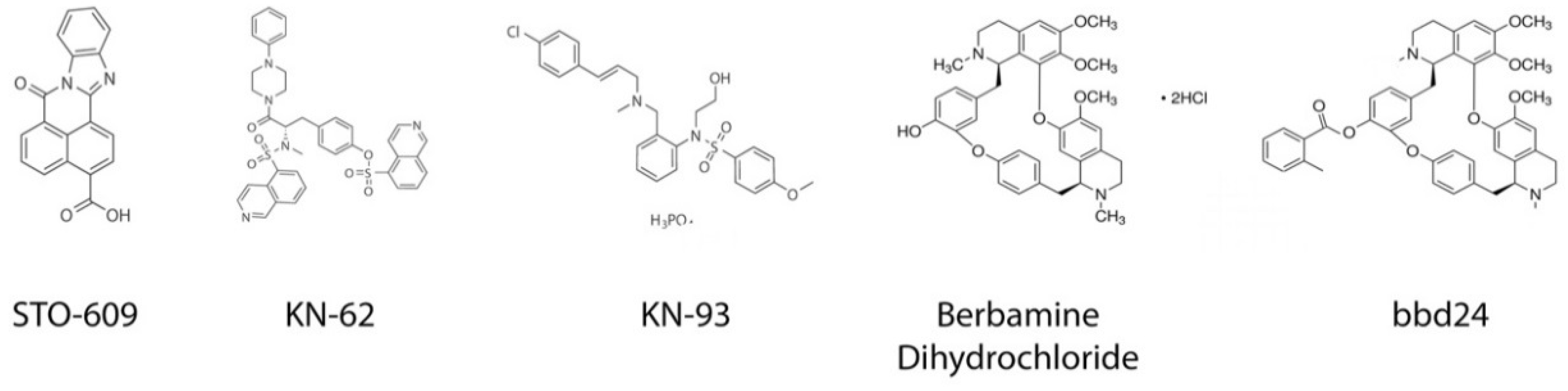
4.1. STO-609
4.2. KN-62/KN-93
4.3. Substrate Based Inhibitors: Autocamtide-3 Derived Peptide Inhibitor (AC3-I) and Autocamtide-2-Related Inhibitory Peptide (AIP)
4.4. CaMKIIN Derived Peptides (CaMKIINtide)
4.5. Berbamine Dihydrochloride
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Okuno, S.; Kitani, T.; Fujisawa, H. Studies on the substrate specificity of Ca2+/calmodulin-dependent protein kinase kinase alpha. J. Biochem. 1997, 122, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Tokumitsu, H.; Inuzuka, H.; Murata-Hori, M.; Hosoya, H.; Kobayashi, R. Identification and characterization of novel components of a Ca2+/calmodulin-dependent protein kinase cascade in HeLa cells. FEBS Lett. 2003, 550, 57–63. [Google Scholar] [CrossRef]
- Hsu, L.S.; Chen, G.D.; Lee, L.S.; Chi, C.W.; Cheng, J.F.; Chen, J.Y. Human Ca2+/calmodulin-dependent protein kinase kinase beta gene encodes multiple isoforms that display distinct kinase activity. J. Biol. Chem. 2001, 276, 31113–31123. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Brickey, D.A.; Glod, J.; Hidaka, H.; Sikela, J.; Soderling, T.R. Activation mechanisms for Ca2+/calmodulin-dependent protein kinase IV. Identification of a brain CaM-kinase IV kinase. J. Biol. Chem. 1994, 269, 28640–28647. [Google Scholar] [PubMed]
- Lee, J.C.; Edelman, A.M. A protein activator of Ca(2+)-calmodulin-dependent protein kinase Ia. J. Biol. Chem. 1994, 269, 2158–2164. [Google Scholar] [PubMed]
- Edelman, A.M.; Mitchelhill, K.I.; Selbert, M.A.; Anderson, K.A.; Hook, S.S.; Stapleton, D.; Goldstein, E.G.; Means, A.R.; Kemp, B.E. Multiple Ca(2+)-calmodulin-dependent protein kinase kinases from rat brain. Purification, regulation by Ca(2+)-calmodulin, and partial amino acid sequence. J. Biol. Chem. 1996, 271, 10806–10810. [Google Scholar] [CrossRef]
- Ohmstede, C.A.; Jensen, K.F.; Sahyoun, N.E. Ca2+/calmodulin-dependent protein kinase enriched in cerebellar granule cells. Identification of a novel neuronal calmodulin-dependent protein kinase. J. Biol. Chem. 1989, 264, 5866–5875. [Google Scholar]
- Racioppi, L.; Means, A.R. Calcium/calmodulin-dependent protein kinase kinase 2: Roles in signaling and pathophysiology. J. Biol. Chem. 2012, 287, 31658–31665. [Google Scholar] [CrossRef]
- Anderson, K.A.; Means, R.L.; Huang, Q.H.; Kemp, B.E.; Goldstein, E.G.; Selbert, M.A.; Edelman, A.M.; Fremeau, R.T.; Means, A.R. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J. Biol. Chem. 1998, 273, 31880–31889. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef]
- Yano, S.; Tokumitsu, H.; Soderling, T.R. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 1998, 396, 584–587. [Google Scholar] [CrossRef]
- Anderson, K.A.; Means, A.R. Defective signaling in a subpopulation of CD4(+) T cells in the absence of Ca(2+)/calmodulin-dependent protein kinase IV. Mol. Cell. Biol. 2002, 22, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Molina-Campos, E.; Marrero-Rosado, B.; Bradford, A.B.; Fox, S.M.; Kovalova, N.; Hannon, H.E. CaMKK-CaMKI signaling pathways differentially control axon and dendrite elongation in cortical neurons. J. Neurosci. 2010, 30, 2807–2809. [Google Scholar] [CrossRef] [PubMed]
- Kitsos, C.M.; Sankar, U.; Illario, M.; Colomer-Font, J.M.; Duncan, A.W.; Ribar, T.J.; Reya, T.; Means, A.R. Calmodulin-dependent protein kinase IV regulates hematopoietic stem cell maintenance. J. Biol. Chem. 2005, 280, 33101–33108. [Google Scholar] [CrossRef] [PubMed]
- Kahl, C.R.; Means, A.R. Regulation of cyclin D1/Cdk4 complexes by calcium/calmodulin-dependent protein kinase I. J. Biol. Chem. 2004, 279, 15411–15419. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Ribar, T.J.; Illario, M.; Means, A.R. Defective survival and activation of thymocytes in transgenic mice expressing a catalytically inactive form of Ca2+/calmodulin-dependent protein kinase IV. Mol. Endocrinol. 1997, 11, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Wayman, G.A.; Lee, Y.S.; Tokumitsu, H.; Silva, A.J.; Soderling, T.R. Calmodulin-kinases: Modulators of neuronal development and plasticity. Neuron 2008, 59, 914–931. [Google Scholar] [CrossRef]
- Lin, F.; Ribar, T.J.; Means, A.R. The Ca2+/calmodulin-dependent protein kinase kinase, CaMKK2, inhibits preadipocyte differentiation. Endocrinology 2011, 152, 3668–3679. [Google Scholar] [CrossRef]
- Witczak, C.A.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. Ca2+/calmodulin-dependent protein kinase kinase-alpha regulates skeletal muscle glucose uptake independent of AMP-activated protein kinase and Akt activation. Diabetes 2007, 56, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Soderling, T.R. Requirements for calcium and calmodulin in the calmodulin kinase activation cascade. J. Biol. Chem. 1996, 271, 5617–5622. [Google Scholar] [CrossRef] [PubMed]
- Green, M.F.; Scott, J.W.; Steel, R.; Oakhill, J.S.; Kemp, B.E.; Means, A.R. Ca2+/Calmodulin-dependent Protein Kinase Kinase beta Is Regulated by Multisite Phosphorylation. J. Biol. Chem. 2011, 286, 28066–28079. [Google Scholar] [CrossRef] [PubMed]
- Wayman, G.A.; Tokumitsu, H.; Soderling, T.R. Inhibitory cross-talk by cAMP kinase on the calmodulin-dependent protein kinase cascade. J. Biol. Chem. 1997, 272, 16073–16076. [Google Scholar] [CrossRef] [PubMed]
- Okuno, S.; Kitani, T.; Fujisawa, H. Regulation of Ca(2+)/calmodulin-dependent protein kinase kinase alpha by cAMP-dependent protein kinase: I. Biochemical analysis. J. Biochem. 2001, 130, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Hatano, N.; Fujimoto, T.; Yurimoto, S.; Kobayashi, R. Generation of autonomous activity of Ca(2+)/calmodulin-dependent protein kinase kinase beta by autophosphorylation. Biochemistry 2011, 50, 8193–8201. [Google Scholar] [CrossRef]
- Davare, M.A.; Saneyoshi, T.; Guire, E.S.; Nygaard, S.C.; Soderling, T.R. Inhibition of calcium/calmodulin-dependent protein kinase kinase by protein 14-3-3. J. Biol. Chem. 2004, 279, 52191–52199. [Google Scholar] [CrossRef]
- Matsushita, M.; Nairn, A.C. Inhibition of the Ca2+/calmodulin-dependent protein kinase I cascade by cAMP-dependent protein kinase. J. Biol. Chem. 1999, 274, 10086–10093. [Google Scholar] [CrossRef]
- Loseth, O.P.; de Lecea, L.; Calbet, M.; Danielson, P.E.; Gautvik, V.; Hovring, P.I.; Walaas, S.I.; Gautvik, K.M. Developmental regulation of two isoforms of Ca(2+)/calmodulin-dependent protein kinase I beta in rat brain. Brain Res. 2000, 869, 137–145. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Zoli, M.; Bertuzzi, G.; Nairn, A.C. Immunochemical localization of calcium/calmodulin-dependent protein kinase I. Synapse 1995, 20, 75–84. [Google Scholar] [CrossRef]
- Kamata, A.; Sakagami, H.; Tokumitsu, H.; Owada, Y.; Fukunaga, K.; Kondo, H. Spatiotemporal expression of four isoforms of Ca2+/calmodulin-dependent protein kinase I in brain and its possible roles in hippocampal dendritic growth. Neurosci. Res. 2007, 57, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Nairn, A.C.; Greengard, P. Purification and characterization of Ca2+/calmodulin-dependent protein kinase I from bovine brain. J. Biol. Chem. 1987, 262, 7273–7281. [Google Scholar] [PubMed]
