De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria

Abstract
:1. Introduction
2. Results
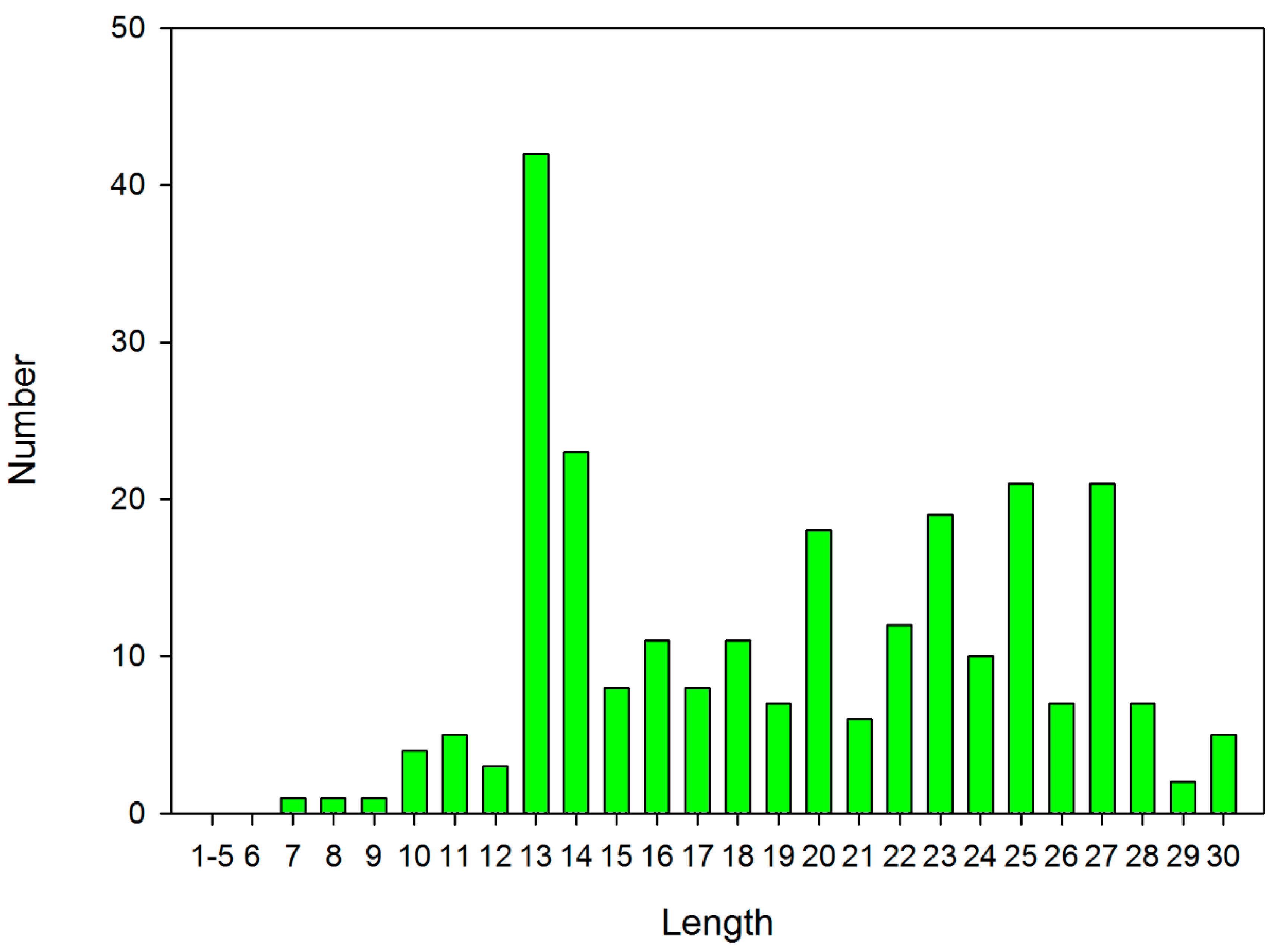
2.1. Selecting the Length for De Novo Design
2.2. SP Models Used for the Design of Peptides
2.3. Peptide Design
2.4. Hemolytic Activity of the Designed Peptides
2.5. Analysis of the Results of In Vitro Tested Peptides
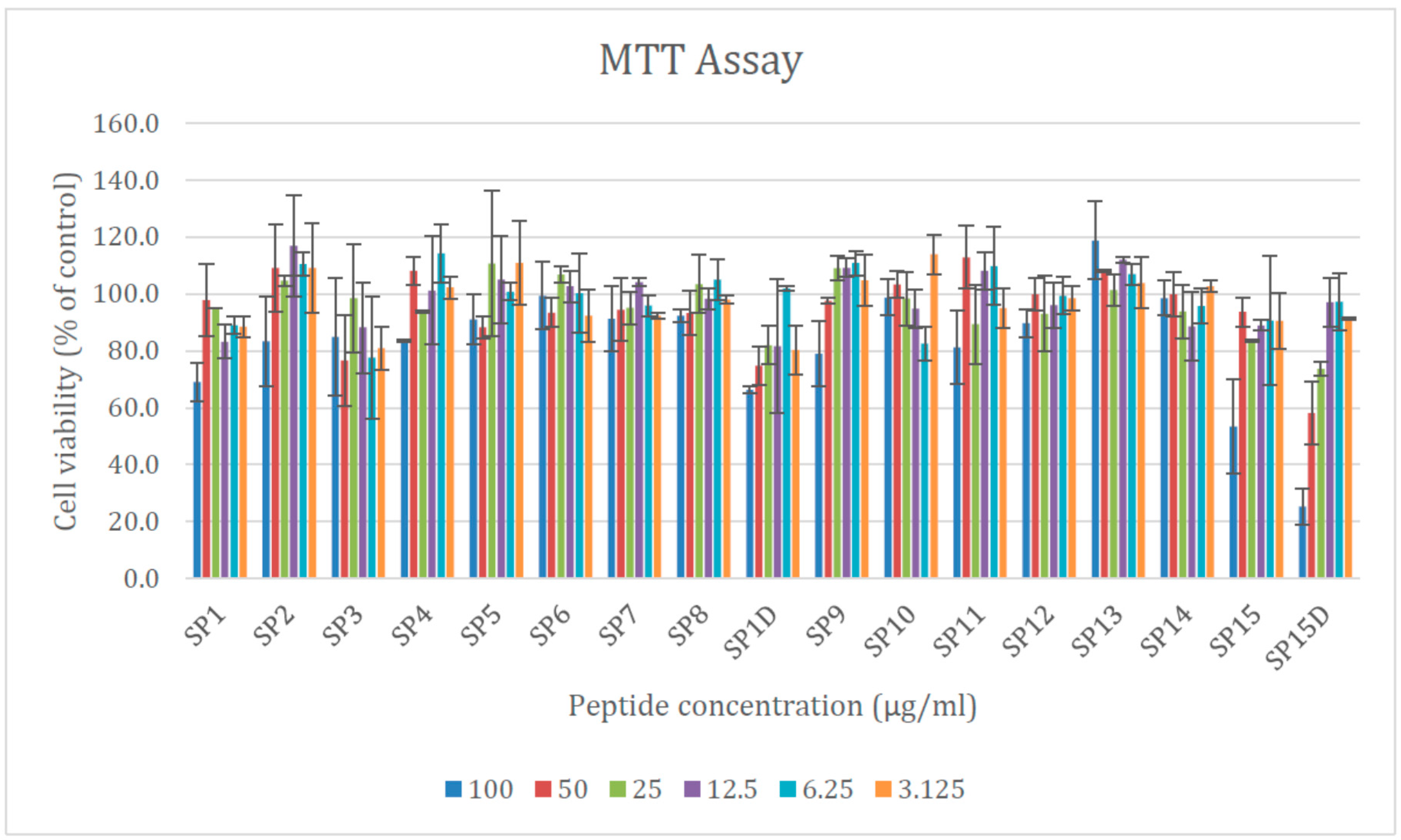
2.6. Cytotoxicity of the Designed AMPs
2.7. The Proteolytic Stability of the Synthesized AMPs
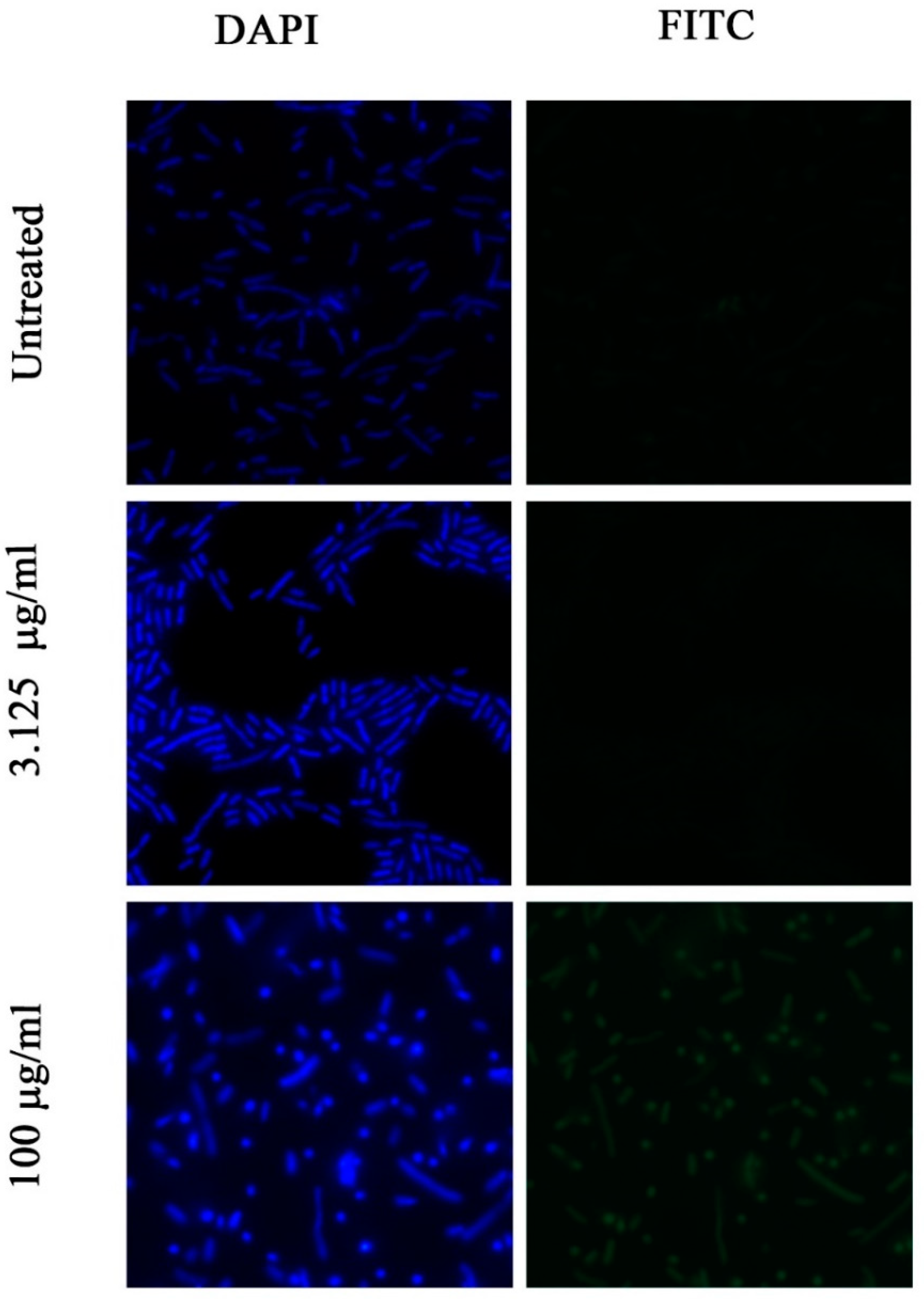
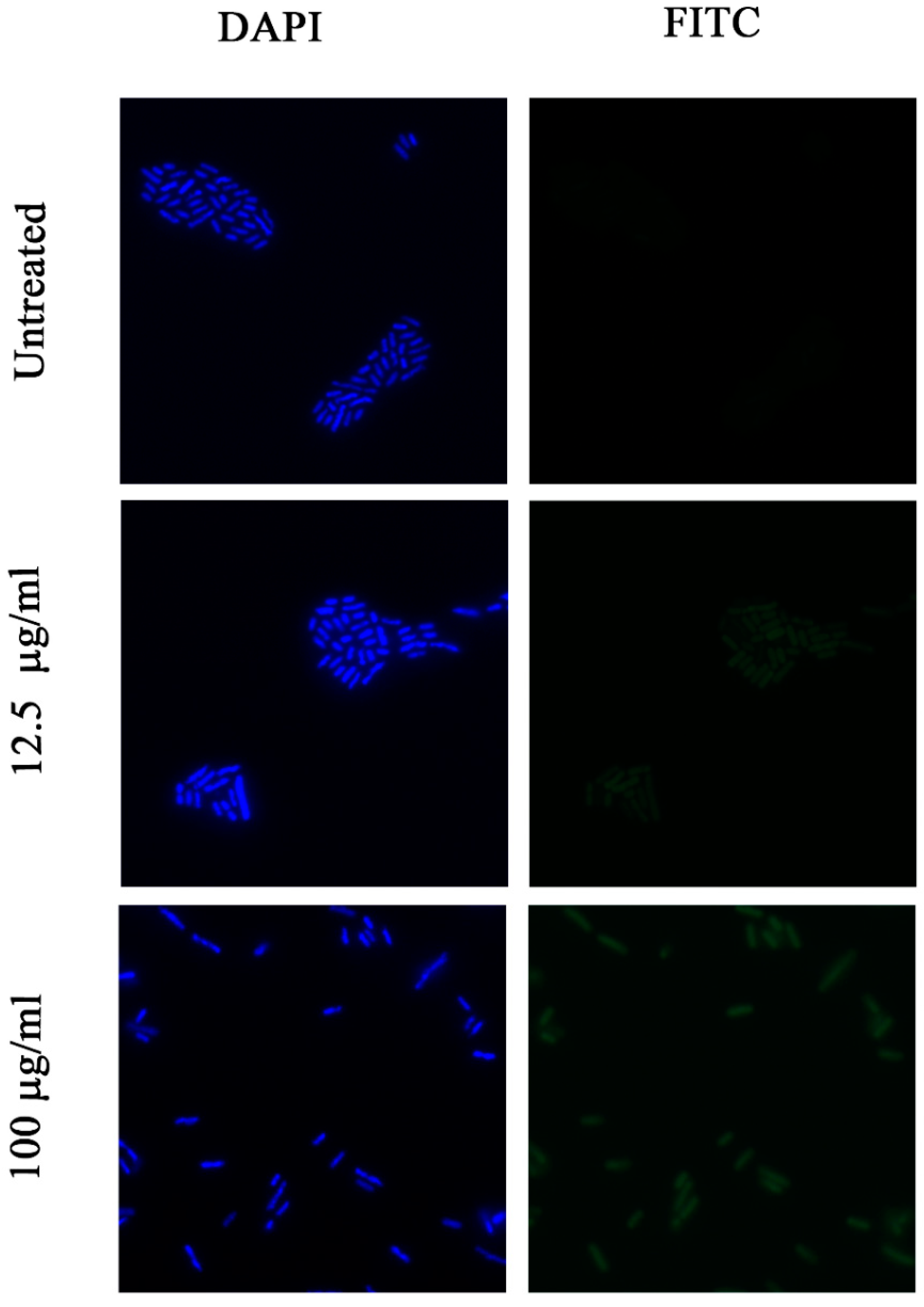
2.8. Investigation of Permeability of the Bacterial Membrane for FITC Dye by Fluorescence Microscopy
3. Discussion
4. Conclusions
5. Methods
5.1. Predictive Models
5.2. Evaluation of the Quality of the Prediction and Definition of the Therapeutic Index
5.3. Peptide Design
5.4. Peptide Synthesis
5.5. Susceptibility Testing against Escherichia coli ATCC 25922
5.6. Susceptibility Testing against other Different Gram-Negative Bacterial Strains
5.7. Hemolytic Activity Assessment
5.8. Cytotoxicity Assay of the Antimicrobial Peptides
5.9. The Proteolytic Stability towards α-Chymotrypsin and Proteinase K Digestion
5.10. Assessment of Membrane Penetrating Properties of Antimicrobial Peptides by Fluorescence Microscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pagès, J.M.; Masi, M.; Barbe, J. Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol. Med. 2005, 11, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An Enhanced Database of Structure and Antimicrobial/Cytotoxic Activity of Natural and Synthetic Peptides. Nucl. Acids Res. 2016, 44, D1104–D1112. [Google Scholar] [CrossRef]
- Greber, K.E.; Dawgul, M. Antimicrobial Peptides Under Clinical Trials. Curr. Top. Med. Chem. 2017, 17, 620–628. [Google Scholar] [CrossRef]
- Li, J.; Koh, J.-J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieco, P.; Luca, V.; Auriemma, L.; Carotenuto, A.; Saviello, M.R.; Campiglia, P.; Barra, D.; Novellino, E.; Mangoni, M.L. Alanine scanning analysis and structure–function relationships of the frog-skin antimicrobial peptide temporin-1Ta. J. Pept. Sci. 2011, 17, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Hänchen, A.; Rausch, S.; Landmann, B.; Toti, L.; Nusser, A.; Süssmuth, R.D. Alanine scan of the peptide antibiotic feglymycin: Assessment of amino acid side chains contributing to antimicrobial activity. ChemBioChem 2013, 14, 625–632. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing Antimicrobial Peptides: Form Follows Function. Nat. Rev. Drug Discov. 2011, 11, 37–51. [Google Scholar] [CrossRef]
