Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review


Abstract
:1. Microtubules as a Target for Treating Cancer
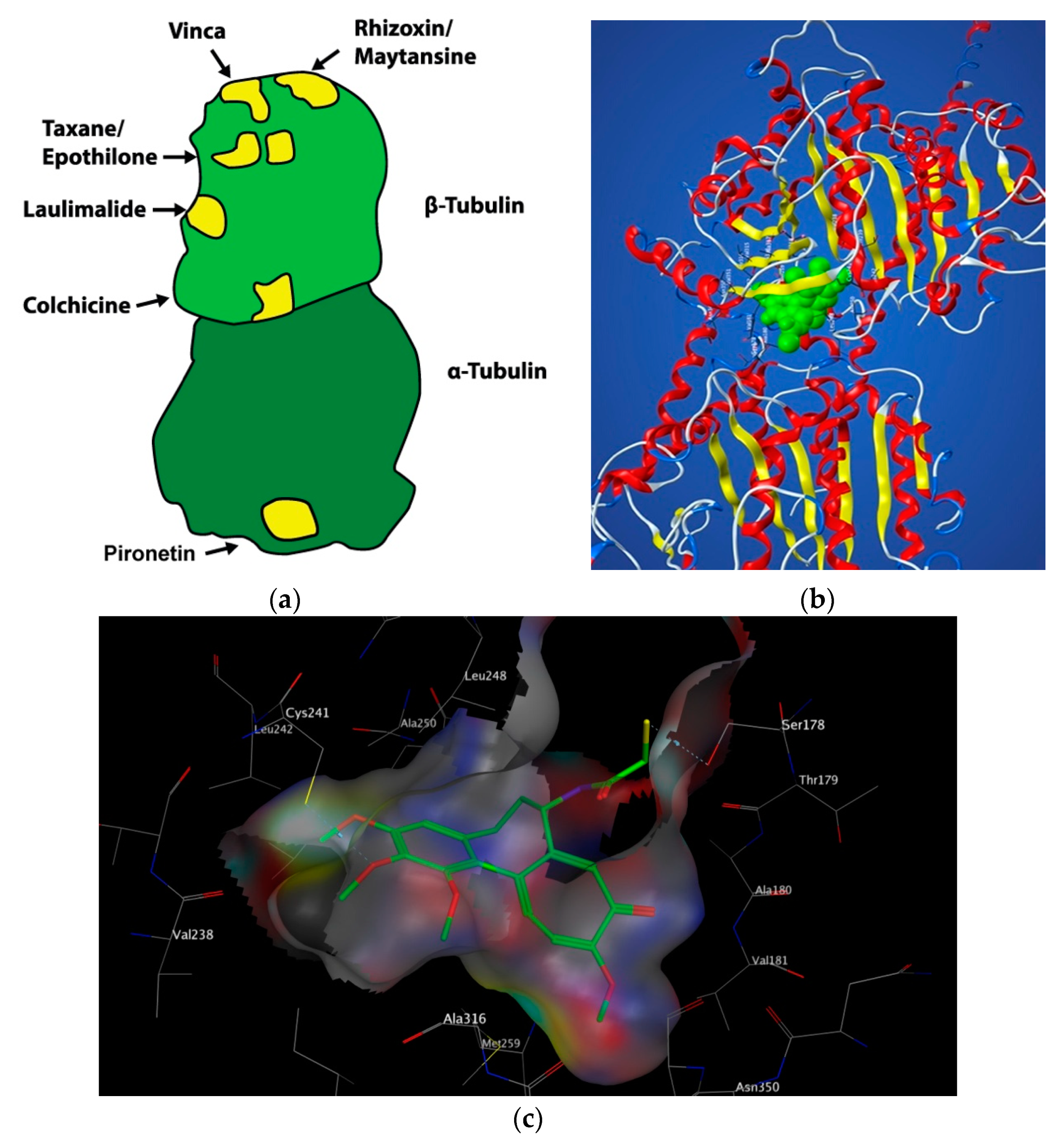
2. Colchicine: Historical Uses, Structure and Mechanism of Action
3. Targeting the Colchicine-Binding Site for Anti-Cancer Therapy
4. Colchicine-Binding Site Inhibitors Known before 2017
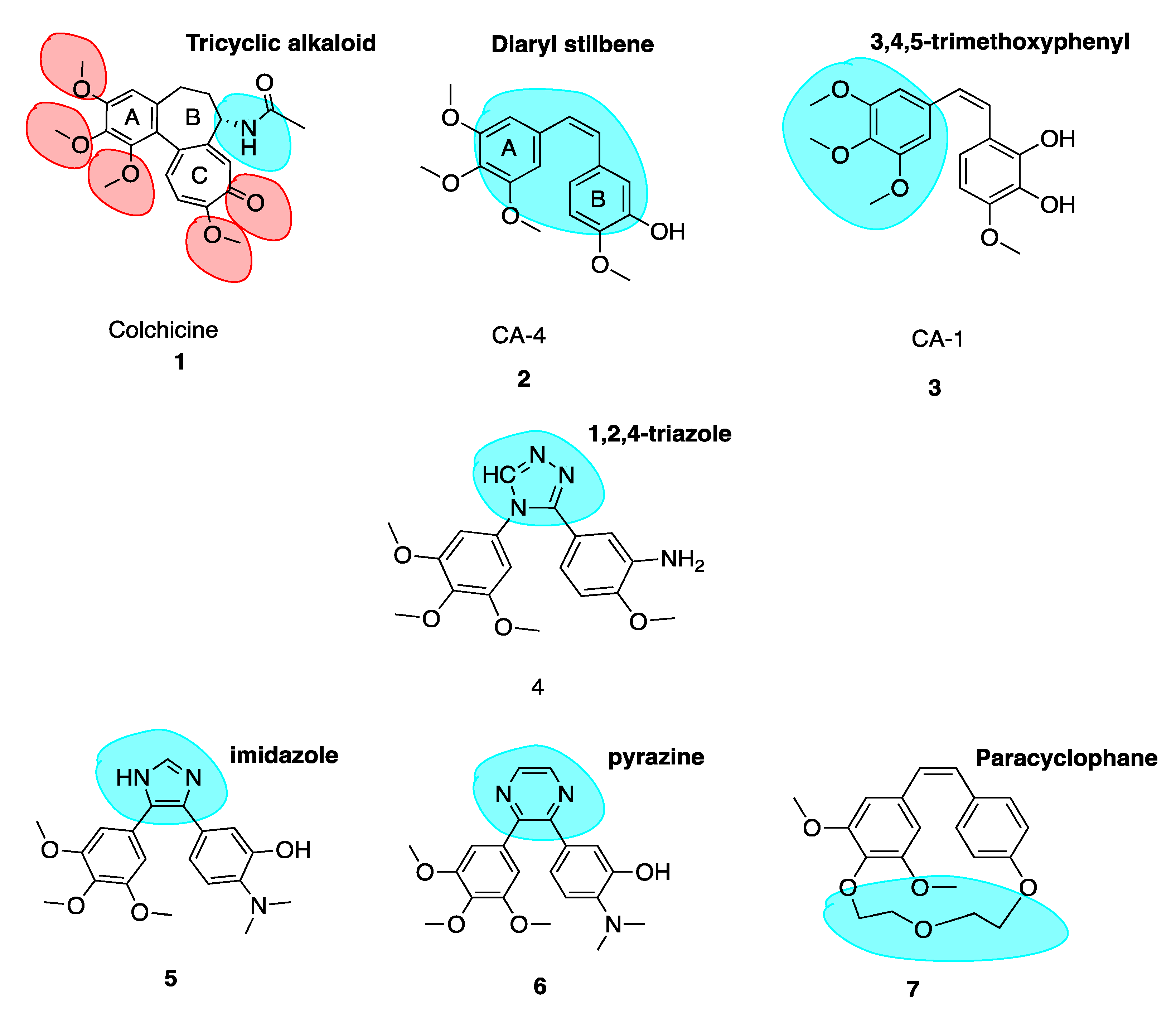
4.1. Combretastatin A-4 and Analogues
4.2. Successful Modifications of Combretastatin A-4
- (1)
- 3,4,5-trimethoxyphenyl-subsituted ring A.
- (2)
- 3-hydroxy-4-methoxyphenyl-substituted ring B.
- (3)
- cis double bond separating the two phenyl rings.
- (1)
- Three-membered ring bridges,
- (2)
- Six-membered ring bridges, and
- (3)
- Immobilized trimethoxyphenyl moieties.