- Joseph, J.D.; Means, A.R. Identification and characterization of two Ca2+/CaM-dependent protein kinases required for normal nuclear division in Aspergillus nidulans. J. Biol. Chem. 2000, 275, 38230–38238. [Google Scholar] [CrossRef] [PubMed]
- Skelding, K.A.; Rostas, J.A.; Verrills, N.M. Controlling the cell cycle: The role of calcium/calmodulin-stimulated protein kinases I and II. Cell Cycle 2011, 10, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wayman, G.A.; Kaech, S.; Grant, W.F.; Davare, M.; Impey, S.; Tokumitsu, H.; Nozaki, N.; Banker, G.; Soderling, T.R. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J. Neurosci. 2004, 24, 3786–3794. [Google Scholar] [CrossRef] [PubMed]
- Condon, J.C.; Pezzi, V.; Drummond, B.M.; Yin, S.; Rainey, W.E. Calmodulin-dependent kinase I regulates adrenal cell expression of aldosterone synthase. Endocrinology 2002, 143, 3651–3657. [Google Scholar] [CrossRef]
- Takemoto-Kimura, S.; Ageta-Ishihara, N.; Nonaka, M.; Adachi-Morishima, A.; Mano, T.; Okamura, M.; Fujii, H.; Fuse, T.; Hoshino, M.; Suzuki, S.; et al. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK-III/CaMKIgamma. Neuron 2007, 54, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.M.; Guire, E.S.; Saneyoshi, T.; Soderling, T.R. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. J. Neurosci. 2005, 25, 1281–1290. [Google Scholar] [CrossRef]
- Ang, E.S.; Zhang, P.; Steer, J.H.; Tan, J.W.; Yip, K.; Zheng, M.H.; Joyce, D.A.; Xu, J. Calcium/calmodulin-dependent kinase activity is required for efficient induction of osteoclast differentiation and bone resorption by receptor activator of nuclear factor kappa B ligand (RANKL). J. Cell. Physiol. 2007, 212, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Haribabu, B.; Hook, S.S.; Selbert, M.A.; Goldstein, E.G.; Tomhave, E.D.; Edelman, A.M.; Snyderman, R.; Means, A.R. Human Calcium-Calmodulin Dependent Protein-Kinase-I—Cdna Cloning, Domain-Structure and Activation by Phosphorylation at Threonine-177 by Calcium-Calmodulin Dependent Protein-Kinase-I Kinase. EMBO J. 1995, 14, 3679–3686. [Google Scholar] [CrossRef]
- Takemoto-Kimura, S.; Terai, H.; Takamoto, M.; Ohmae, S.; Kikumura, S.; Segi, E.; Arakawa, Y.; Furuyashiki, T.; Narumiya, S.; Bito, H. Molecular cloning and characterization of CLICK-III/CaMKIgamma, a novel membrane-anchored neuronal Ca2+/calmodulin-dependent protein kinase (CaMK). J. Biol. Chem. 2003, 278, 18597–18605. [Google Scholar] [CrossRef] [PubMed]
- Senga, Y.; Ishida, A.; Shigeri, Y.; Kameshita, I.; Sueyoshi, N. The Phosphatase-Resistant Isoform of CaMKI, Ca(2)(+)/Calmodulin-Dependent Protein Kinase Idelta (CaMKIdelta), Remains in Its “Primed” Form without Ca(2)(+) Stimulation. Biochemistry 2015, 54, 3617–3630. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.G.; Bourke, A.M.; O’Leary, H.; Zaegel, V.; Lasda, E.; Mize-Berge, J.; Quillinan, N.; Tucker, C.L.; Coultrap, S.J.; Herson, P.S.; et al. Analysis of the CaMKIIalpha and beta splice-variant distribution among brain regions reveals isoform-specific differences in holoenzyme formation. Sci. Rep. 2018, 8, 5448. [Google Scholar] [CrossRef] [PubMed]
- Giese, K.P.; Fedorov, N.B.; Filipkowski, R.K.; Silva, A.J. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 1998, 279, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Soderling, T.R.; Derkach, V.A. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000, 23, 75–80. [Google Scholar] [CrossRef]
- Von Hertzen, L.S.; Giese, K.P. Alpha-isoform of Ca2+/calmodulin-dependent kinase II autophosphorylation is required for memory consolidation-specific transcription. Neuroreport 2005, 16, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.W. Regulation of synaptic transmission by presynaptic CaMKII and BK channels. Mol. Neurobiol. 2008, 38, 153–166. [Google Scholar] [CrossRef]
- Fink, C.C.; Bayer, K.U.; Myers, J.W.; Ferrell, J.E.; Schulman, H.; Meyer, T. Selective regulation of neurite extension and synapse formation by the beta but not the of isoform of CaMKII. Neuron 2003, 39, 283–297. [Google Scholar] [CrossRef]
- Jones, K.T. Intracellular calcium in the fertilization and development of mammalian eggs. Clin. Exp. Pharmacol. Physiol. 2007, 34, 1084–1089. [Google Scholar] [CrossRef]
- Shin, M.K.; Kim, M.K.; Bae, Y.S.; Jo, I.; Lee, S.J.; Chung, C.P.; Park, Y.J.; Min do, S. A novel collagen-binding peptide promotes osteogenic differentiation via Ca2+/calmodulin-dependent protein kinase II/ERK/AP-1 signaling pathway in human bone marrow-derived mesenchymal stem cells. Cell. Signal. 2008, 20, 613–624. [Google Scholar] [CrossRef]
- Munevar, S.; Gangopadhyay, S.S.; Gallant, C.; Colombo, B.; Sellke, F.W.; Morgan, K.G. CaMKIIT287 and T305 regulate history-dependent increases in alpha agonist-induced vascular tone. J. Cell. Mol. Med. 2008, 12, 219–226. [Google Scholar] [CrossRef]
- Maier, L.S.; Bers, D.M. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007, 73, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xu, C.; Ran, D.; Wang, Y.; Zhao, H.; Gu, J.; Liu, X.; Bian, J.; Yuan, Y.; Liu, Z. CaMK mediates cadmium induced apoptosis in rat primary osteoblasts through MAPK activation and endoplasmic reticulum stress. Toxicology 2018, 406–407, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Rostas, J.A.; Hoffman, A.; Murtha, L.A.; Pepperall, D.; McLeod, D.D.; Dickson, P.W.; Spratt, N.J.; Skelding, K.A. Ischaemia- and excitotoxicity-induced CaMKII-Mediated neuronal cell death: The relative roles of CaMKII autophosphorylation at T286 and T253. Neurochem. Int. 2017, 104, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Rostas, J.A.P.; Spratt, N.J.; Dickson, P.W.; Skelding, K.A. The role of Ca2+-calmodulin stimulated protein kinase II in ischaemic stroke—A potential target for neuroprotective therapies. Neurochem. Int. 2017, 107, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Skelding, K.A.; Spratt, N.J.; Fluechter, L.; Dickson, P.W.; Rostas, J.A.P. alpha CaMKII is differentially regulated in brain regions that exhibit differing sensitivities to ischemia and excitotoxicity. J. Cerebr. Blood Flow Metab. 2012, 32, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, O.S.; Deindl, S.; Sung, R.J.; Nairn, A.C.; Kuriyan, J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell 2005, 123, 849–860. [Google Scholar] [CrossRef]
- Hanson, P.I.; Meyer, T.; Stryer, L.; Schulman, H. Dual Role of Calmodulin in Autophosphorylation of Multifunctional Cam Kinase May Underlie Decoding of Calcium Signals. Neuron 1994, 12, 943–956. [Google Scholar] [CrossRef]
- Rostas, J.A.; Skelding, K.A.; Verrills, N.M.; Suzuki, P.W.; Dickson, T. Camkii Binding Partners Vary with Cell Type and Phosphorylation State. J. Neurochem. 2009, 110, 40–41. [Google Scholar]
- Skelding, K.A.; Suzuki, T.; Gordon, S.; Xue, J.; Verrills, N.M.; Dickson, P.W.; Rostas, J.A. Regulation of CaMKII by phospho-Thr253 or phospho-Thr286 sensitive targeting alters cellular function. Cell. Signal. 2010, 22, 759–769. [Google Scholar] [CrossRef]
- Meyer, T.; Hanson, P.I.; Stryer, L.; Schulman, H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science 1992, 256, 1199–1202. [Google Scholar] [CrossRef] [PubMed]