- Torrent, M.; Di Tommaso, P.; Pulido, D.; Nogués, M.V.; Notredame, C.; Boix, E.; Andreu, D. AMPA: An Automated Web Server for Prediction of Protein Antimicrobial Regions. Bioinformatics 2012, 28, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Lejon, T.; Hilpert, K.; Fjell, C.D.; Cherkasov, A.; Hancock, R.E.W. Evaluating Different Descriptors for Model Design of Antimicrobial Peptides with Enhanced Activity toward P. aeruginosa. Chem. Biol. Drug. Des. 2007, 70, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Taboureau, O.; Olsen, O.H.; Nielsen, J.D.; Raventos, D.; Mygind, P.H.; Kristensen, H.H. Design of Novispirin Antimicrobial Peptides by Quantitative Structure-Activity Relationship. Chem. Biol. Drug Des. 2006, 68, 48–57. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, C.H.; Hu, D.; Ulmschneider, M.B.; Ulmschneider, J.P. Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 2016, 7, 13535. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, S.; Koh, J.-J.; Zou, H.; Lakshminarayanan, R.; Bai, Y.; Pervushin, K.; Zhou, L.; Verma, C.; Beuerman, R.W. A novel fragment based strategy for membrane active antimicrobials against MRSA. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1023–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, S.; Lakshminarayanan, R.; Bai, Y.; Pervushin, K.; Verma, C.; Beuerman, R.W. Molecular simulations suggest how a branched antimicrobial peptide perturbs a bacterial membrane and enhances permeability. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.S.; Li, J.; Tan, Y.S.N.; Nguyen, M.; Pal, A.; Ouaray, Z.; Yadahalli, S.; Kannan, S. The multifaceted roles of molecular dynamics simulations in drug discovery. Curr. Pharm. Des. 2016, 22, 3585–3600. [Google Scholar] [PubMed]
- Saravanan, R.; Li, X.; Lim, K.; Mohanram, H.; Peng, L.; Mishra, B.; Basu, A.; Lee, J.; Bhattacharjya, S.; Leong, S.S. Design of short membrane selective antimicrobial peptides containing tryptophan and arginine residues for improved activity, salt-resistance, and biocompatibility. Biotechnol. Bioeng. 2014, 111, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Mohanram, H.; Bhattacharjya, S. b-boomerang antimicrobial and antiendotoxic peptides: Lipidation and disulfide bond effects on activity and structure. Pharmaceuticals 2014, 7, 482–501. [Google Scholar] [CrossRef]
- Jeong, J.-H.; Kim, J.-S.; Choi, S.-S.; Kim, Y. NMR structural studies of antimicrobial peptides: LPcin analogs. Biophys. J. 2016, 110, 423–430. [Google Scholar] [CrossRef]