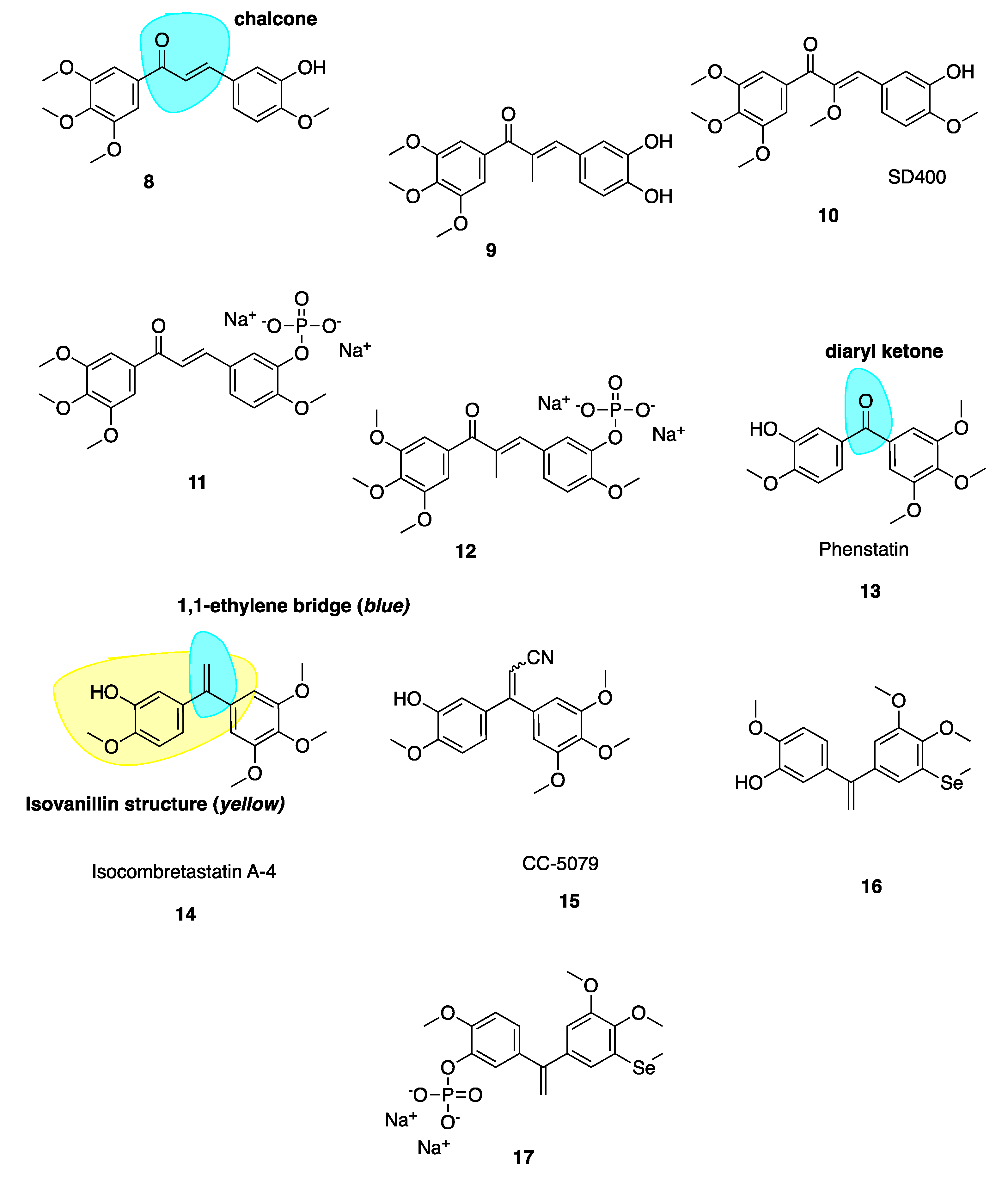
4.2.1. Potent Chalcone Derivatives of CA-4
4.2.2. Phenstatin and Derivatives
4.2.3. IsoCA-4 and Selenium CA-4 Derivatives
4.3. New Combretastatin A-4 Analogues Reported between 2017 and 2019
4.3.1. New Combretastatin A-4 Analogues of 2017
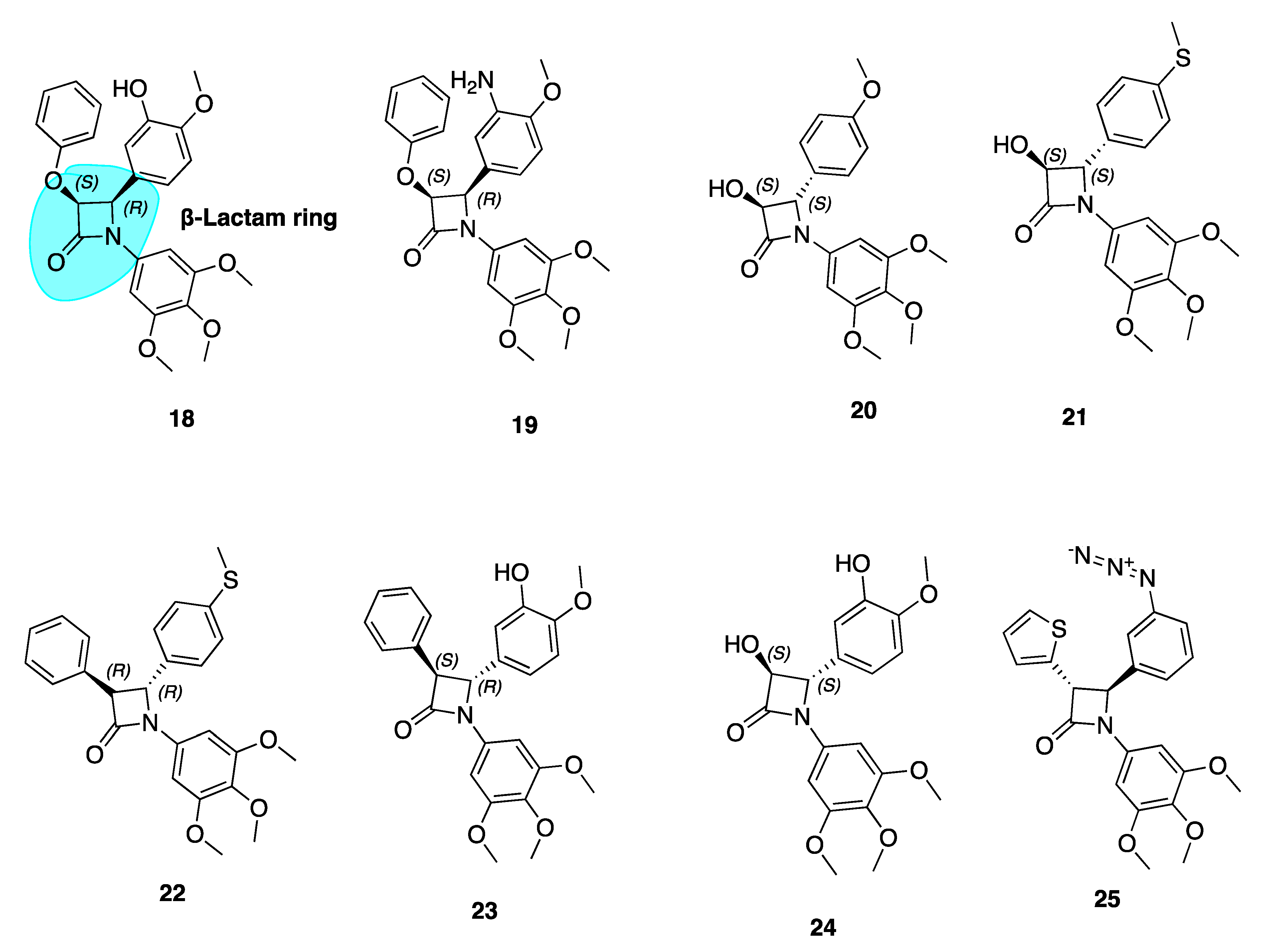
β-Lactam Cis Restricted Analogues
β-Lactam CA-4 Analogues with an Azide Substituted B Ring
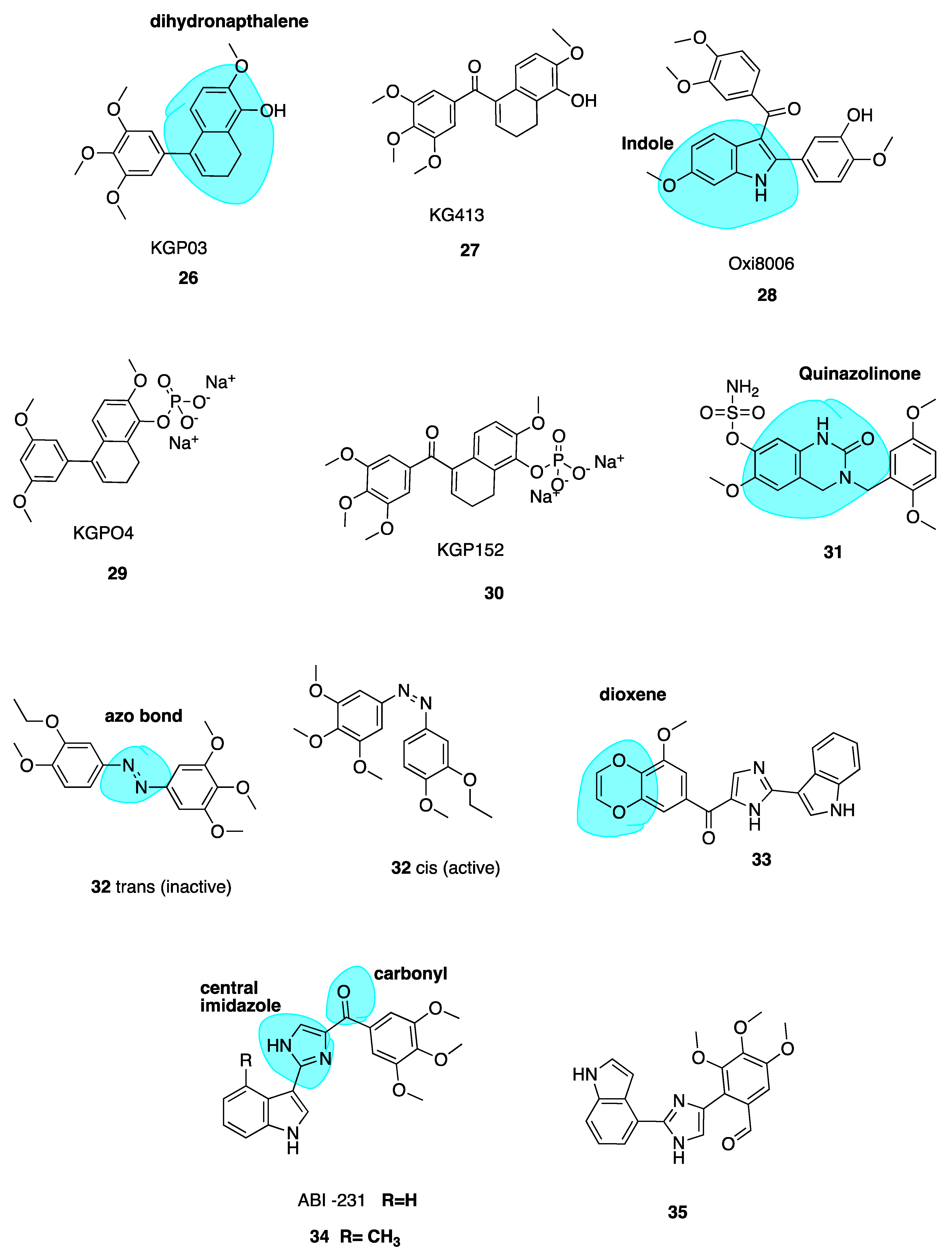
4.3.2. New Combretastatin A-4 Analogues of 2018
Dihydronapthalene Derivatives
Quinazolinone Derivatives
Photo-Responsive Azo CA-4 Analogues
Replacement of the Trimethoxyphenyl Moiety
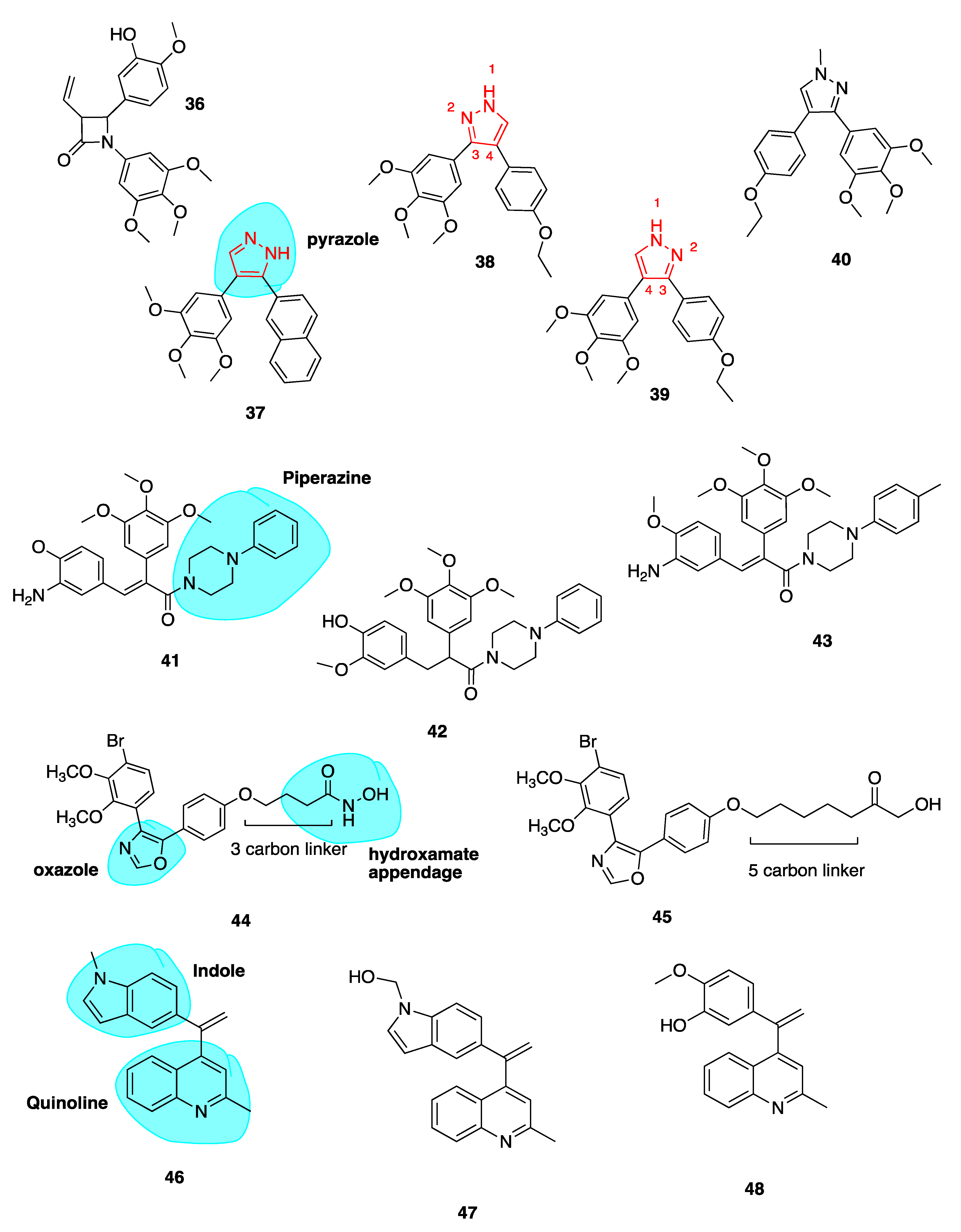
4.3.3. New Combretastatin A-4 Analogues of 2019
ABI-231 Analogues
3-Vinyl Substituted β-Lactams
Pyrazole Analogues
Piperazine Conjugates
Oxazole-Bridged Analogues
Quinoline and Indole Derivatives of isoCA-4
Heterocyclic isoCA-4 Derivatives
Novel Benzosuberene Analogues
1,2,4-Triazole-3-Carboxamide Derivatives