- Strack, S.; Colbran, R.J. Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. J. Biol. Chem. 1998, 273, 20689–20692. [Google Scholar] [CrossRef] [PubMed]
- Strack, S.; Choi, S.; Lovinger, D.M.; Colbran, R.J. Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J. Biol. Chem. 1997, 272, 13467–13470. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Schulman, H. Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J. Biol. Chem. 1992, 267, 17216–17224. [Google Scholar] [PubMed]
- Patton, B.L.; Miller, S.G.; Kennedy, M.B. Activation of type II calcium/calmodulin-dependent protein kinase by Ca2+/calmodulin is inhibited by autophosphorylation of threonine within the calmodulin-binding domain. J. Biol. Chem. 1990, 265, 11204–11212. [Google Scholar]
- Meador, W.E.; Means, A.R.; Quiocho, F.A. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science 1993, 262, 1718–1721. [Google Scholar] [CrossRef]
- Braun, A.P.; Schulman, H. The multifunctional calcium/calmodulin-dependent protein kinase: From form to function. Annu. Rev. Physiol. 1995, 57, 417–445. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Jaffe, H.; Vinade, L.; Dosemeci, A. Identification of novel phosphorylation sites on postsynaptic density proteins. Biochem. Biophys. Res. Commun. 2004, 321, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Molloy, S.S.; Kennedy, M.B. Autophosphorylation of type II Ca2+/calmodulin-dependent protein kinase in cultures of postnatal rat hippocampal slices. Proc. Natl. Acad. Sci. USA 1991, 88, 4756–4760. [Google Scholar] [CrossRef]
- Collins, M.O.; Yu, L.; Coba, M.P.; Husi, H.; Campuzano, I.; Blackstock, W.P.; Choudhary, J.S.; Grant, S.G. Proteomic analysis of in vivo phosphorylated synaptic proteins. J. Biol. Chem. 2005, 280, 5972–5982. [Google Scholar] [CrossRef] [PubMed]
- Migues, P.V.; Lehmann, I.T.; Fluechter, L.; Cammarota, M.; Gurd, J.W.; Sim, A.T.R.; Dickson, P.W.; Rostas, J.A.P. Phosphorylation of CaMKII at Thr253 occurs in vivo and enhances binding to isolated postsynaptic densities. J. Neurochem. 2006, 98, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakagami, H.; Kondo, H. Cloning and sequencing of a gene encoding the beta polypeptide of Ca2+/calmodulin-dependent protein kinase IV and its expression confined to the mature cerebellar granule cells. Brain Res. Mol. Brain Res. 1993, 19, 215–218. [Google Scholar] [CrossRef]
- Mosialos, G.; Hanissian, S.H.; Jawahar, S.; Vara, L.; Kieff, E.; Chatila, T.A. A Ca2+/calmodulin-dependent protein kinase, CaM kinase-Gr, expressed after transformation of primary human B lymphocytes by Epstein-Barr virus (EBV) is induced by the EBV oncogene LMP1. J. Virol. 1994, 68, 1697–1705. [Google Scholar]
- Wu, J.Y.; Gonzalez-Robayana, I.J.; Richards, J.S.; Means, A.R. Female fertility is reduced in mice lacking Ca2+/calmodulin-dependent protein kinase IV. Endocrinology 2000, 141, 4777–4783. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Means, A.R. Ca(2+)/calmodulin-dependent protein kinase IV is expressed in spermatids and targeted to chromatin and the nuclear matrix. J. Biol. Chem. 2000, 275, 7994–7999. [Google Scholar] [CrossRef]
- Kimura, Y.; Corcoran, E.E.; Eto, K.; Gengyo-Ando, K.; Muramatsu, M.A.; Kobayashi, R.; Freedman, J.H.; Mitani, S.; Hagiwara, M.; Means, A.R.; et al. A CaMK cascade activates CRE-mediated transcription in neurons of Caenorhabditis elegans. EMBO Rep. 2002, 3, 962–966. [Google Scholar] [CrossRef] [Green Version]
- Bleier, J.; Toliver, A. Exploring the Role of CaMKIV in Homeostatic Plasticity. J. Neurosci. 2017, 37, 11520–11522. [Google Scholar] [CrossRef]
- Takemura, M.; Mishima, T.; Wang, Y.; Kasahara, J.; Fukunaga, K.; Ohashi, K.; Mizuno, K. Ca2+/Calmodulin-dependent Protein Kinase IV-mediated LIM Kinase Activation Is Critical for Calcium Signal-induced Neurite Outgrowth. J. Biol. Chem. 2009, 284, 28554–28562. [Google Scholar] [CrossRef]
- Wei, F.; Qiu, C.S.; Liauw, J.; Robinson, D.A.; Ho, N.; Chatila, T.; Zhuo, M. Calcium calmodulin-dependent protein kinase IV is required for fear memory. Nat. Neurosci. 2002, 5, 573–579. [Google Scholar] [CrossRef]
- Racioppi, L.; Means, A.R. Calcium/calmodulin-dependent kinase IV in immune and inflammatory responses: Novel routes for an ancient traveller. Trends Immunol. 2008, 29, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Selbert, M.A.; Anderson, K.A.; Huang, Q.H.; Goldstein, E.G.; Means, A.R.; Edelman, A.M. Phosphorylation and activation of Ca(2+)-calmodulin-dependent protein kinase IV by Ca(2+)-calmodulin-dependent protein kinase Ia kinase. Phosphorylation of threonine 196 is essential for activation. J. Biol. Chem. 1995, 270, 17616–17621. [Google Scholar] [CrossRef] [PubMed]
- Chatila, T.; Anderson, K.A.; Ho, N.; Means, A.R. A unique phosphorylation-dependent mechanism for the activation of Ca2+/calmodulin-dependent protein kinase type IV/GR. J. Biol. Chem. 1996, 271, 21542–21548. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Okuno, S.; Kitani, T.; Fujisawa, H. Inactivation of calmodulin-dependent protein kinase IV by autophosphorylation of serine 332 within the putative calmodulin-binding domain. J. Biol. Chem. 1996, 271, 6903–6910. [Google Scholar] [CrossRef] [PubMed]
- Subbannayya, Y.; Syed, N.; Barbhuiya, M.A.; Raja, R.; Marimuthu, A.; Sahasrabuddhe, N.; Pinto, S.M.; Manda, S.S.; Renuse, S.; Manju, H.C.; et al. Calcium calmodulin dependent kinase kinase 2—A novel therapeutic target for gastric adenocarcinoma. Cancer Biol. Ther. 2015, 16, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Marcelo, K.L.; Rajapakshe, K.; Coarfa, C.; Dean, A.; Wilganowski, N.; Robinson, H.; Sevick, E.; Bissig, K.D.; Goldie, L.C.; et al. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology 2015, 62, 505–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.M.; Wang, H.J.; Han, B.; Meng, X.Q.; Chen, M.H.; Yang, D.B.; Sun, Y.; Li, Y.L.; Jiang, C.L. CAMKK2, Regulated by Promoter Methylation, is a Prognostic Marker in Diffuse Gliomas. CNS Neurosci. Ther. 2016, 22, 518–524. [Google Scholar] [CrossRef]
- Gocher, A.M.; Azabdaftari, G.; Euscher, L.M.; Dai, S.; Karacosta, L.G.; Franke, T.F.; Edelman, A.M. Akt activation by Ca(2+)/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J. Biol. Chem. 2017, 292, 14188–14204. [Google Scholar] [CrossRef] [PubMed]
- Karacosta, L.G.; Foster, B.A.; Azabdaftari, G.; Feliciano, D.M.; Edelman, A.M. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J. Biol. Chem. 2012, 287, 24832–24843. [Google Scholar] [CrossRef]
- Shima, T.; Mizokami, A.; Miyagi, T.; Kawai, K.; Izumi, K.; Kumaki, M.; Ofude, M.; Zhang, J.; Keller, E.T.; Namiki, M. Down-regulation of calcium/calmodulin-dependent protein kinase kinase 2 by androgen deprivation induces castration-resistant prostate cancer. Prostate 2012, 72, 1789–1801. [Google Scholar] [CrossRef] [Green Version]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigo, D.E.; Howe, M.K.; Wittmann, B.M.; Brunner, A.M.; Cushman, I.; Wang, Q.; Brown, M.; Means, A.R.; McDonnell, D.P. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011, 71, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Cui, C.; Wang, C.; Wu, G.; Chen, H.; Lu, Z.; Chen, X.; Wang, L.; Huang, J.; Geng, H.; et al. CAMKs support development of acute myeloid leukemia. J. Hematol. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Ueda, T.; Nasu, K.; Yamashita, S.; Toyofuku, M.; Narahara, H. Targeting calcium/calmodulin-dependence kinase I and II as a potential anti-proliferation remedy for endometrial carcinomas. Cancer Lett. 2009, 277, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Gardner, H.P.; Ha, S.I.; Reynolds, C.; Chodosh, L.A. The caM kinase, Pnck, is spatially and temporally regulated during murine mammary gland development and may identify an epithelial cell subtype involved in breast cancer. Cancer Res. 2000, 60, 5571–5577. [Google Scholar] [PubMed]