- Jenssen, H.; Fjell, C.D.; Cherkasov, A.; Hancock, R.E. QSAR Modeling and Computer-Aided Design of Antimicrobial Peptides. J. Pept. Sci. 2008, 14, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Jenssen, H.; Hilpert, K.; Cheung, W.A.; Pante, N.; Hancock, R.E.; Cherkasov, A. Identification of Novel Antibacterial Peptides by Chemoinformatics and Machine Learning. J. Med. Chem. 2009, 52, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Hilpert, K.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Mullaly, S.C.; Volkmer, R.; Hancock, R.E. Use of Artificial Intelligence in the Design of Small Peptide Antibiotics Effective against a Broad Spectrum of Highly Antibiotic-Resistant Superbugs. ACS Chem. Biol. 2009, 4, 65–74. [Google Scholar] [CrossRef]
- Torrent, M.; Andreu, D.; Nogues, V.M.; Boix, E. Connecting Peptide Physicochemical and Antimicrobial Properties by a Rational Prediction Model. PLoS ONE 2011, 6, e16968. [Google Scholar] [CrossRef]
- Mooney, C.; Haslam, N.J.; Holton, T.A.; Pollastri, G.; Shields, D.C. Peptidelocator: Prediction of Bioactive Peptides in Protein Sequences. Bioinformatics 2013, 29, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Porto, W.F.; Pires, A.S.; Franco, O.L. Cs-Amppred: An Updated SVM Model for Antimicrobial Activity Prediction in Cysteine-Stabilized Peptides. PLoS ONE 2012, 7, e51444. [Google Scholar] [CrossRef]
- Ng, X.Y.; Rosdi, B.A.; Shahrudin, S. Prediction of Antimicrobial Peptides Based on Sequence Alignment and Support Vector Machine-Pairwise Algorithm Utilizing LZ-Complexity. Biomed. Res. Int. 2015, 2015, 212715. [Google Scholar] [CrossRef]
- Khosravian, M.; Faramarzi, F.K.; Beigi, M.M.; Behbahani, M.; Mohabatkar, H. Predicting Antibacterial Peptides by the Concept of Chou’s Pseudo-Amino Acid Composition and Machine Learning Methods. Protein Pept. Lett. 2013, 20, 180–186. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting Antimicrobial Peptides with Improved Accuracy by Incorporating the Compositional, Physico-Chemical and Structural Features into Chou’s General PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef]
- Lira, F.; Perez, P.S.; Baranauskas, J.A.; Nozawa, S.R. Prediction of Antimicrobial Activity of Synthetic Peptides by a Decision Tree Model. Appl. Environ. Microbiol. 2013, 79, 3156–3159. [Google Scholar] [CrossRef] [Green Version]
- Khamis, A.M.; Essack, M.; Gao, X.; Bajic, V.B. Distinct Profiling of Antimicrobial Peptide Families. Bioinformatics 2015, 31, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. Iamp-2l: A Two-Level Multi-Label Classifier for Identifying Antimicrobial Peptides and Their Functional Types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Maccari, G.; Di Luca, M.; Nifosí, R.; Cardarelli, F.; Signore, G.; Boccardi, C.; Bifone, A. Antimicrobial Peptides Design by Evolutionary Multiobjective Optimization. PLoS Comput. Biol. 2013, 9, e1003212. [Google Scholar] [CrossRef]