5. CBSIs Derived from Sources Other Than Combretastatins
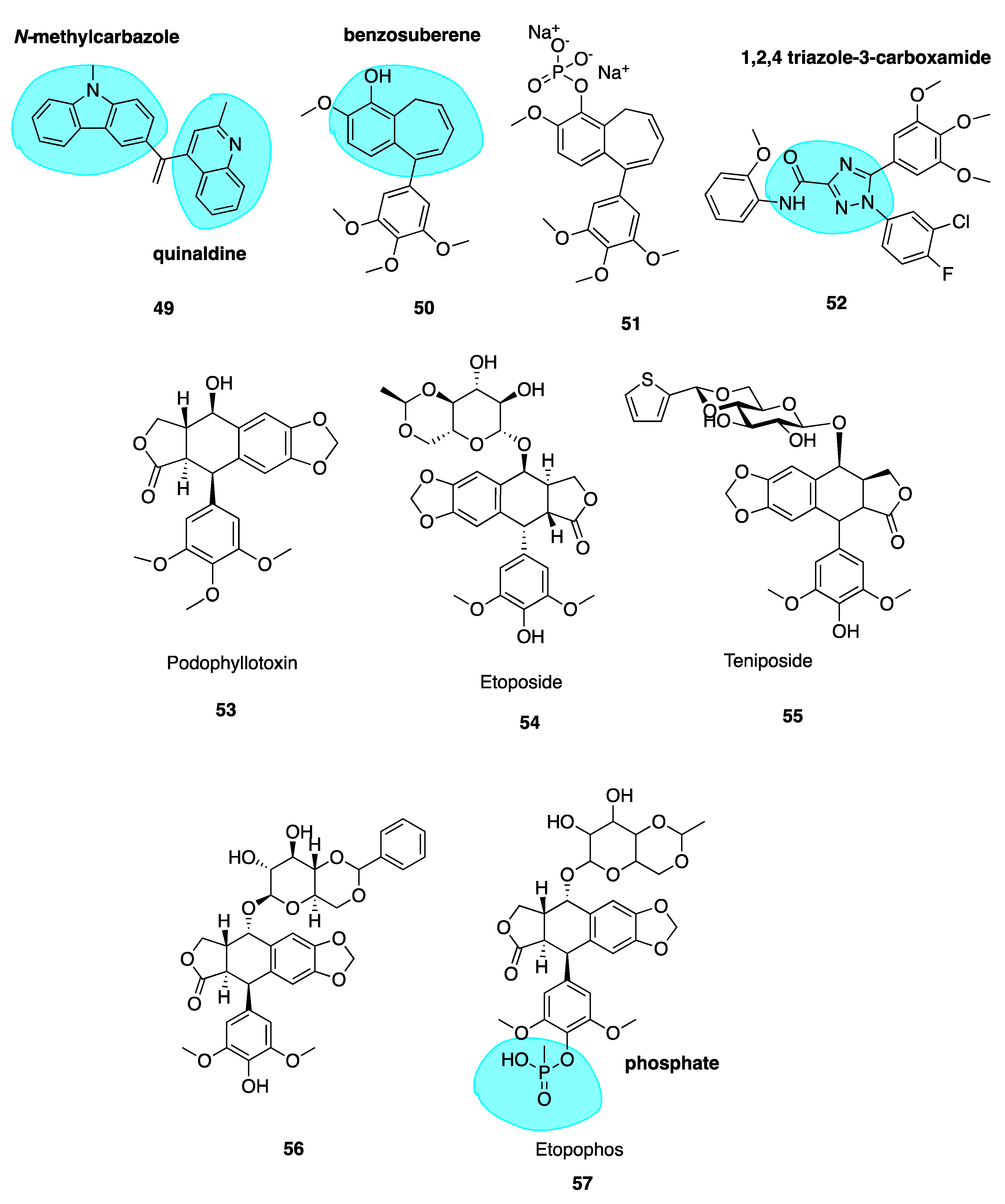
5.1. Podophyllotoxin and Analogues
5.2. Chalcones
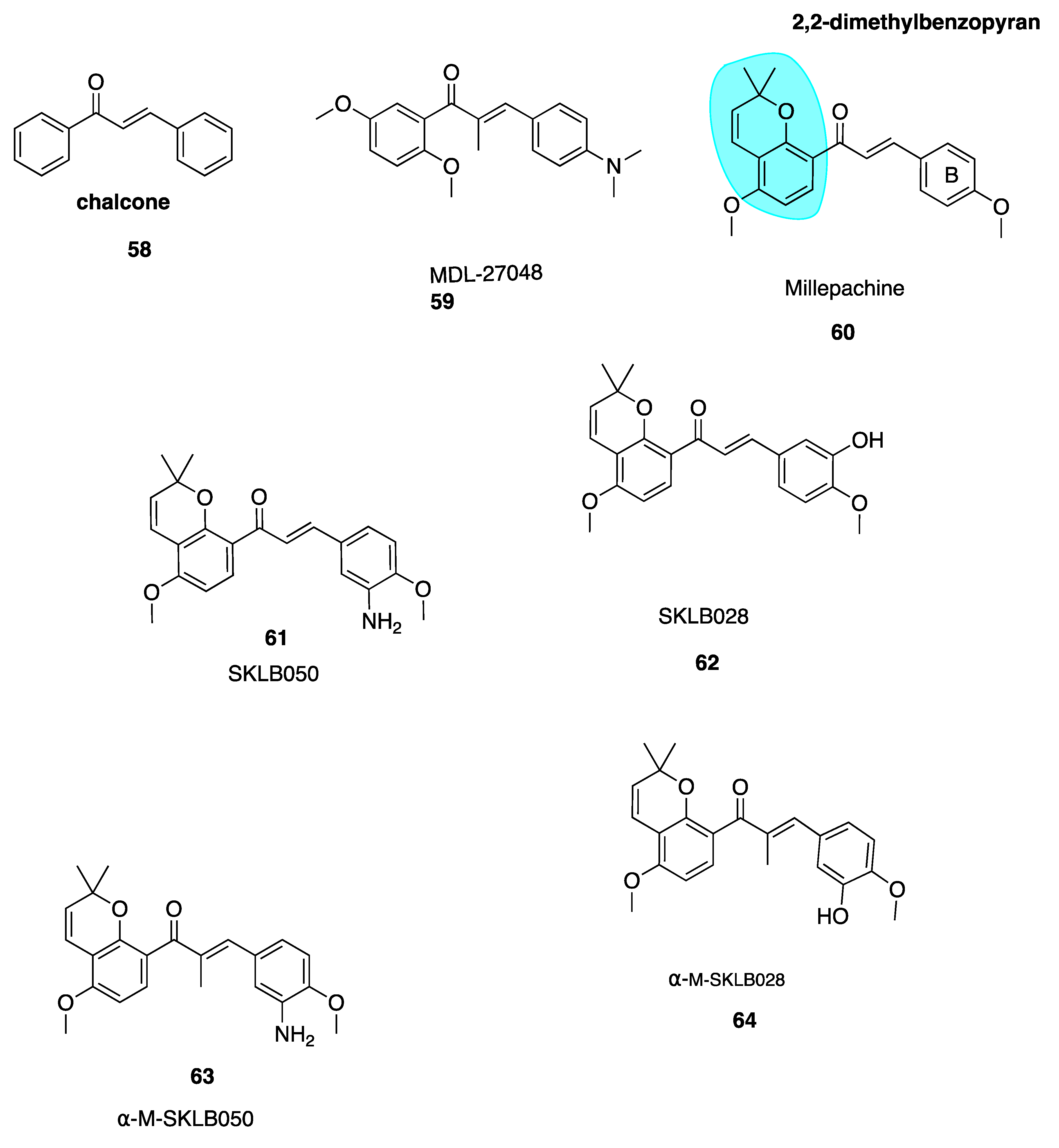
5.2.1. Millepachine and Derivatives
5.2.2. New Quinolone Chalcones: CBSIs and Inhibitors of MRP1 Function
5.2.3. Quinoline-Chalcone Derivatives of 2019
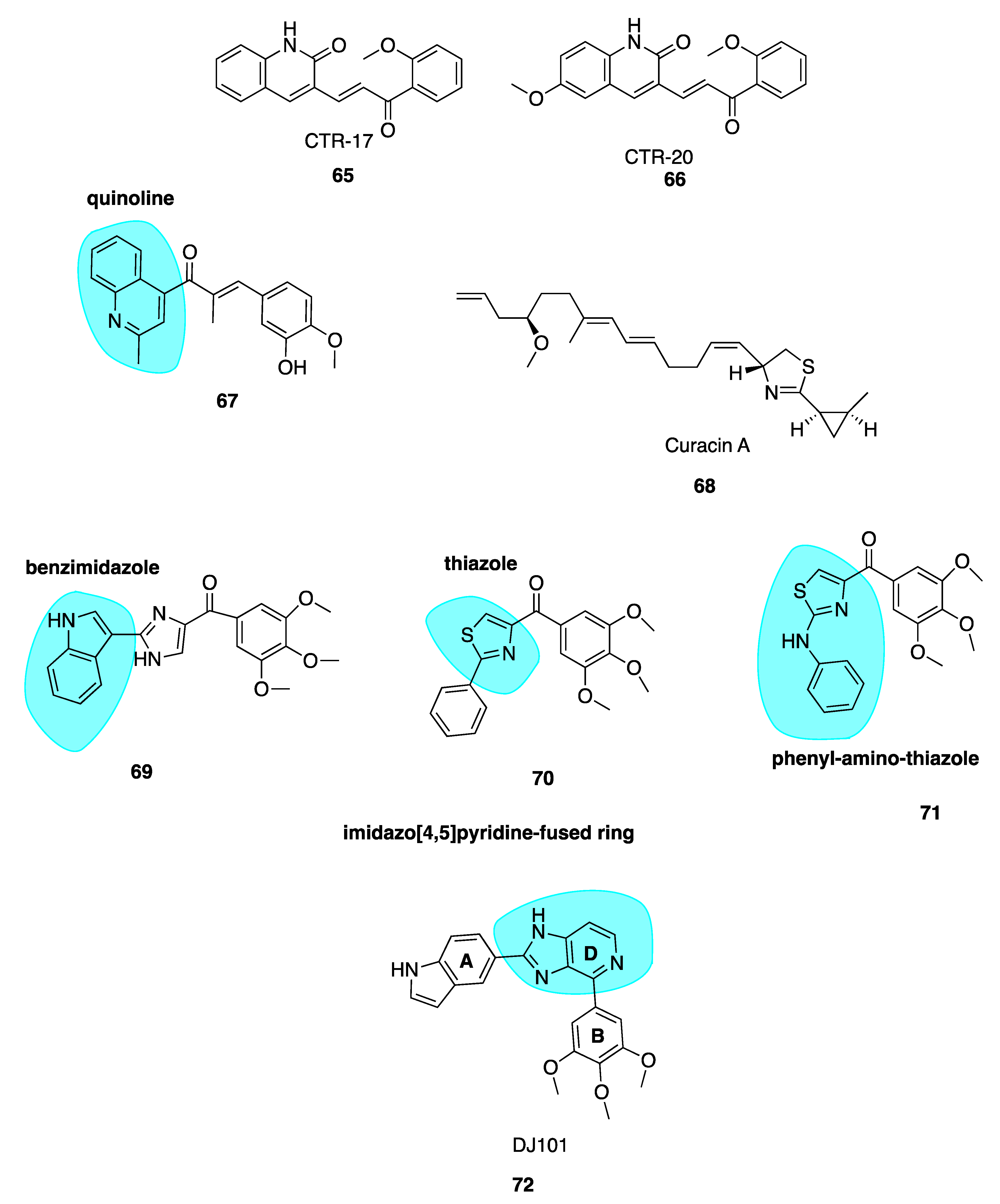
5.3. Curacin A
5.4. Imidazo[4,5]pyridine DJ95 (DJ101)
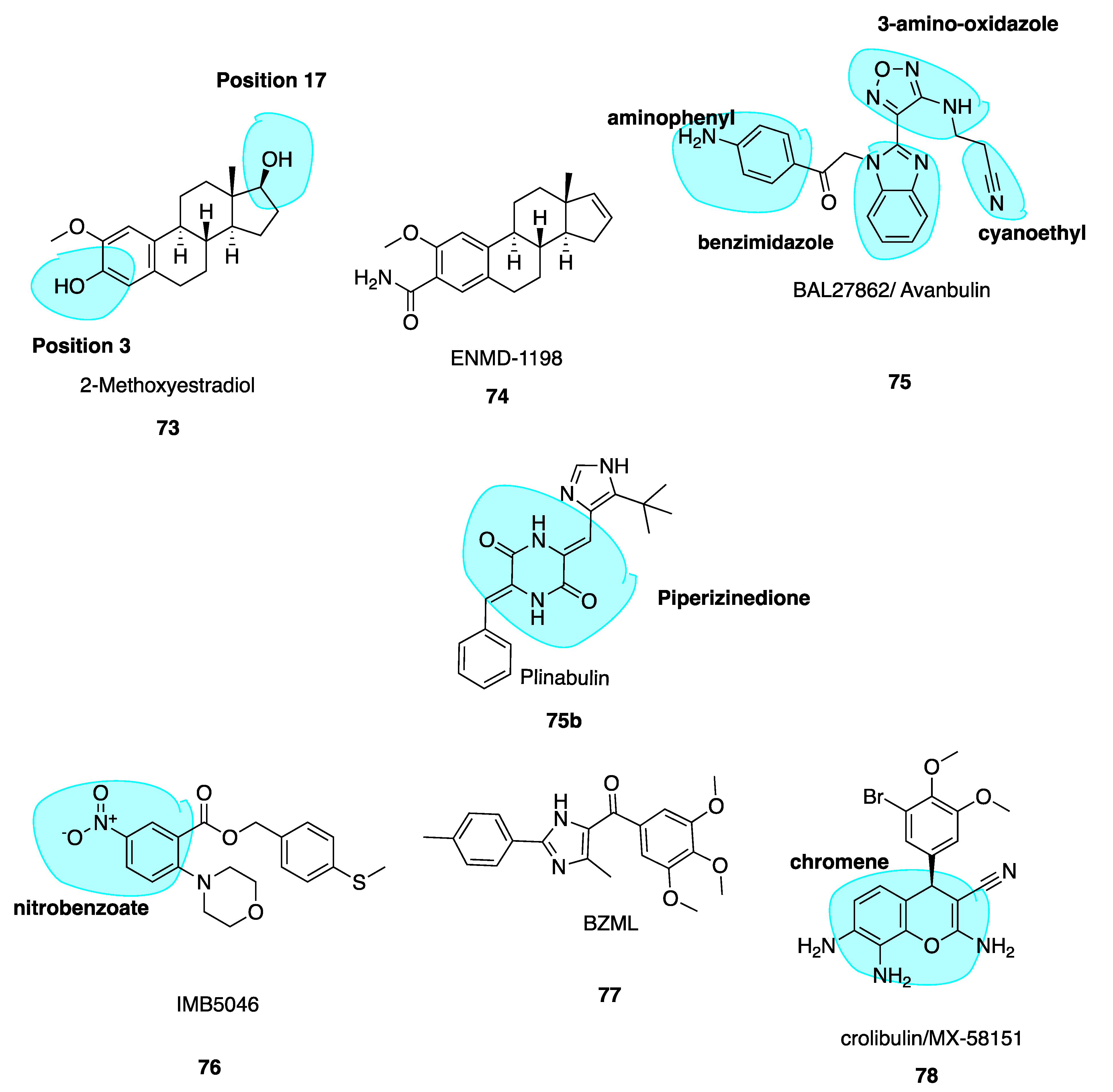
5.5. 2-Methoxyestradiol and ENMD1198
5.6. Avanbulin (BAL27862) (75)
5.7. Nitrobenzoate IMB5046
5.8. Imidazole BZML
5.9. Crolibulin (MX-58151)
5.10. Novel Nicotinonitrile Analogues of Crolibulin and CA-4
5.11. Indenes CP248 and CP461
6. CBSIs in Clinical Trials and Clinical Use
6.1. Combretastatin A-4 Analogues in the Clinic
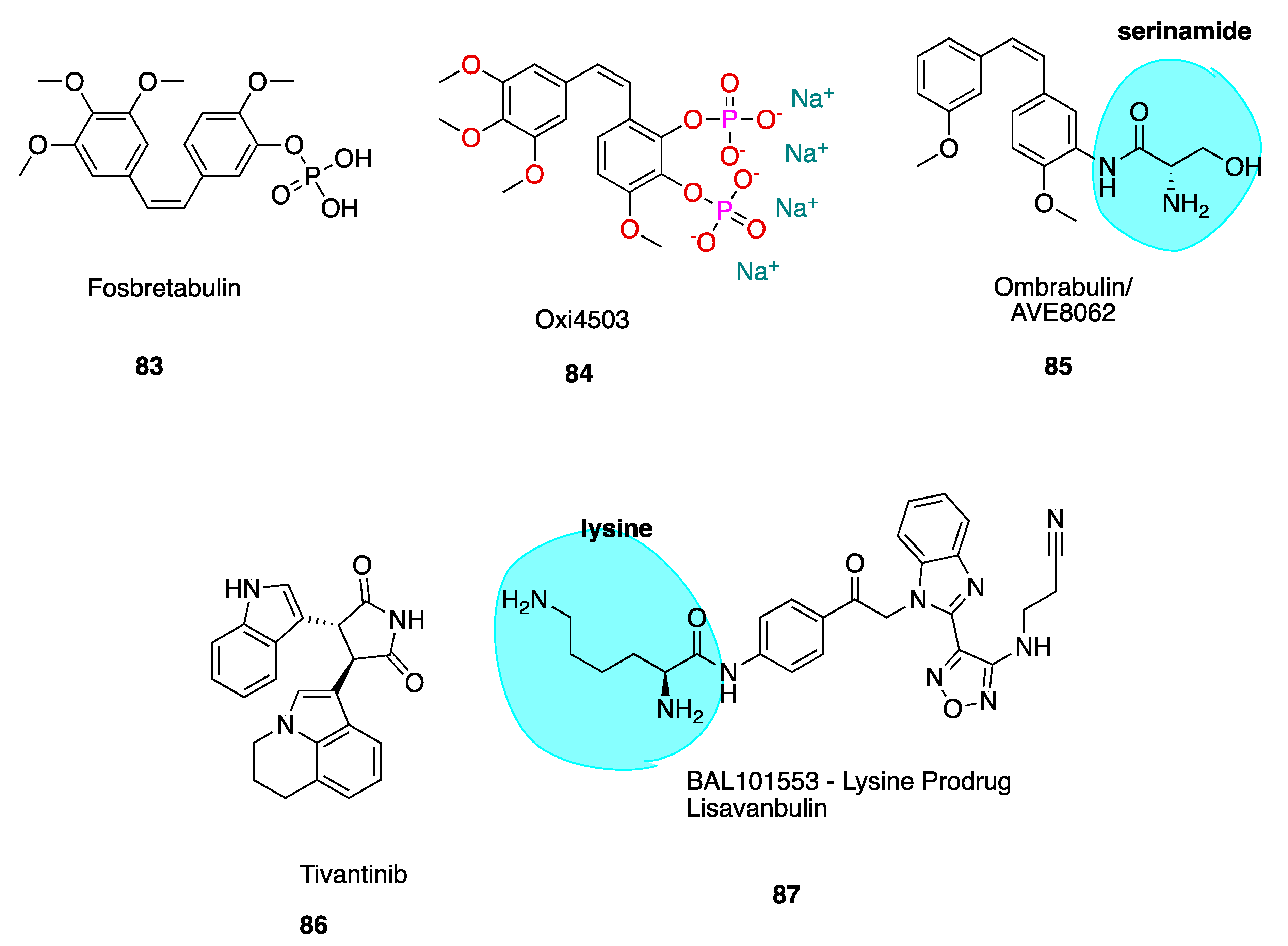
6.1.1. Fosbretabulin—A CA-4 Analogue Prodrug
6.1.2. Oxi4503—A CA-1 Prodrug
6.1.3. Ombrabulin/AVE8062—An CA-4 Amino Analogue Prodrug