- Wu, S.; Lv, Z.J.; Wang, Y.; Sun, L.; Jiang, Z.M.; Xu, C.J.; Zhao, J.; Sun, X.J.; Li, X.X.; Hu, L.J.; et al. Increased Expression of Pregnancy Up-Regulated Non-Ubiquitous Calmodulin Kinase Is Associated with Poor Prognosis in Clear Cell Renal Cell Carcinoma. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.; De Brasi, C.; Ferri, C.; Bengio, R.; Bianchini, M.; Larripa, I. CAMKIIgamma, HSP70 and HSP90 transcripts are differentially expressed in chronic myeloid leukemia cells from patients with resistant mutated disease. Leuk. Lymphoma 2014, 55, 2101–2108. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Collins, S.J. Activated Ca2+/calmodulin-dependent protein kinase IIgamma is a critical regulator of myeloid leukemia cell proliferation. Cancer Res. 2008, 68, 3733–3742. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; An, P.; Quan, X.J.; Zhang, J.; Zhou, Z.Y.; Zou, L.P.; Luo, H.S. Ca(2+)/calmodulin-dependent protein kinase II regulates colon cancer proliferation and migration via ERK1/2 and p38 pathways. World J. Gastroenterol. 2017, 23, 6111–6118. [Google Scholar] [CrossRef]
- Chi, M.; Evans, H.; Gilchrist, J.; Mayhew, J.; Hoffman, A.; Pearsall, E.A.; Jankowski, H.; Brzozowski, J.S.; Skelding, K.A. Phosphorylation of calcium/calmodulin-stimulated protein kinase II at T286 enhances invasion and migration of human breast cancer cells. Sci. Rep. 2016, 6, 33132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daft, P.G.; Yuan, K.; Warram, J.M.; Klein, M.J.; Siegal, G.P.; Zayzafoon, M. Alpha-CaMKII plays a critical role in determining the aggressive behavior of human osteosarcoma. Mol. Cancer Res. 2013, 11, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Chung, L.W.; Siegal, G.P.; Zayzafoon, M. alpha-CaMKII controls the growth of human osteosarcoma by regulating cell cycle progression. Lab. Investig. 2007, 87, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.; Xu, X.; Wang, Y.; Zhou, Y.; Zhang, C.; Yang, Y.; Yang, Y.; Xu, H.; Xu, R.; Wang, K. Ca2+/calmodulin-dependent protein kinase IIgamma enhances stem-like traits and tumorigenicity of lung cancer cells. Oncotarget 2015, 6, 16069–16083. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Pan, Y.; Wang, Y.; Hu, L.; Cao, S.; Chu, M.; Dai, J.; Shu, Y.; Xu, L.; Chen, J.; et al. Genome-wide association study of survival in early-stage non-small cell lung cancer. Ann. Surg. Oncol. 2015, 22, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Han, G.; Cao, Y.; Wang, Y.; Gong, H. Calcium/calmodulindependent protein kinase II enhances metastasis of human gastric cancer by upregulating nuclear factorkappaB and Aktmediated matrix metalloproteinase9 production. Mol. Med. Rep. 2014, 10, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- Tamura, N.; Tai, Y.; Sugimoto, K.; Kobayashi, R.; Konishi, R.; Nishioka, M.; Masaki, T.; Nagahata, S.; Tokuda, M. Enhanced expression and activation of Ca(2+)/calmodulin-dependent protein kinase IV in hepatocellular carcinoma. Cancer 2000, 89, 1910–1916. [Google Scholar] [CrossRef]
- Fu, H.; He, H.C.; Han, Z.D.; Wan, Y.P.; Luo, H.W.; Huang, Y.Q.; Cai, C.; Liang, Y.X.; Dai, Q.S.; Jiang, F.N.; et al. MicroRNA-224 and its target CAMKK2 synergistically influence tumor progression and patient prognosis in prostate cancer. Tumour Biol. 2015, 36, 1983–1991. [Google Scholar] [CrossRef]
- Ma, Z.; Wen, D.; Wang, X.; Yang, L.; Liu, T.; Liu, J.; Zhu, J.; Fang, X. Growth inhibition of human gastric adenocarcinoma cells in vitro by STO-609 is independent of calcium/calmodulin-dependent protein kinase kinase-beta and adenosine monophosphate-activated protein kinase. Am. J. Transl. Res. 2016, 8, 1164–1171. [Google Scholar]
- Davare, M.A.; Saneyoshi, T.; Soderling, T.R. Calmodulin-kinases regulate basal and estrogen stimulated medulloblastoma migration via Rac1. J. Neurooncol. 2011, 104, 65–82. [Google Scholar] [CrossRef]
- Rodriguez-Mora, O.G.; LaHair, M.M.; McCubrey, J.A.; Franklin, R.A. Calcium/calmodulin-dependent kinase I and calcium/calmodulin-dependent kinase kinase participate in the control of cell cycle progression in MCF-7 human breast cancer cells. Cancer Res. 2005, 65, 5408–5416. [Google Scholar] [CrossRef]
- Williams, C.L.; Phelps, S.H.; Porter, R.A. Expression of Ca2+/calmodulin-dependent protein kinase types II and IV, and reduced DNA synthesis due to the Ca2+/calmodulin-dependent protein kinase inhibitor KN-62 (1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenyl piperazine) in small cell lung carcinoma. Biochem. Pharmacol. 1996, 51, 707–715. [Google Scholar] [PubMed]
- Russo, E.; Salzano, M.; De Falco, V.; Mian, C.; Barollo, S.; Secondo, A.; Bifulco, M.; Vitale, M. Calcium/Calmodulin-dependent protein kinase II and its endogenous inhibitor alpha in medullary thyroid cancer. Clin. Cancer Res. 2014, 20, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zhuang, L.; Zhang, B.; Wang, F.; Chen, X.; Xia, C.; Zhang, B. DAG/PKCdelta and IP3/Ca(2)(+)/CaMK IIbeta Operate in Parallel to Each Other in PLCgamma1-Driven Cell Proliferation and Migration of Human Gastric Adenocarcinoma Cells, through Akt/mTOR/S6 Pathway. Int. J. Mol. Sci. 2015, 16, 28510–28522. [Google Scholar] [CrossRef]
- Mamaeva, O.A.; Kim, J.; Feng, G.; McDonald, J.M. Calcium/calmodulin-dependent kinase II regulates notch-1 signaling in prostate cancer cells. J. Cell. Biochem. 2009, 106, 25–32. [Google Scholar] [CrossRef]
- Wang, Q.; Symes, A.J.; Kane, C.A.; Freeman, A.; Nariculam, J.; Munson, P.; Thrasivoulou, C.; Masters, J.R.; Ahmed, A. A novel role for Wnt/Ca2+ signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PLoS ONE 2010, 5, e10456. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zhang, J.; Ma, X.; Kim, B.W.; Wang, H.; Li, J.; Pan, Y.; Xu, Y.; Ding, L.; Yang, L.; et al. Stabilization of the c-Myc Protein by CAMKIIgamma Promotes T Cell Lymphoma. Cancer Cell 2017. [Google Scholar] [CrossRef]
- Hoffman, A.; Carpenter, H.; Kahl, R.; Watt, L.F.; Dickson, P.W.; Rostas, J.A.P.; Verrills, N.M.; Skelding, K.A. Dephosphorylation of CaMKII at T253 controls the metaphase-anaphase transition. Cell. Signal. 2014, 26, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Huang, S.M.; Chen, C.C.; Tsai, C.F.; Yeh, W.L.; Chou, S.J.; Hsieh, W.T.; Lu, D.Y. Ghrelin induces cell migration through GHS-R, CaMKII, AMPK, and NF-kappaB signaling pathway in glioma cells. J. Cell. Biochem. 2011, 112, 2931–2941. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Sontheimer, H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J. Biol. Chem. 2010, 285, 11188–11196. [Google Scholar] [CrossRef]