- Bhadra, P.; Yan, J.; Li, J.; Fong, S.; Siu, S.W.I. AmPEP: Sequence-based Prediction of Antimicrobial Peptides using Distribution Patterns of Amino Acid Properties and Random Forest. Sci. Rep. 2018, 8, 1697. [Google Scholar] [CrossRef] [PubMed]
- Youmans, M.; Spainhour, C.; Qiu, P. Long Short-Term Memory Recurrent Neural Networks for Antibacterial Peptide Identification. In Proceedings of the 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Kansas City, MO, USA, 13–16 November 2017; pp. 498–502. [Google Scholar]
- Juretic, D.; Vukicevic, D.; Ilic, N.; Antcheva, N.; Tossi, A. Computational Design of Highly Selective Antimicrobial Peptides. J. Chem. Inf. Model. 2009, 49, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Hu, L.; Liu, G.; Jiang, N.; Chen, X.; Xu, J.; Zheng, W.; Li, L.; Tan, M.; Chen, Z.; et al. Prediction of Antimicrobial Peptides Based on Sequence Alignment and Feature Selection Methods. PLoS ONE 2011, 6, e18476. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.N.; Ferre, R.; Feliu, L.; Bardaji, E.; Planas, M.; Castanho, M.A. Prediction of Antibacterial Activity from Physicochemical Properties of Antimicrobial Peptides. PLoS ONE 2011, 6, e28549. [Google Scholar] [CrossRef] [PubMed]
- Freire, J.M.; Dias, A.S.; Flores, L.; Veiga, A.S.; Castanho, M.A. Mining Viral Proteins for Antimicrobial and Cell-Penetrating Drug Delivery Peptides. Bioinformatics 2015, 31, 2252–2256. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.Y.; Lin, T.P.; Shih, L.Y.; Wang, C.K. Analysis and Prediction of the Critical Regions of Antimicrobial Peptides Based on Conditional Random Fields. PLoS ONE 2015, 10, e0119490. [Google Scholar] [CrossRef]
- Toropova, M.A.; Veselinovic, A.M.; Veselinovic, J.B.; Stojanovic, D.B.; Toropov, A.A. QSAR Modeling of the Antimicrobial Activity of Peptides as a Mathematical Function of a Sequence of Amino Acids. Comput. Biol. Chem. 2015, 59, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Toropov, A.A.; Toropova, A.P.; Raska, I., Jr.; Benfenati, E.; Gini, G. QSAR Modeling of Endpoints for Peptides Which Is Based on Representation of the Molecular Structure by a Sequence of Amino Acids. Struct. Chem. 2012, 23, 1891–1904. [Google Scholar] [CrossRef]
- Lata, S.; Sharma, B.K.; Raghava, G.P. Analysis and Prediction of Antibacterial Peptides. BMC Bioinf. 2007, 8, 263. [Google Scholar] [CrossRef] [PubMed]