6.2. Non-Combretastatin A-4 Analogues in Clinical Trials
6.2.1. Tivantinib
6.2.2. Plinabulin (BPI-2358, formerly NPI-2358)
6.2.3. Lisavanbulin (BAL101553)
6.2.4. Crolibulin
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukaemia |
| CA-1 | Combretastatin A-1 |
| CA-4 | Combretastatin A-4 |
| CA-4P | Combretastatin A-4 phosphate |
| CBS | Colchicine-binding site |
| CBSI | Colchicine-binding site inhibitor |
| DAMA-colchicine | N-deacetyl-N-(2-mercaptoacetyl) colchicine |
| DAPI | 4′,6-diamidino-2-phenylindole |
| EGFR | Epidermal growth factor receptor |
| FITC | Fluorescein isothiocyanate |
| GDP | Guanosine diphosphate |
| GEP-NETs | Gastro-entero-pancreatic neuroendocrine tumours |
| GTP | Guanosine triphosphate |
| HCC | Hepatocellular carcinoma |
| HDAC | Histone deacetylase enzyme |
| KRAS | Kirsten rat sarcoma viral analogue homologue |
| MDR | Multi-drug resistance |
| MDS | Myelodysplastic syndrome |
| MTA | Microtubule-targeting agent |
| NLRP3 | nucleotide-binding domain (NOD)-like receptor protein |
| NSCLC | Non-small-cell lung carcinoma |
| P-gp | P-glycoprotein |
| PPTP | Paediatric preclinical testing program |
| SAR | Structure–activity relationship |
| UGT | UDP-glucuronyl transferase |
| VDA | Vascular-disrupting agent |
| VEGFR | Vascular endothelial growth factor receptor |
References
- Adams, J. Essentials of Cell Biology. In Learn Science at Scitable; O’Connor, C., Ed.; NPG Education: Cambridge, UK, 2014; Available online: https://www.nature.com/scitable/ebooks/essentials-of-cell-biology-14749010/118240354/ (accessed on 9 September 2019).
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Downing, K.H. Structural Basis for the Interaction of Tubulin with Proteins and Drugs That Affect Microtubule Dynamics. Ann. Rev. Cell Biol. 2000, 16, 89–111. [Google Scholar] [CrossRef]
- Cooper, G.M.; Hausman, R.E.; Hausman, R.E. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK9876/ (accessed on 29 September 2019).