- Shin, H.J.; Lee, S.; Jung, H.J. A curcumin derivative hydrazinobenzoylcurcumin suppresses stem-like features of glioblastoma cells by targeting Ca(2+) /calmodulin-dependent protein kinase II. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Monaco, S.; Rusciano, M.R.; Maione, A.S.; Soprano, M.; Gomathinayagam, R.; Todd, L.R.; Campiglia, P.; Salzano, S.; Pastore, L.; Leggiero, E.; et al. A novel crosstalk between calcium/calmodulin kinases II and IV regulates cell proliferation in myeloid leukemia cells. Cell. Signal. 2015, 27, 204–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Suzuki, M.; Satoh, H.; Kihara-Negishi, F.; Nakano, H.; Oikawa, T. Effects of PU.1-induced mouse calcium-calmodulin-dependent kinase I-like kinase (CKLiK) on apoptosis of murine erythroleukemia cells. Exp. Cell Res. 2004, 294, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Tombes, R.M.; Krystal, G.W. Identification of novel human tumor cell-specific CaMK-II variants. Biochim. Biophys. Acta 1997, 1355, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.F.; Xiao, C.; Roa, W.H.; Krammer, P.H.; Hao, C. Calcium/calmodulin-dependent protein kinase II regulation of c-FLIP expression and phosphorylation in modulation of Fas-mediated signaling in malignant glioma cells. J. Biol. Chem. 2003, 278, 7043–7050. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Yang, B.F.; Song, J.H.; Schulman, H.; Li, L.; Hao, C. Inhibition of CaMKII-mediated c-FLIP expression sensitizes malignant melanoma cells to TRAIL-induced apoptosis. Exp. Cell Res. 2005, 304, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, T.; Meng, Z.P.; Gan, Y.C.; Xu, X.H.; Lou, G.Y.; Li, H.Z.; Gan, X.X.; Zhou, H.; Tang, J.F.; et al. CaMKII gamma, a critical regulator of CML stem/progenitor cells, is a target of the natural product berbamine. Blood 2012, 120, 4829–4839. [Google Scholar] [CrossRef] [PubMed]
- House, S.J.; Ginnan, R.G.; Armstrong, S.E.; Singer, H.A. Calcium/calmodulin-dependent protein kinase II-delta isoform regulation of vascular smooth muscle cell proliferation. Am. J. Physiol. Cell Physiol. 2007, 292, C2276–C2287. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.A.; Xie, L.; Li, H.; Li, W.; He, J.B.; Sanders, P.N.; Carter, A.B.; Backs, J.; Anderson, M.E.; Grumbach, I.M. The multifunctional Ca2+/calmodulin-dependent kinase II regulates vascular smooth muscle migration through matrix metalloproteinase 9. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1953–H1964. [Google Scholar] [CrossRef] [PubMed]
- Mercure, M.Z.; Ginnan, R.; Singer, H.A. CaM kinase II delta2-dependent regulation of vascular smooth muscle cell polarization and migration. Am. J. Physiol. Cell Physiol. 2008, 294, C1465–C1475. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Kahn, A.M. Insulin-inhibited and stimulated cultured vascular smooth muscle cell migration are related to divergent effects on protein phosphatase-2A and autonomous calcium/calmodulin-dependent protein kinase II. Atherosclerosis 2008, 196, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Daft, P.G.; Yang, Y.; Napierala, D.; Zayzafoon, M. The Growth and Aggressive Behavior of Human Osteosarcoma Is Regulated by a CaMKII-Controlled Autocrine VEGF Signaling Mechanism. PLoS ONE 2015, 10, e0121568. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Meng, Z.; Jin, L.; Xiao, Z.; Wang, X.; Tsark, W.M.; Ding, L.; Gu, Y.; Zhang, J.; Kim, B.; et al. CAMK2gamma in intestinal epithelial cells modulates colitis-associated colorectal carcinogenesis via enhancing STAT3 activation. Oncogene 2017, 36, 4060–4071. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Ma, X.; Du, J.; Wang, X.; He, M.; Gu, Y.; Zhang, J.; Han, W.; Fang, Z.; Gan, X.; et al. CAMK2gamma antagonizes mTORC1 activation during hepatocarcinogenesis. Oncogene 2017, 36, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Takai, N.; Nishida, M.; Miyazaki, T.; Nasu, K.; Miyakawa, I. CaMKIV expression is associated with clinical stage and PCNA-labeling index in endometrial carcinoma. Int. J. Mol. Med. 2003, 11, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Ueda, T.; Nishida, M.; Nasu, K.; Miyakawa, I. The relationship between oncogene expression and clinical outcome in endometrial carcinoma. Curr. Cancer Drug Targets 2004, 4, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Inuzuka, H.; Ishikawa, Y.; Ikeda, M.; Saji, I.; Kobayashi, R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 2002, 277, 15813–15818. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Gilot, D.; Langouet, S.; Fardel, O. Activation of the aryl hydrocarbon receptor by the calcium/calmodulin-dependent protein kinase kinase inhibitor 7-oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid (STO-609). Drug Metab. Dispos. 2008, 36, 2556–2563. [Google Scholar] [CrossRef] [PubMed]
- York, B.; Li, F.; Lin, F.; Marcelo, K.L.; Mao, J.; Dean, A.; Gonzales, N.; Gooden, D.; Maity, S.; Coarfa, C.; et al. Pharmacological inhibition of CaMKK2 with the selective antagonist STO-609 regresses NAFLD. Sci. Rep. 2017, 7, 11793. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Li, T.; Ma, X.; Wang, X.; Van Ness, C.; Gan, Y.; Zhou, H.; Tang, J.; Lou, G.; Wang, Y.; et al. Berbamine inhibits the growth of liver cancer cells and cancer-initiating cells by targeting Ca(2)(+)/calmodulin-dependent protein kinase II. Mol. Cancer Ther. 2013, 12, 2067–2077. [Google Scholar] [CrossRef]
- Ishida, A.; Kameshita, I.; Okuno, S.; Kitani, T.; Fujisawa, H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem. Biophys. Res. Commun. 1995, 212, 806–812. [Google Scholar] [CrossRef]
- Tokumitsu, H.; Chijiwa, T.; Hagiwara, M.; Mizutani, A.; Terasawa, M.; Hidaka, H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1990, 265, 4315–4320. [Google Scholar] [PubMed]
- Hidaka, H.; Ishikawa, T. Molecular pharmacology of calmodulin pathways in the cell functions. Cell Calcium 1992, 13, 465–472. [Google Scholar] [CrossRef]
- Kahl, C.R.; Means, A.R. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev. 2003, 24, 719–736. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, J.; Chartier, D.; Leblanc, N. Inhibitors of calmodulin-dependent protein kinase are nonspecific blockers of voltage-dependent K+ channels in vascular myocytes. J. Pharmacol. Exp. Ther. 1999, 290, 1165–1174. [Google Scholar] [PubMed]
- Rezazadeh, S.; Claydon, T.W.; Fedida, D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J. Pharmacol. Exp. Ther. 2006, 317, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Blair, L.A.; Marshall, J. CaMKII-independent effects of KN93 and its inactive analog KN92: Reversible inhibition of L-type calcium channels. Biochem. Biophys. Res. Commun. 2006, 345, 1606–1610. [Google Scholar] [CrossRef]
- Rokhlin, O.W.; Taghiyev, A.F.; Bayer, K.U.; Bumcrot, D.; Koteliansk, V.E.; Glover, R.A.; Cohen, M.B. Calcium/calmodulin-dependent kinase II plays an important role in prostate cancer cell survival. Cancer Biol. Ther. 2007, 6, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Rokhlin, O.W.; Guseva, N.V.; Taghiyev, A.F.; Glover, R.A.; Cohen, M.B. KN-93 inhibits androgen receptor activity and induces cell death irrespective of p53 and Akt status in prostate cancer. Cancer Biol. Ther. 2010, 9, 224–235. [Google Scholar] [CrossRef]