- Lata, S.; Mishra, N.K.; Raghava, G.P. AntiBP2: Improved Version of Antibacterial Peptide Prediction. BMC Bioinf. 2010, 11, S19. [Google Scholar] [CrossRef]
- Nagarajan, D.; Nagarajan, T.; Roy, N.; Kulkarni, O.; Ravichandran, S.; Mishra, M.; Chakravortty, D.; Chandra, N. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem. 2018, 293, 3492–3509. [Google Scholar] [CrossRef] [Green Version]
- Hincapié, O.; Giraldo, P.; Orduz, S. In silico design of polycationic antimicrobial peptides active against Pseudomonas aeruginosa and Staphylococcus aureus. Antonie Van Leeuwenhoek 2018, 111, 1871. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Managadze, G.; Grigolava, M.; Makhatadze, G.I.; Pirtskhalava, M. Predictive model of linear AMPs active against Gram-negative bacteria. J. Chem. Inf. Model. 2018, 58, 1141–1151. [Google Scholar] [CrossRef]
- Vishnepolsky, B.; Pirtskhalava, M. Comment on: ‘Empirical Comparison of Web-Based Antimicrobial Peptide Prediction Tools’. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Pirtskhalava, M. Prediction of Linear Cationic Antimicrobial Peptides Based on Characteristics Responsible for Their Interaction with the Membranes. J. Chem. Inf. Model. 2014, 54, 1512–1523. [Google Scholar] [CrossRef]
- Nicolau, D.P. Carbapenems: A potent class of antibiotics. Expert Opin. Pharm. 2008, 9, 23–37. [Google Scholar] [CrossRef]
- Holton, T.A.; Pollastri, G.; Shields, D.C.; Mooney, C. CPPpred: Prediction of cell penetrating peptides. Bioinformatics 2013, 29, 3094–3096. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Why and how are peptide–lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta (BBA) Biomembr. 1999, 1462, 1–10. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta (BBA) Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Yang, L.; Weiss, T.M.; Lehrer, R.; Huang, H.W. Crystallization of antimicrobial pores in membranes: Mgainin and ptegrin. Biophys. J. 2000, 79, 2002–2009. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.V.R.; Yedery, R.D.; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L. Antibacterial peptides isolated from insects. J. Pept. Sci. 2000, 6, 497–511. [Google Scholar] [CrossRef]
- Brötz, H.; Bierbaum, G.; Leopold, K.; Reynolds, P.E.; Sahl, H.-G. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 1998, 42, 154–160. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Sugishita, K.-I.; Ishibe, N.; Ueha, M.; Nakata, S.; Miyajima, K.; Epand, R.M. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry 1998, 37, 11856–11863. [Google Scholar] [CrossRef]