- Wilson, L.; Jordan, M.A. Microtubule Dynamics: Taking Aim at a Moving Target. Chem. Biol. 1995, 2, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Rieder, C.L.; Maiato, H. Stuck in Division or Passing Through: What Happens When Cells Cannot Satisfy the Spindle Assembly Checkpoint. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.-H.; Steinmetz, M.O. A New Tubulin-Binding Site and Pharmacophore for Microtubule-Destabilizing Anticancer Drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, E.N.; Patnaik, J.; Mullins, M.R.; Lemasters, J.J. Free Tubulin Modulates Mitochondrial Membrane Potential in Cancer Cells. Cancer Res. 2010, 70, 10192–10201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahern, M.J.; Reid, C.; Gordon, T.P.; McCredlE, M.; Brooks, P.M.; Jones, M. Does Colchicine Work? The Results of the First Controlled Study in Acute Gout. Aust. N. Z. J. Med. 1987, 17, 301–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, U.; Schumann, C.; Schmidt, K.L. Current Aspects of Colchicine Therapy—Classical Indications and New Therapeutic Uses. Eur. J. Med. Res. 2001, 6, 150–160. [Google Scholar] [PubMed]
- Ben-Chetrit, E.; Levy, M. Colchicine Prophylaxis in Familial Mediterranean Fever: Reappraisal after 15 Years. Semin. Arthritis Rheum. 1991, 20, 241–246. [Google Scholar] [CrossRef]
- Yurdakul, S.; Mat, C.; Tüzün, Y.; Özyazgan, Y.; Hamuryudan, V.; Uysal, Ö.; Şenocak, M.; Yazici, H. A Double-Blind Trial of Colchicine in Behçet’s Syndrome. Arthiritis Rheum. 2001, 44, 2686–2692. [Google Scholar] [CrossRef]
- Vaidya, K.; Gonzalo Martínez, S.P. The Role of Colchicine in Acute Coronary Syndromes. Clin. Therepeutics 2019, 41, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, G.J.; Celermajer, D.S.; Patel, S. The Nlrp3 Inflammasome and the Emerging Role of Colchicine to Inhibit Atherosclerosis-Associated Inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Rymer, J.A.; Newby, L.K. Failure to Launch: Targeting Inflammation in Acute Coronary Syndromes. J. Am. Coll. Cardiol. 2017, 2, 484–497. [Google Scholar]
- Finkelstein, Y.; Aks, S.E.; Hutson, J.R.; Juurlink, D.N.; Nguyen, P.; Dubnov-Raz, G.; Pollak, U.; Koren, G.; Bentur, Y. Colchicine Poisoning: The Dark Side of an Ancient Drug. Clin. Toxicol. 2010, 45, 407–414. [Google Scholar] [CrossRef]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Structure of the Alpha-Beta Tubulin Dimer by Electron Crystallography. Nature 1998, 391, 199–204. [Google Scholar] [CrossRef]
- Andreu, J.M.; Perez-Ramirez, B.; Gorbunoff, M.J.; Ayala, D.; Timasheff, S.N. Role of the Colchicine Ring a and Its Methoxy Groups in the Binding to Tubulin and Microtubule Inhibition. Biochemistry 1998, 37, 8356–8368. [Google Scholar] [CrossRef]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into Tubulin Regulation from a Complex with Colchicine and a Stathmin-Like Domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The Novel Microtubule-Destabilizing Drug Bal27862 Binds to the Colchicine Site of Tubulin with Distinct Effects on Microtubule Organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef]
- Dong, M.; Liu, F.; Zhou, H.; Zhai, S.; Yan, B. Novel Natural Product- and Privileged Scaffold-Based Tubulin Inhibitors Targeting the Colchicine Binding Site. Molecules 2016, 21, 1375. [Google Scholar] [CrossRef] [Green Version]
- Hearn, B.R.; Shaw, S.J.; Myles, D.C. Comprehensive Medicinal Chemistry II. In 7.04 Microtubule Targeting Agents; 7.04.2.2.1 Colchicine and Analogs; Taylor, J.B., Triggle, D.J., Eds.; 2007; Available online: https://www.sciencedirect.com/science/article/pii/B008045044X002054 (accessed on 15 September 2019).
- NCI National Cancer Institute. Angiogenesis Inhibitors. Available online: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/angiogenesis-inhibitors-fact-sheet#why-is-angiogenesis-important-in-cancer (accessed on 16 November 2019).
- Denekamp, J. Vascular Endothelium as the Vulnerable Element in Tumours. Acta Radiol. Oncol. 1984, 23, 217–225. [Google Scholar] [CrossRef]
- Thorpe, P.E.; Chaplin, D.J.; Blakey, D.C. The First International Conference on Vascular Targeting: Meeting Overview. Cancer Res. 2003, 63, 1144–1147. [Google Scholar] [PubMed]
- Denekamp, J. Endothelial Cell Proliferation as a Novel Approach to Targeting Tumour Therapy. Br. J. Cancer 1982, 45, 136–139. [Google Scholar] [CrossRef] [Green Version]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Stengel, C.; Newman, S.P.; Leese, M.P.; Potter, B.V.L.; Reed, M.J.; Purohit, A. Class 111 Beta-Tubulin Expression and in Vitro Resistance to Microtubule Targeting Agents. Br. J. Cancer 2010, 102, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Watt, J.M.; Breyer-Brandwijk, M.G. Medicinal Poisonous Plants Southern Eastern Africa, 2nd ed.; Livingstone: Edinburgh, UK, 1962. [Google Scholar]
- Kanthou, C.; Tozer, G.M. The Tumor Vascular Targeting Agent Combretastatin a–4-Phosphate Induces Reorganization of the Actin Cytoskeleton and Early Membrane Blebbing in Human Endothelial Cells. Blood 2002, 99, 2060–2069. [Google Scholar] [CrossRef]
- Hua, J.; Sheng, Y.; Pinney, K.G.; Garner, C.M.; Kane, R.R.; Prezioso, J.A.; Pettit, G.R.; Chaplin, D.J.; Edvardsen, K. Oxi4503, a Novel Vascular Targeting Agent: Effects on Blood Flow and Antitumor Activity in Comparison to Combretastatin a-4 Phosphate. Anticancer Res. 2003, 23, 1433–1440. [Google Scholar] [PubMed]
- Pettit, G.R.; Singh, S.B.; Hamel, E.; Lin, C.M.; Alberts, D.S.; Garcia-Kendall, D. Isolation and Structure of the Strong Cell Growth and Tubulin Inhibitor Combretastatin a-4. Experientia 1989, 45, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Chaplin, D.J.; Horsman, M.R. Vascular-Targeting Therapies for Treatment of Malignant Disease. Cancer 2004, 100, 2491–2499. [Google Scholar] [CrossRef]
- McGown, A.T.; Fox, B.W. Differential Cytotoxicity of Combretastatins A1 and A4 in Two Daunorubicin-Resistant P388 Cell Lines. Cancer Chemother. Pharmacol. 1990, 26, 79–81. [Google Scholar] [CrossRef]
- Baguley, B.C.; Holdaway, K.M.; Thomsen, L.L.; Zhuang, L.; Zwi, L.J. Inhibition of Growth of Colon 38 Adenocarcinoma by Vinblastine and Colchicine: Evidence for a Vascular Mechanism. Eur. J. Cancer Clin. Oncol. 1991, 27, 482–487. [Google Scholar] [CrossRef]
- Hill, S.A.; Lonergan, S.J.; Denekamp, J.; Chaplin, D.J. Vinca Alkaloids: Anti-Vascular Effects in a Murine Tumour. Eur. J. Cancer 1993, 29, 1320–1324. [Google Scholar] [CrossRef]
- Dark, G.G.; Hill, S.A.; Prise, V.E.; Tozer, G.M.; Pettit, G.R.; Chaplin, D.J. Combretastatin a-4, an Agent That Displays Potent and Selective Toxicity toward Tumor Vasculature. Cancer Res. 1997, 57, 1829–1834. [Google Scholar] [PubMed]
- Chaplin, D.J.; Hill, S.A. The Development of Combretastatin A4 Phosphate as a Vascular Targeting Agent. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 1491–1496. [Google Scholar] [CrossRef]
- Kremmidiotis, G.; Leske, A.F.; Lavranos, T.C.; Beaumont, D.; Gasic, J.; Hall, A.; Callaghan, M.; Matthews, C.A.; Flynn, B. Bnc105: A Novel Tubulin Polymerization Inhibitor That Selectively Disrupts Tumor Vasculature and Displays Single-Agent Antitumor Efficacy. Mol. Cancer Ther. 2010, 9, 1562–1573. [Google Scholar] [CrossRef] [Green Version]
- Siemann, D.W.; Shi, W. Dual Targeting of Tumor Vasculature: Combining Avastin and Vascular Disrupting Agents (Ca4p or Oxi4503). Anticancer Res. 2008, 28, 2027–2031. [Google Scholar]
- Tozer, G.M.; Kanthou, C.; Lewis, G.; Prise, V.E.; Vojnovic, B.; Hill, S.A. Tumour Vascular Disrupting Agents: Combating Treatment Resistance. Br. J. Radiol. 2008, 81, 12–20. [Google Scholar] [CrossRef]
- Pettit, G.R.; Rhodes, M.R.; Herald, D.L.; Hamel, E.; Schmidt, J.M.; Pettit, R.K. Antineoplastic Agents. 445. Synthesis and Evaluation of Structural Modifications of (Z)- and (E)-Combretastatin a-4. J. Med. Chem. 2005, 48, 4087–4099. [Google Scholar] [CrossRef]
- Ohsumi, K.; Hatanaka, T.; Fujita, K.; Nakagawa, R.; Fukuda, Y.; Nihei, Y.; Suga, Y.; Morinaga, Y.; Akiyama, Y.; Tsuji, T. Syntheses and Antitumor Activity of Cis-Restricted Combretastatins: 5-Membered Heterocyclic Analogues. Bioorg. Med. Chem. Lett. 1998, 8, 3153–3158. [Google Scholar] [CrossRef]