- Yang, B.F.; Xiao, C.; Li, H.; Yang, S.J. Resistance to Fas-mediated apoptosis in malignant tumours is rescued by KN-93 and cisplatin via downregulation of c-FLIP expression and phosphorylation. Clin. Exp. Pharmacol. Physiol. 2007, 34, 1245–1251. [Google Scholar] [CrossRef]
- Si, J.T.; Mueller, L.; Collins, S.J. CaMKII regulates retinoic acid receptor transcriptional activity and the differentiation of myeloid leukemia cells. J. Clin. Investig. 2007, 117, 1412–1421. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H. CaMKII Inhibitor KN-62 Blunts Tumor Response to Hypoxia by Inhibiting HIF-1alpha in Hepatoma Cells. Korean J. Physiol. Pharmacol. 2010, 14, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Peyrollier, K.; Bourguignon, L.Y. The influence of hyaluronan-CD44 interaction on topoisomerase II activity and etoposide cytotoxicity in head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 2007, 133, 281–288. [Google Scholar] [CrossRef]
- Aoyama, M.; Grabowski, D.R.; Holmes, K.A.; Rybicki, L.A.; Bukowski, R.M.; Ganapathi, M.K.; Ganapathi, R. Cell cycle phase specificity in the potentiation of etoposide-induced DNA damage and apoptosis by KN-62, an inhibitor of calcium-calmodulin-dependent enzymes. Biochem. Pharmacol. 2001, 61, 49–54. [Google Scholar] [CrossRef]
- Obata, N.H.; Okazaki, K.; Maeda, O.; Kikkawa, F.; Tomoda, Y.; Hidaka, H. Effect of KN-62, Ca2+/calmodulin-dependent protein kinase II inhibitor, on adriamycin resistance of human ovarian cancer cells. Biochem. Biophys. Res. Commun. 1995, 215, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Ishikawa, T.; Inui, M.; Tada, M.; Goshima, K.; Okamoto, T.; Hidaka, H. KN-62, a specific Ca++/calmodulin-dependent protein kinase inhibitor, reversibly depresses the rate of beating of cultured fetal mouse cardiac myocytes. J. Pharmacol. Exp. Ther. 1994, 270, 1319–1324. [Google Scholar] [PubMed]
- Anderson, M.E.; Braun, A.P.; Wu, Y.; Lu, T.; Wu, Y.; Schulman, H.; Sung, R.J. KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J. Pharmacol. Exp Ther. 1998, 287, 996–1006. [Google Scholar]
- Ito, I.; Hidaka, H.; Sugiyama, H. Effects of KN-62, a specific inhibitor of calcium/calmodulin-dependent protein kinase II, on long-term potentiation in the rat hippocampus. Neurosci. Lett. 1991, 121, 119–121. [Google Scholar] [CrossRef]
- Hanson, P.I.; Kapiloff, M.S.; Lou, L.L.; Rosenfeld, M.G.; Schulman, H. Expression of a multifunctional Ca2+/calmodulin-dependent protein kinase and mutational analysis of its autoregulation. Neuron 1989, 3, 59–70. [Google Scholar] [CrossRef]
- Braun, A.P.; Schulman, H. A non-selective cation current activated via the multifunctional Ca(2+)-calmodulin-dependent protein kinase in human epithelial cells. J. Physiol. 1995, 488 Pt 1, 37–55. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, Z.; Chen, B.; Koval, O.M.; Singh, M.V.; Guan, X.; Hund, T.J.; Kutschke, W.; Sarma, S.; Grumbach, I.M.; et al. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc. Natl. Acad. Sci. USA 2009, 106, 5972–5977. [Google Scholar] [CrossRef] [Green Version]
- Backs, J.; Backs, T.; Neef, S.; Kreusser, M.M.; Lehmann, L.H.; Patrick, D.M.; Grueter, C.E.; Qi, X.; Richardson, J.A.; Hill, J.A.; et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA 2009, 106, 2342–2347. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhao, D.; Li, Y.L.; Sun, Y.; Lei, X.H.; Zhang, J.N.; Wu, M.M.; Li, R.Y.; Zhao, Z.F.; Zhang, Z.R.; et al. Regulation of ASIC1 by Ca2+/calmodulin-dependent protein kinase II in human glioblastoma multiforme. Oncol. Rep. 2013, 30, 2852–2858. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Holt, M.; Philipova, R.; Moss, S.; Schulman, H.; Hidaka, H.; Whitaker, M. Calcium/calmodulin-dependent phosphorylation and activation of human Cdc25-C at the G2/M phase transition in HeLa cells. J. Biol. Chem. 1999, 274, 7958–7968. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.H.; Mukherji, S.; Soderling, T.R. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc. Natl. Acad. Sci. USA 1998, 95, 10890–10895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, B.H.; Mukherji, S.; Soderling, T.R. Calcium/calmodulin-dependent protein kinase II inhibitor protein: Localization of isoforms in rat brain. Neuroscience 2001, 102, 767–777. [Google Scholar] [CrossRef]
- Zhang, J.; Li, N.; Yu, J.; Zhang, W.; Cao, X. Molecular cloning and characterization of a novel calcium/calmodulin-dependent protein kinase II inhibitor from human dendritic cells. Biochem. Biophys. Res. Commun. 2001, 285, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, N.; Liu, X.; Zheng, Y.; Cao, X. A novel endogenous human CaMKII inhibitory protein suppresses tumor growth by inducing cell cycle arrest via p27 stabilization. J. Biol. Chem. 2008, 283, 11565–11574. [Google Scholar] [CrossRef]
- Ma, S.; Yang, Y.; Wang, C.; Hui, N.; Gu, L.; Zhong, H.; Cai, Z.; Wang, Q.; Zhang, Q.; Li, N.; et al. Endogenous human CaMKII inhibitory protein suppresses tumor growth by inducing cell cycle arrest and apoptosis through down-regulation of the phosphatidylinositide 3-kinase/Akt/HDM2 pathway. J. Biol. Chem. 2009, 284, 24773–24782. [Google Scholar] [CrossRef] [PubMed]
- Heinze, K.; Kritsch, D.; Mosig, A.S.; Durst, M.; Hafner, N.; Runnebaum, I.B. Functional Analyses of RUNX3 and CaMKIINalpha in Ovarian Cancer Cell Lines Reveal Tumor-Suppressive Functions for CaMKIINalpha and Dichotomous Roles for RUNX3 Transcript Variants. Int. J. Mol. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Coultrap, S.J.; Bayer, K.U. Improving a natural CaMKII inhibitor by random and rational design. PLoS ONE 2011, 6, e25245. [Google Scholar] [CrossRef]
- Gomez-Monterrey, I.; Sala, M.; Rusciano, M.R.; Monaco, S.; Maione, A.S.; Iaccarino, G.; Tortorella, P.; D’Ursi, A.M.; Scrima, M.; Carotenuto, A.; et al. Characterization of a selective CaMKII peptide inhibitor. Eur. J. Med. Chem. 2013, 62, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Xie, J.; Perkins, A.; Ma, Y.; Yang, F.; Wu, J.; Wang, Y.; Xu, R.Z.; Huang, W.; Horne, D.A.; et al. Novel synthetic derivatives of the natural product berbamine inhibit Jak2/Stat3 signaling and induce apoptosis of human melanoma cells. Mol. Oncol. 2012, 6, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Tan, Y.; Wu, G.; Liu, L.; Wang, Y.; Luo, Y.; Shi, J.; Huang, H. Berbamine overcomes imatinib-induced neutropenia and permits cytogenetic responses in Chinese patients with chronic-phase chronic myeloid leukemia. Int. J. Hematol. 2011, 94, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, Q.; Zhang, Y.; Liu, K.; Yu, P.; Liu, K.; Luan, J.; Duan, H.; Lu, Z.; Wang, F.; et al. Suppression of growth, migration and invasion of highly-metastatic human breast cancer cells by berbamine and its molecular mechanisms of action. Mol. Cancer 2009, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Dong, Q.; Yu, Y.; Zhao, X.; Gan, X.; Wu, D.; Lu, Q.; Xu, X.; Yu, X.F. Berbamine: A novel inhibitor of bcr/abl fusion gene with potent anti-leukemia activity. Leuk. Res. 2006, 30, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.L.; Liang, Y.; Xu, L.; Zhao, X.Y. The antiproliferation effect of berbamine on k562 resistant cells by inhibiting NF-kappaB pathway. Anat. Rec. 2009, 292, 945–950. [Google Scholar] [CrossRef]