- Dagan, A.; Efron, L.; Gaidukov, L.; Mor, A.; Ginsburg, H. In vitro antiplasmodium effects of dermaseptin S4 derivatives. Antimicrob. Agents Chemother. 2002, 46, 1059–1066. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 1996, 35, 11361–11368. [Google Scholar] [CrossRef]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E.W. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef] [PubMed]
- Lázár, V.; Martins, A.; Spohn, R.; Daruka, L.; Grézal, G.; Fekete, G.; Számel, M.; Jangir, P.K.; Kintses, B.; Csörgő, B.; et al. Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat. Microbiol. 2018, 3, 718–731. [Google Scholar] [CrossRef] [Green Version]
- Shagaghi, N.; Bhave, M.; Palombo, E.A.; Clayton, A.H.A. Revealing the sequence of interactions of PuroA peptide with Candida albicans cells by live-cell imaging. Sci. Rep. 2017, 7, 43542. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta (BBA) Biomembr. 2006, 758, 1184–1202. [Google Scholar] [CrossRef]
- Patrzykat, A.; Friedrich, C.L.; Zhang, L.; Mendoza, V.; Hancock, R.E.W. Sublethal concentrations of pleurocidin-derived antimicrobial peptides inhibit macromolecular synthesis in Escherichia coli. Antimicrob. Agents Chemother. 2002, 46, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Ester, M.; Kriegel, H.; Sander, J.; Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD-96), Portland, Oregon, 2–4 August 1996; AAAI Press: Palo Alto, CA, USA, 1996; pp. 226–231. [Google Scholar]
- Moon, C.P.; Fleming, K.G. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc. Natl. Acad. Sci. USA 2011, 108, 10174–10177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senes, A.; Chadi, D.C.; Law, P.B.; Walters, R.F.S.; Nanda, V.; DeGrado, W.F. E(z), a Depth-dependent Potential for Assessing the Energies of Insertion of Amino Acid Side-chains into Membranes: Derivation and Applications to Determining the Orientation of Transmembrane and Interfacial Helices. J. Mol. Biol. 2007, 366, 436–448. [Google Scholar] [CrossRef]
- Uversky, V.; Gillespie, J.; Fink, A. Why are “natively unfolded” proteins unstructured under physiological conditions? Proteins Struct. Funct. Gen. 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Kessel, A.; Ben-Tal, N. Introduction to Proteins: Structure, Function and Motion (Chapman & Hall/CRC Mathematical and Computational Biology), 1st ed.; CRC Press, Taylor & Francis Group: London, UK, 2011. [Google Scholar]
- The UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 14, 24482. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protocol. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean Values ± SD of Attributes | Cluster H1 (MHCIORLS a) | Cluster H2 (MA a) | Cluster H3 (MCIA a) |