- Lee, L.; Davis, R.; Vanderham, J.; Hills, P.; Mackay, H.; Brown, T.; Mooberry, S.L.; Lee, M. 1,2,3,4-Tetrahydro-2-Thioxopyrimidine Analogs of Combretastatin-A4. Eur. J. Med. Chem. 2008, 43, 2011–2015. [Google Scholar] [CrossRef]
- Gaspari, R.; Prota, A.E.; Bargsten, K.; Cavalli, A.; Steinmetz, M.O. Structural Basis of Cis- and Trans-Combretastatin Binding to Tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Gomtsyan, A. Heterocycles in Drugs and Drug Discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Nguyen, T.T.B.; Lomberget, T.; Tran, N.C.; Colomb, E.; Nachtergaele, L.; Thoret, S.; Dubois, J.; Guillaume, J.; Abdayem, R.; Haftek, M.; et al. Synthesis and Biological Evaluation of Novel Heterocyclic Derivatives of Combretastatin a-4. Bioorg. Med. Chem. Lett. 2012, 22, 7227–7231. [Google Scholar] [CrossRef] [PubMed]
- Herdman, C.A.; Strecker, T.E.; Tanpure, R.P.; Chen, Z.; Winters, A.; Gerberich, J.; Liu, L.; Hamel, E.; Mason, R.P.; Chaplin, D.J.; et al. Synthesis and Biological Evaluation of Benzocyclooctene-Based and Indene-Based Anticancer Agents That Function as Inhibitors of Tubulin Polymerization. MedChemComm 2016, 7, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Fürst, R.; Zupkó, I.; Berényi, Á.; Ecker, G.F.; Rinner, U. Synthesis and Antitumor-Evaluation of Cyclopropyl-Containing Combretastatin Analogs. Bioorg. Med. Chem. Lett. 2009, 19, 6948–6951. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Woods, K.W.; Li, Q.; Barr, K.J.; McCroskey, R.W.; Hannick, S.M.; Gherke, L.; Credo, R.B.; Hui, Y.-H.; Marsh, K.; et al. Potent, Orally Active Heterocycle-Based Combretastatin a-4 Analogues: Synthesis, Structure−Activity Relationship, Pharmacokinetics, and in Vivo Antitumor Activity Evaluation. J. Med. Chem. 2002, 45, 1697–1711. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Pérez-Melero, C.; Peláez, R.; Medarde, M. Stilbenophane Analogues of Deoxycombretastatin a-4. J. Org. Chem. 2005, 70, 6544–6547. [Google Scholar] [CrossRef] [PubMed]
- Daniel, T.; Siyaram, P.; James, M. Review of Cytotoxic Ca4 Analogues That Do Not Target Microtubules: Implications for Ca4 Development. Mini-Rev. Med. Chem. 2017, 17, 1507–1514. [Google Scholar]
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More Than Just Vascular Targeting Agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef]
- Siemann, D.W.; Chaplin, D.J.; Walicke, P.A. A Review and Update of the Current Status of the Vasculature Disabling Agent Combretastatin-A4 Phosphate (Ca4p). Expert Opin. Investig. Drugs 2009, 18, 189–197. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [Green Version]
- Ducki, S. The Development of Chalcones as Promising Anticancer Agents. Investig. Drugs J. 2007, 10, 42–46. [Google Scholar]
- Ducki, S.; Rennison, D.; Woo, M.; Kendall, A.; Chabert, J.F.D.; McGown, A.T.; Lawrence, N.J. Combretastatin-Like Chalcones as Inhibitors of Microtubule Polymerization. Part 1: Synthesis and Biological Evaluation of Antivascular Activity. Bioorg. Med. Chem. 2009, 17, 7698–7710. [Google Scholar] [CrossRef]
- Pettit, G.R.; Toki, B.; Herald, D.L.; Verdier-Pinard, P.; Boyd, M.R.; Hamel, E.; Pettit, R.K. Antineoplastic Agents. 379. Synthesis of Phenstatin Phosphate 1a. J. Med. Chem. 1998, 41, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Le Broc-Ryckewaert, D.; Pommery, N.; Pommery, J.; Ghinet, A.; Farce, A.; Wiart, J.-F.; Gautret, P.; Rigo, B.; Hénichart, J.-P. In Vitro Metabolism of Phenstatin: Potential Pharmacological Consequences. Drug Metab. Lett. 2011, 5, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Winn, B.A.; Shi, Z.; Carlson, G.J.; Wang, Y.; Nguyen, B.L.; Kelly, E.M.; Ross, R.D.T.; Hamel, E.; Chaplin, D.J.; Trawick, M.L.; et al. Bioreductively Activatable Prodrug Conjugates of Phenstatin Designed to Target Tumor Hypoxia. Bioorg. Med. Chem. Lett. 2017, 27, 636–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messaoudi, S.; Tréguier, B.; Hamze, A.; Provot, O.; Peyrat, J.-F.; De Losada, J.R.; Liu, J.-M.; Bignon, J.; Wdzieczak-Bakala, J.; Thoret, S.; et al. Isocombretastatins a Versus Combretastatins A: The Forgotten Isoca-4 Isomer as a Highly Promising Cytotoxic and Antitubulin Agent. J. Med. Chem. 2009, 52, 4538–4542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-H.; Wu, L.; Raymon, H.K.; Chen, R.S.; Corral, L.; Shirley, M.A.; Krishna Narla, R.; Gamez, J.; Muller, G.W.; Stirling, D.I.; et al. The Synthetic Compound Cc-5079 Is a Potent Inhibitor of Tubulin Polymerization and Tumor Necrosis Factor-Α Production with Antitumor Activity. Cancer Res. 2006, 66, 951–959. [Google Scholar] [CrossRef] [Green Version]
- Vu, H.N.; Miller, W.J.; O’Connor, S.A.; He, M.; Schafer, P.H.; Payvandi, F.; Muller, G.W.; Stirling, D.I.; Libutti, S.K. Cc-5079: A Small Molecule with Mkp1, Antiangiogenic, and Antitumor Activity. J. Surg. Res. 2010, 164, 116–125. [Google Scholar] [CrossRef]
- Wen, Z.; Xu, J.; Wang, Z.; Qi, H.; Xu, Q.; Bai, Z.; Zhang, Q.; Bao, K.; Wu, Y.; Zhang, W. 3-(3,4,5-Trimethoxyphenylselenyl)-1h-Indoles and Their Selenoxides as Combretastatin a-4 Analogs: Microwave-Assisted Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2015, 90, 184–194. [Google Scholar] [CrossRef]
- Zuo, D.; Guo, D.; Jiang, X.; Guan, Q.; Qi, H.; Xu, J.; Li, Z.; Yang, F.; Zhang, W.; Wu, Y. 3-(3-Hydroxy-4-Methoxyphenyl)-4-(3,4,5-Trimethoxyphenyl)-1,2,5-Selenadiazole (G-1103), a Novel Combretastatin a-4 Analog, Induces G2/M Arrest and Apoptosis by Disrupting Tubulin Polymerization in Human Cervical Hela Cells and Fibrosarcoma Ht-1080 Cells. Chem.-Biol. Interact. 2015, 227, 7–17. [Google Scholar] [CrossRef]
- Guan, Q.; Yang, F.; Guo, D.; Xu, J.; Jiang, M.; Liu, C.; Bao, K.; Wu, Y.; Zhang, W. Synthesis and Biological Evaluation of Novel 3,4-Diaryl-1,2,5-Selenadiazol Analogues of Combretastatin a-4. Eur. J. Med. Chem. 2014, 87, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, E.; Hamel, E.; Bai, R.; Burnett, J.; Tozatti, C.; Bogo, D.; Perdomo, R.; Antunes, A.; Marques, M.; de Matos, M.; et al. Synthesis and Evaluation of Diaryl Sulfides and Diaryl Selenide Compounds for Antitubulin and Cytotoxic Activity. Bioorg. Med. Chem. Lett. 2013, 23, 4669–4673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.; An, B.; Lou, L.; Zhang, J.; Yan, J.; Huang, L.; Li, X.; Yin, S. Design, Synthesis, and Biological Evaluation of Novel Selenium-Containing Isocombretastatins and Phenstatins as Antitumor Agents. J. Med. Chem. 2017, 60, 7300–7314. [Google Scholar] [CrossRef] [PubMed]
- Greene, T.F.; Wang, S.; Greene, L.M.; Nathwani, S.M.; Pollock, J.K.; Malebari, A.M.; McCabe, T.; Twamley, B.; O’Boyle, N.M.; Zisterer, D.M.; et al. Synthesis and Biochemical Evaluation of 3-Phenoxy-1,4-Diarylazetidin-2-Ones as Tubulin-Targeting Antitumor Agents. J. Med. Chem. 2016, 59, 90–113. [Google Scholar] [CrossRef]
- Banik, I.; Becker, F.F.; Banik, B.K. Stereoselective Synthesis of Β-Lactams with Polyaromatic Imines: Entry to New and Novel Anticancer Agents. J. Med. Chem. 2003, 46, 12–15. [Google Scholar] [CrossRef]
- Sun, L.; Vasilevich, N.I.; Fuselier, J.A.; Hocart, S.J.; Coy, D.H. Examination of the 1,4-Disubstituted Azetidinone Ring System as a Template for Combretastatin a-4 Conformationally Restricted Analogue Design. Bioorg. Med. Chem. Lett. 2004, 14, 2041–2046. [Google Scholar] [CrossRef]
- Wang, S.; Malebari, A.M.; Greene, T.F.; O’Boyle, N.M.; Fayne, D.; Nathwani, S.M.; Twamley, B.; McCabe, T.; Keely, N.O.; Zisterer, D.M.; et al. 3-Vinylazetidin-2-Ones: Synthesis, Antiproliferative and Tubulin Destabilizing Activity in Mcf-7 and Mda-Mb-231 Breast Cancer Cells. Pharmaceuticals 2019, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Carr, M.; Greene, L.M.; Keely, N.O.; Knox, A.J.S.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis, Biochemical and Molecular Modelling Studies of Antiproliferative Azetidinones Causing Microtubule Disruption and Mitotic Catastrophe. Eur. J. Med. Chem. 2011, 46, 4595–4607. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Carr, M.; Greene, L.M.; Bergin, O.; Nathwani, S.M.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis and Evaluation of Azetidinone Analogues of Combretastatin a-4 as Tubulin Targeting Agents. J. Med. Chem. 2010, 53, 8569–8584. [Google Scholar] [CrossRef]