- Hou, Z.B.; Lu, K.J.; Wu, X.L.; Chen, C.; Huang, X.E.; Yin, H.T. In vitro and in vivo antitumor evaluation of berbamine for lung cancer treatment. Asian Pac. J. Cancer Prev. 2014, 15, 1767–1769. [Google Scholar] [CrossRef]
- Wang, G.Y.; Lv, Q.H.; Dong, Q.; Xu, R.Z.; Dong, Q.H. Berbamine induces Fas-mediated apoptosis in human hepatocellular carcinoma HepG2 cells and inhibits its tumor growth in nude mice. J. Asian Nat. Prod. Res. 2009, 11, 219–228. [Google Scholar] [CrossRef]
- Kirkwood, N.K.; O’Reilly, M.; Derudas, M.; Kenyon, E.J.; Huckvale, R.; van Netten, S.M.; Ward, S.E.; Richardson, G.P.; Kros, C.J. d-Tubocurarine and Berbamine: Alkaloids That Are Permeant Blockers of the Hair Cell’s Mechano-Electrical Transducer Channel and Protect from Aminoglycoside Toxicity. Front. Cell. Neurosci. 2017, 11, 262. [Google Scholar] [CrossRef]
- Liang, Y.; Xu, R.Z.; Zhang, L.; Zhao, X.Y. Berbamine, a novel nuclear factor kappaB inhibitor, inhibits growth and induces apoptosis in human myeloma cells. Acta Pharmacol. Sin. 2009, 30, 1659–1665. [Google Scholar] [CrossRef]
- Parhi, P.; Suklabaidya, S.; Kumar Sahoo, S. Enhanced anti-metastatic and anti-tumorigenic efficacy of Berbamine loaded lipid nanoparticles in vivo. Sci. Rep. 2017, 7, 5806. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.S.; Sebag, S.C.; Paschke, J.D.; Wongrakpanich, A.; Ebeid, K.; Anderson, M.E.; Grumbach, I.M.; Salem, A.K. Cationic CaMKII Inhibiting Nanoparticles Prevent Allergic Asthma. Mol. Pharm. 2017, 14, 2166–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wongrakpanich, A.; Morris, A.S.; Geary, S.M.; Joiner, M.A.; Salem, A.K. Surface-modified particles loaded with CaMKII inhibitor protect cardiac cells against mitochondrial injury. Int. J. Pharm. 2017, 520, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CaMK Family Member | Cancer | Sample Type | Expression | Reference |
|---|---|---|---|---|
| CaMKKβ | Prostate | Prostate cancer TMA (n = 84); NHT TMA with hormone naïve, NHT <3 months, 3–6 months, or >6 months, or castrate-resistant (n = 107) | Increased protein expression in prostate cancer compared to PIN and BPH and in castrate-resistant cancer. Reduced expression following NHT. | [91] |
| Prostate cancer progression (n = 5) | Increased protein expression in prostate cancer compared to normal prostate and with increasing Gleason score | [89] | ||
| Normal prostate and prostate cancer TMA (n = 80), cancer, adjacent normal and metastases TMA (n = 95) | Increased protein expression in primary prostate cancer and bone metastasis compared to normal prostate | [90] | ||
| Gastric | Gastric adenocarcinoma and normal oesophagus TMA (n = 98) | Increased protein expression in gastric tumours compared to normal oesophagus | [85] | |
| Liver | Hepatocellular carcinoma transcriptome profile microarray (n = 247); matched normal and tumour (n = 22). | Increased expression in liver cancer and CAMKK2high correlates with poor disease-free survival. CaMKKβ protein upregulated in tumour compared to adjacent normal tissue. | [86] | |
| Glioma | Human glioma and normal brain tissue (n = 147 for expression and n = 101 for methylation); Whole genome mRNA expression microarray (n = 305 diffuse glioma samples, n = 151 methylation microarray, n = 275 GBM) | CAMKK2 mRNA and protein is more highly expressed in high-grade gliomas compared to low-grade. Increased expression and CAMKK2high correlates with poor overall survival. CAMKK2 is differentially methylated between low and high grade glioma. | [87] | |
| Ovarian | High grade serous papillary ovarian cystadenocarcinoma and high-grade ovarian carcinoma with mucinous features (n = 4) | Increased protein expression in high grade serous papillary cystadenocarcinoma and high-grade ovarian cancer with mucinous features compared to non-malignant stromal tissue. | [88] | |
| CaMKI | AML | TCGA AML database (n = 186) | CAMK1Dhigh correlates with poor overall survival | [93] |
| Endometrial cancer | Endometrial carcinoma and normal endometria (n = 31 and n = 20) | Protein expression is associated with PCNA-labeling, stage, histological grade, the presence of invasion and outcome | [94] | |
| Breast cancer | Primary breast ductal carcinoma (n = 35) | PNCK mRNA is more highly expressed in a subset (8/23) of human breast tumours compared to benign breast tissue | [95] | |
| ccRCC | ccRCC and adjacent normal tissue (n = 92) and primary ccRCC tissue (n-248) | PNCK mRNA and protein expression higher in tumour compared to normal. Patients with PNCKhigh have shorter overall survival | [96] | |
| CaMKII | CML | Peripheral blood (n = 15 at diagnosis; n = 30 in chronic phase with remission; n = 26 in chronic phase treatment-resistant; n = 30 in advanced phase; n = 20 healthy) | CaMKIIγ upregulated at diagnosis and in treatment resistance | [97] |
| AML | Peripheral blood samples (n = 16) | Total and phosphorylation of CaMKIIγ at T287 increased in AML | [98] | |
| Endometrial cancer | Endometrial carcinoma and normal endometria (n = 31 and n = 20) | Protein expression is associated with PCNA-labeling, stage, histological grade, the presence of invasion and outcome | [94] | |
| Colon cancer | Paracancerous tissue, well-differentiated and poorly differentiated colon cancer (n = 5, n = 6, n = 6) | CaMKII protein expression increased in colon cancer compared to paracancerous tissue, and increased with poor differentiation | [99] | |
| Breast cancer | GOBO Breast Cancer Database (n = 1881); Normal, primary and metastatic breast cancer TMA (n = 40, n = 70, and n = 10) | CAMK2high associated with worse overall and distant metastasis free survival. Total CaMKII protein and T286/7 phosphorylation is increased in primary breast cancer and metastases | [100] | |
| Osteosarcoma | Chondroblastic, osteoblastic and fibroblastic subtypes (n = 114) | Phosphorylation of αCaMKII at T286 is increased in osteosarcoma compared to normal osteoblasts and mesenchymal stromal cells | [101] | |
| Primary osteosarcoma tumours (n = 4) | Phosphorylation of αCaMKII at T286 is increased in osteosarcoma | [102] | ||
| Lung cancer | Oncomine databases (n = 187, n = 226, n = 130) | CAMK2Ghigh associated with worse overall survival | [103] | |
| GWAS in NSCLC patients (n = 354) | Rs10023113 in CAMK2D associated with survival | [104] | ||
| Gastric cancer | Non-metastatic and metastatic gastric cancer tissues (n = 10, and n = 10) | Phosphorylation at T286 is increased in metastatic compared to non-metastatic tissue | [105] | |
| CaMKIV | AML | TCGA AML database (n = 186) | CAMK4high correlates with poor overall survival | [93] |
| HCC | Normal liver, chronic hepatitis, cirrhosis, and HCC (n = 4, n = 6, n = 4, n = 12) | CaMKIV protein expression and activation increased in HCC compared to normal liver and cirrhosis | [106] |
| Target | Cancer | Cell Line(s) | Method of Manipulation | Effect | Reference |
|---|---|---|---|---|---|