|---|---|---|---|
| M ± σ | 1.42 ± 0.37 | 0.33 ± 0.13 | 1.07 ± 0.24 |
| H ± σ | 0.09 ± 0.56 | −0.14 ± 0.73 | −0.71 ± 0.31 |
| C ± σ | 6.9 ± 1.93 | 3.87 ± 3.12 | 2.64 ± 0.79 |
| I ± σ | 13.75 ± 0.58 | 10.73 ± 2.51 | 10.81 ± 0.48 |
| D ± σ | 16.74 ± 4.2 | 23.9 ± 6.39 | 15 ± 4.79 |
| O ± σ | 100.76 ± 26.29 | 82.42 ± 43.36 | 95.96 ± 28.04 |
| R ± σ | −0.32 ± 0.29 | −0.26 ± 0.24 | 0.19 ± 0.22 |
| L ± σ | 0.31 ± 0.08 | 0.34 ± 0.1 | 0.32 ± 0.08 |
| A ± σ | 2.25 ± 10.54 | 2.68 ± 7.27 | 13.05 ± 16.16 |
| S ± σ | 13.84 ± 3.35 | 17.39 ± 6.09 | 16.72 ± 4 |
| TP | TP + FN | FP | TN + FP | Sn | Sp | AC | PPV | ||
|---|---|---|---|---|---|---|---|---|---|
| Training Set | Cluster H1 | 50 | 120 | 13 | 120 | 0.79 | |||
| Cluster H2 | 31 | 120 | 4 | 120 | 0.89 | ||||
| Cluster H3 | 25 | 120 | 8 | 120 | 0.76 | ||||
| All Clusters | 106 | 120 | 25 | 120 | 0.88 | 0.79 | 0.84 | 0.81 | |
| Cluster H1 | 14 | 43 | 2 | 43 | 0.88 | ||||
| Test Set | Cluster H2 | 11 | 43 | 3 | 43 | 0.79 | |||
| Cluster H3 | 7 | 43 | 5 | 43 | 0.58 | ||||
| All Clusters | 32 | 43 | 10 | 43 | 0.74 | 0.77 | 0.76 | 0.75 |
| Name | Sequence | MIC (µg/mL) | STP (Peptide/Protease M Ratio) | LC10 (µg/mL) | TI ** | ||||
|---|---|---|---|---|---|---|---|---|---|
| At NaCl | Without NaCl | Proteinase K | α-chymotrypsin | ||||||
| 1000:1 Ratio | 500:1 Ratio | 1000:1 Ratio | 500:1 Ratio | ||||||
| SP1 | AIKIRKLFKKLLR | 12.5–25 | 3.125–6.25 | D | NT | D | NT | >100 | >16 |
| SP2 | GIKIRKLFKKLLR | 6.25–12.5 | 3.125–6.25 | D | NT | D | NT | >100 | >16 |
| SP3 | GWAKLITKAIKKI | 25–50 | 12.5–25 | PD | PD | D | NT | 50–100 | 4 |
| SP4 | GIKFFLKKLKKHI | 25–50 | 6.25–12.5 | PD | PD | D | NT | >100 | >8 |
| SP5 | IRPAKLRWFKKIK | >100 | 12.5–25 | D | NT | D | NT | >100 | >4 |
| SP6 | RLFIKKLKFITRR | 25–50 | 3.125–6.25 | PD | D | D | NT | >100 | >16 |
| SP7 | NAMRGAKRVWRHI | >100 | 50–100 | PD | PD | D | NT | >100 | >1 |
| SP8 | KFRKFGKQVWVRL | 12.5–25 | 3.125–6.25 | PD | D | D | NT | >100 | >16 |
| SP1D * | aikirklfkkllr | 12.5–25 | 3.125–6.25 | ND | ND | ND | ND | 25–50 | 4–8 |
| SP9 | KVWSRLRKIFSTR | 6.25–12.5 | 3.125–6.25 | D | NT | D | NT | 50–100 | 8–16 |
| SP10 | AKVLKISRRAFRK | >100 | 25–50 | D | NT | D | NT | >100 | >2 |
| SP11 | IRRWRLHWFRRAI | 12.5–25 | 3.125–6.25 | PD | D | D | NT | >100 | >16 |
| SP12 | IRRRIRLIVRRQI | 12.5–25 | 1.56–3.125 | ND | PD | D | NT | >100 | >32 |
| SP13 | HFKIRKRFVKKLV | >100 | 6.25–12.5 | PD | D | D | NT | >100 | >16 |
| SP14 | RWIRWVWRKKLRI | 12.5–25 | 3.125–6.25 | PD | D | D | NT | 50–100 | 8–16 |
| SP15 * | RWIRWVWRKKLR | 3.125–6.25 | 0.78–1.56 | PD | PD | PD | PD | >100 | >64 |