| CaMKK | Prostate | LNCaP | Pharmacological inhibition (STO-609) | Decreased proliferation | [89] |
| LNCaP, VCqP, C4-2B, 22Rv1 | siRNA and pharmacological inhibition (STO-609) | Decreased proliferation | [91] | ||
| LNCaP | siRNA and pharmacological inhibition (STO-609) | Decreased migration and invasion | [92] | ||
| LNCaP | CaMKKβ overexpression | Increased migration | [92] | ||
| LNCaP | CaMKKβ overexpression | Decreased proliferation | [90] | ||
| DU145 | CaMKKβ siRNA | Decreased proliferation | [107] | ||
| Gastric | AGS, KATO-III, SNU-16, N87 | CaMKKβ siRNA | Decreased proliferation | [85] | |
| SNU-1, N87 | CaMKKβ siRNA and pharmacological inhibition (STO-609) | Decreased proliferation and induced apoptosis | [108] | ||
| HCC | PHM1, SK-Hep1, HepG2 | CaMKKβ siRNA and pharmacological inhibition (STO-609) | Decreased proliferation | [86] | |
| Glioma | U-87MG | CaMKKβ siRNA | Decreased proliferation, migration and invasion | [87] | |
| Ovarian | SKOV-3, OVCAR-3 | CaMKKβ siRNA and pharmacological inhibition (STO-609) | Decreased proliferation and induced apoptosis | [88] | |
| Breast cancer | MCF-7 | CaMKKα and CaMKKβ siRNA | Arrested cells in G1 | [110] | |
| Medulloblastoma | DOAY | Expression of dominant negative CaMKK mutant | Decreased migration | [109] | |
| CaMKI | AML | MV-4-11, Kasumi-1 | shRNA and CaMKI overexpression | Downregulation decreased proliferation; Overexpression of kinase dead mutant decreased colony formation | [93] |
| Breast cancer | MCF-7 | siRNA | Arrested cells in G1 | [110] | |
| Medulloblastoma | DOAY | Expression of dominant negative CaMKI mutant | Decreased migration | [109] | |
| CaMKII | Osteosarcoma | MG-63, 143B, HOS | CaMKIIα shRNA and overexpression | Knockdown decreased proliferation, migration and invasion. Overexpression increased proliferation, migration, invasion | [101] |
| MG-63, 154B | Wild-type and K42M kinase dead CaMKIIα overexpression | K42M kinase dead overexpression reduced growth | [102] | ||
| AML | KG1, KCL22, THP-1, Kasumi-1 | Overexpression of kinase dead truncated CaMKIIγ, CaMKIIγ shRNA, pharmacological inhibition (KN-62, KN-93, KN-92) | Kinase dead overexpression, shRNA and pharmacological inhibition decreased colony formation and proliferation. | [98] | |
| Lung cancer | SCC-9, NCI-H345, NCI-H128, NCI-H146, NCI-H69 | Pharmacological inhibition (KN-62) | Slowed progression through S phase and decreased proliferation | [111] | |
| Medullary thyroid cancer | TT, MZ-CRC1 | Pharmacological inhibition (antCaNtide) | Decreased cell proliferation | [112] | |
| Colon cancer | HCT116 | Pharmacological inhibition (KN-92, KN-93) | Decreased proliferation, migration and invasion | [99] | |
| Gastric cancer | BGC-823 | Pharmacological inhibition (KN-93) and CaMKIIβ shRNA | Decreased cell proliferation and migration, induced apoptosis | [113] | |
| BGC-823 | Pharmacological inhibition (KN-62) and H282R constitutively active CaMKIIα overexpression | Pharmacological inhibition decreased cell proliferation. Overexpression of constitutively active increased cell proliferation, migration and invasion | [105] | ||
| Prostate cancer | C4-2B, LNCaP, PC3, DU145 | Pharmacological inhibition (KN-93) | Decreased proliferation | [114] | |
| 1542-CP3TX | Pharmacological inhibition (AIP) | Decreased cell migration | [115] | ||
| T cell lymphoma | H9 | CaMKIIγ knockout by CRISPR/Cas | Decreased proliferation and colony formation | [116] | |
| Breast cancer | MDA-MB-231, MCF-7 | Overexpression of CaMKIIα, T286D (phosphomimic) and T286V (phosphonull), Pharmacological inhibition (KN-92, KN-93, AIP) | Overexpression of WT and phosphomimic forms increased cell proliferation, migration and invasion. Pharmacological inhibition decreased migration and invasion | [100] | |
| MDA-MB-231 | Overexpression of CaMKIIα, T286D (phosphomimic) and T253D (phosphomimic) | Overexpression of WT and T286D increased proliferation. Overexpression of T253D decreased proliferation | [117] | ||
| Glioma | C6, U-251MG | Pharmacological inhibition (KN-93) | Decreased migration | [118] | |
| D54, H8a | Pharmacological inhibition (AIP) | Decreased migration | [119] | ||
| U-87MG | CaMKIIγ siRNA, pharmacological inhibition (KN-93) | Decreased proliferation, invasion and neurosphere formation | [120] | ||
| CaMKIV | AML | Lin− AML, MV-4-11, Kasumi-1 | CaMKIV and K75M overexpression and CaMKIV shRNA | CaMKIV-K75M overexpression and shRNA knockdown decreased colony formation. shRNA knockdown induced apoptosis and decreased proliferation. | [93] |
| U937 | CaMKIV wild-type and K71M kinase dead mutant overexpression | Cells arrested in G0/G1 following WT, but not K71M, overexpression | [121] | ||
| HCC | PHM1, SK-Hep1 | CaMKIV siRNA | Inhibited colony formation and proliferation | [86] |
| Pharmacological Agent | Cancer | Model | Treatment Schedule | Outcome | Reference |
|---|---|---|---|---|---|
| STO-609 | Prostate | Subcutaneous C4-2B xenograft in full and castrated nude mice | 10 µmol/kg STO-609 or vehicle intraperitoneally three times/week | Reduction in tumour growth, which was enhanced in castrated mice | [91] |
| HCC | DEN-induced hepatic cancer model | 30 µg/kg STO-609 or vehicle intraperitoneally twice/week for 4 weeks | Reduction in tumour growth | [86] | |
| KN-93 | Osteosarcoma | Subcutaneous and intratibial MG-63 xenograft in nude mice | 1 mg/kg saline or KN-93 intraperitoneally every other day for 6 weeks | Reduction in tumour growth | [102] |
| Intratibial 143B xenograft in nude mice | Osmotic pump delivery of 5 µg/µL KN-93, 10 µg/µL CBO-P11 or vehicle set to release 0.25 µL/h for 2 weeks | Reduction in tumour growth alone and in combination with CBO-P11 | [131] | ||
| Berbamine | HCC | Subcutaneous Huh7 or SK-Hep-1 xenograft in NOD-SCID mice | 100 mg/kg berbamine orally twice day for 5 consecutive days, 2 days withdrawal, and then repeated once | Reduction in tumour growth | [139] |
| CML | Subcutaneous K562 and primary CML cells from a patient at blast crisis xenograft in nude mice | 100 mg/kg berbamine, imatinib or vehicle orally three time daily for 10 days | Reduction in tumour growth | [126] | |
| T cell lymphoma | MNU-induced lymphoma model and subcutaneous H9 xenograft in NSG mice | 50 m 100 or 150 mg/kg berbamine, or vehicle, orally administered to mice 2 times a day for 14 days, 14 days withdrawal, cycle repeated; Xenograft study: 150 mg/kg berbamine or vehicle oral twice a day | Reduction in tumour growth in both models | [116] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzozowski, J.S.; Skelding, K.A. The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals 2019, 12, 8. https://doi.org/10.3390/ph12010008
Brzozowski JS, Skelding KA. The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals. 2019; 12(1):8. https://doi.org/10.3390/ph12010008
Chicago/Turabian StyleBrzozowski, Joshua S., and Kathryn A. Skelding. 2019. "The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention" Pharmaceuticals 12, no. 1: 8. https://doi.org/10.3390/ph12010008
APA StyleBrzozowski, J. S., & Skelding, K. A. (2019). The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals, 12(1), 8. https://doi.org/10.3390/ph12010008