| SP15D * | rwirwvwrkklr | 0.78–1.56 | 0.39–0.78 | ND | ND | ND | ND | >100 | >128 |
| Isolate # | Organism ID | Phenotype | MIC (µg/mL) Meropenem | MIC (µg/mL) SP1 | MIC (µg/mL) SP1 | MIC (µg/mL) SP3 | MIC (µg/mL) SP4 |
|---|---|---|---|---|---|---|---|
| ATCC 27853 | P. aeruginosa | CLSI Control | 1 | 4 | 4 | 8 | 8 |
| J4228 | P. aeruginosa | R: Meropenem | >64 | 8 | 8 | 16 | 16 |
| BB2013-100 | P. aeruginosa | FQR | 1 | 32 | 32 | 32 | 32 |
| Josh 28 | A. baumannii | Susceptible | 32 | 4 | 4 | 4 | 2 |
| Josh 230 | A. baumannii | OXA-48 | 1 | 16 | 16 | 16 | 4 |
| BB2012-181 | E. cloacae | R: Meropenem | 16 | >32 | 32 | 16 | >32 |
| BB2013-32 | E. cloacae | FQR | 0.5 | >32 | >32 | 16 | 16 |
| St. L P63 | E. aerogenes | NDM-1 | 16 | >32 | 32 | 16 | 16 |
| St. L P23 | E. asburiae/cloacae | NDM-1 | 16 | >32 | >32 | 32 | >32 |
| BB2009-209 | K. pneumoniae | KPC-2 | 32 | >32 | >32 | >32 | >32 |
| J3702 | K. pneumoniae | Susceptible | ≤0.125 | >32 | >32 | 16 | 32 |
| Oschner KP-1 | K. pneumoniae | KPC-3 | 16 | >32 | >32 | >32 | >32 |
| BW25113 (7636) | E. coli | WT, Tol parent strain | ≤0.125 | 8 | 8 | 4 | 8 |
| JW55034 (11430) | E. coli | Tol neg | ≤0.125 | 8 | 8 | 4 | 8 |
| BB2013-30 | E. coli | R:carbapenem | 1 | 16 | 16 | 8 | 16 |
| ARLG-1012 | E. coli | NDM | 64 | 8 | 8 | 4 | 8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vishnepolsky, B.; Zaalishvili, G.; Karapetian, M.; Nasrashvili, T.; Kuljanishvili, N.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Grigolava, M.; et al. De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria. Pharmaceuticals 2019, 12, 82. https://doi.org/10.3390/ph12020082
Vishnepolsky B, Zaalishvili G, Karapetian M, Nasrashvili T, Kuljanishvili N, Gabrielian A, Rosenthal A, Hurt DE, Tartakovsky M, Grigolava M, et al. De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria. Pharmaceuticals. 2019; 12(2):82. https://doi.org/10.3390/ph12020082
Chicago/Turabian StyleVishnepolsky, Boris, George Zaalishvili, Margarita Karapetian, Tornike Nasrashvili, Nato Kuljanishvili, Andrei Gabrielian, Alex Rosenthal, Darrell E. Hurt, Michael Tartakovsky, Maya Grigolava, and et al. 2019. "De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria" Pharmaceuticals 12, no. 2: 82. https://doi.org/10.3390/ph12020082
APA StyleVishnepolsky, B., Zaalishvili, G., Karapetian, M., Nasrashvili, T., Kuljanishvili, N., Gabrielian, A., Rosenthal, A., Hurt, D. E., Tartakovsky, M., Grigolava, M., & Pirtskhalava, M. (2019). De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria. Pharmaceuticals, 12(2), 82. https://doi.org/10.3390/ph12020082