- Carr, M.; Greene, L.M.; Knox, A.J.S.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Lead Identification of Conformationally Restricted Β-Lactam Type Combretastatin Analogues: Synthesis, Antiproliferative Activity and Tubulin Targeting Effects. Eur. J. Med. Chem. 2010, 45, 5752–5766. [Google Scholar] [CrossRef]
- Malebari, A.M.; Greene, L.M.; Nathwani, S.M.; Fayne, D.; O’Boyle, N.M.; Wang, S.; Twamley, B.; Zisterer, D.M.; Meegan, M.J. Β-Lactam Analogues of Combretastatin a-4 Prevent Metabolic Inactivation by Glucuronidation in Chemoresistant Ht-29 Colon Cancer Cells. Eur. J. Med. Chem. 2017, 130, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Rustin, G.J.S.; Galbraith, S.M.; Anderson, H.; Stratford, M.; Folkes, L.K.; Sena, L.; Gumbrell, L.; Price, P.M. Phase I Clinical Trial of Weekly Combretastatin A4 Phosphate: Clinical and Pharmacokinetic Results. J. Clin. Oncol. 2003, 21, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Zelcer, N.; Allen, J.D.; Yao, D.; Boyd, G.; Maliepaard, M.; Friedberg, T.H.; Smyth, J.F.; Jodrell, D.I. Glucuronidation as a Mechanism of Intrinsic Drug Resistance in Colon Cancer Cells: Contribution of Drug Transport Proteins. Biochem. Pharmacol. 2004, 67, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.-J.; Fu, L.; Liu, Y.-C.; Wang, J.-W.; Wang, Y.-Q.; Han, B.-K.; Li, X.-R.; Zhang, C.; Li, F.; Song, J.; et al. Structure-Activity Relationship Studies of Β-Lactam-Azide Analogues as Orally Active Antitumor Agents Targeting the Tubulin Colchicine Site. Sci. Rep. 2017, 7, 12788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonough, M.T.; Strecker, T.E.; Hamel, E.; Hall, J.J.; Chaplin, D.J.; Trawick, M.L.; Pinney, K.G. Synthesis and Biological Evaluation of Indole-Based, Anti-Cancer Agents Inspired by the Vascular Disrupting Agent 2-(3′-Hydroxy-4′-Methoxyphenyl)-3-(3″,4″,5″-Trimethoxybenzoyl)-6-Methoxyindole (Oxi8006). Bioorg. Med. Chem. 2013, 21, 6831–6843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, A.; Kumar, G.B.; Vishnuvardhan, M.V.P.S.; Shaik, A.B.; Reddy, V.S.; Mahesh, R.; Bin Sayeeda, I.; Kapure, J.S. Synthesis of Phenstatin/Isocombretastatin–Chalcone Conjugates as Potent Tubulin Polymerization Inhibitors and Mitochondrial Apoptotic Inducers. Org. Biomol. Chem. 2015, 13, 3963–3981. [Google Scholar] [CrossRef]
- Maguire, C.; Chen, Z.; Mocharla, V.; Sriram, M.; Hamel, E.; Zhou, H.; Lopez, R.; Wang, Y.; Mason, R.; Chaplin, D.; et al. Synthesis of Dihydronaphthalene Analogues Inspired by Combretastatin a-4 and Their Biological Evaluation as Anticancer Agents. MedChemComm 2018, 9, 1649–1662. [Google Scholar] [CrossRef]
- Dohle, W.; Jourdan, F.L.; Menchon, G.; Prota, A.E.; Foster, P.A.; Mannion, P.; Hamel, E.; Thomas, M.P.; Kasprzyk, P.G.; Ferrandis, E.; et al. Quinazolinone-Based Anticancer Agents: Synthesis, Antiproliferative Sar, Antitubulin Activity, and Tubulin Co-Crystal Structure. J. Med. Chem. 2018, 61, 1031–1044. [Google Scholar] [CrossRef]
- Rastogi, S.K.; Zhao, Z.; Barrett, S.L.; Shelton, S.D.; Zafferani, M.; Anderson, H.E.; Blumenthal, M.O.; Jones, L.R.; Wang, L.; Li, X.; et al. Photoresponsive Azo-Combretastatin a-4 Analogues. Eur. J. Med. Chem. 2018, 143, 1–7. [Google Scholar] [CrossRef]
- Sheldon, J.E.; Dcona, M.M.; Lyons, C.E.; Hackett, J.C.; Hartman, M.C.T. Photoswitchable Anticancer Activity Via Trans–Cis Isomerization of a Combretastatin a-4 Analog. Org. Biomol. Chem. 2016, 14, 40–49. [Google Scholar] [CrossRef]
- Engdahl, A.J.; Torres, E.A.; Lock, S.E.; Engdahl, T.B.; Mertz, P.S.; Streu, C.N. Synthesis, Characterization, and Bioactivity of the Photoisomerizable Tubulin Polymerization Inhibitor Azo-Combretastatin A4. Org. Lett. 2015, 17, 4546–4549. [Google Scholar] [CrossRef] [PubMed]
- Broichhagen, J.; Frank, J.A.; Trauner, D. A Roadmap to Success in Photopharmacology. Acc. Chem. Res. 2015, 48, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Velema, W.A.; Szymanski, W.; Feringa, B.L. Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehrentz, T.; Schönberger, M.; Trauner, D. Optochemical Genetics. Angew. Chem. Int. Ed. 2011, 50, 12156–12182. [Google Scholar] [CrossRef]
- Wang, Q.; Arnst, K.E.; Wang, Y.; Kumar, G.; Ma, D.; Chen, H.; Wu, Z.; Yang, J.; White, S.W.; Miller, D.D.; et al. Structural Modification of the 3,4,5-Trimethoxyphenyl Moiety in the Tubulin Inhibitor Veru-111 Leads to Improved Antiproliferative Activities. J. Med. Chem. 2018, 61, 7877–7891. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ahn, S.; Wang, J.; Lu, Y.; Dalton, J.T.; Miller, D.D.; Li, W. Discovery of Novel 2-Aryl-4-Benzoyl-Imidazole (Abi-Iii) Analogues Targeting Tubulin Polymerization as Antiproliferative Agents. J. Med. Chem. 2012, 55, 7285–7289. [Google Scholar] [CrossRef]
- Chen, J.; Li, C.-M.; Wang, J.; Ahn, S.; Wang, Z.; Lu, Y.; Dalton, J.T.; Miller, D.D.; Li, W. Synthesis and Antiproliferative Activity of Novel 2-Aryl-4-Benzoyl-Imidazole Derivatives Targeting Tubulin Polymerization. Bioorg. Med. Chem. 2011, 19, 4782–4795. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, V.K.; Wang, Q.; Setua, S.; Nagesh, P.K.B.; Chauhan, N.; Kumari, S.; Chowdhury, P.; Miller, D.D.; Yallapu, M.M.; Li, W.; et al. Therapeutic Efficacy of a Novel Βiii/Βiv-Tubulin Inhibitor (Veru-111) in Pancreatic Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 29. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.-J.; Wang, J.; Li, W.; Miller, D.D. Structural Optimization of Indole Derivatives Acting at Colchicine Binding Site as Potential Anticancer Agents. ACS Med. Chem. Lett. 2015, 6, 993–997. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Arnst, K.E.; Wang, Y.; Kumar, G.; Ma, D.; White, S.W.; Miller, D.D.; Li, W.; Li, W. Structure-Guided Design, Synthesis, and Biological Evaluation of (2-(1h-Indol-3-Yl)-1h-Imidazol-4-Yl)(3,4,5-Trimethoxyphenyl) Methanone (Abi-231) Analogues Targeting the Colchicine Binding Site in Tubulin. J. Med. Chem. 2019, 62, 6734–6750. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Salvador, M.K.; Preti, D.; Aghazadeh Tabrizi, M.; Brancale, A.; Fu, X.-H.; Li, J.; Zhang, S.-Z.; Hamel, E.; et al. Synthesis and Evaluation of 1,5-Disubstituted Tetrazoles as Rigid Analogues of Combretastatin a-4 with Potent Antiproliferative and Antitumor Activity. J. Med. Chem. 2012, 55, 475–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoli, R.; Oliva, P.; Salvador, M.K.; Camacho, M.E.; Padroni, C.; Brancale, A.; Ferla, S.; Hamel, E.; Ronca, R.; Grillo, E.; et al. Design, Synthesis and Biological Evaluation of Novel Vicinal Diaryl-Substituted 1h-Pyrazole Analogues of Combretastatin a-4 as Highly Potent Tubulin Polymerization Inhibitors. Eur. J. Med. Chem. 2019, 181, 111577. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Maya, A.B.; Pérez-Melero, C.; Salvador, N.; Peláez, R.; Caballero, E.; Medarde, M. New Naphthylcombretastatins. Modifications on the Ethylene Bridge. Bioorg. Med. Chem. 2005, 13, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, M.L.; Flego, M.; Molinari, A.; Cianfriglia, M. Saquinavir Induces Stable and Functional Expression of the Multidrug Transporter P-Glycoprotein in Human Cd4 T-Lymphoblastoid Cemrev Cells. HIV Med. 2003, 4, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Lai, Q.; Wang, Y.; Wang, R.; Lai, W.; Tang, L.; Tao, Y.; Liu, Y.; Zhang, R.; Huang, L.; Xiang, H.; et al. Design, Synthesis and Biological Evaluation of a Novel Tubulin Inhibitor 7a3 Targeting the Colchicine Binding Site. Eur. J. Med. Chem. 2018, 156, 162–179. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Ana, G.; Kelly, P.M.; Nathwani, S.M.; Noorani, S.; Fayne, D.; Bright, S.A.; Twamley, B.; Zisterer, D.M.; Meegan, M.J. Synthesis and Evaluation of Antiproliferative Microtubule-Destabilising Combretastatin A-4 Piperazine Conjugates. Org. Biomol. Chem. 2019, 17, 6184–6200. [Google Scholar] [CrossRef]
- Choi, J.-H.; Kwon, H.J.; Yoon, B., II; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef]
- Boyle, N.M.O.; Meegan, M.J. Designed Multiple Ligands for Cancer Therapy. Curr. Med. Chem. 2011, 18, 4722–4737. [Google Scholar] [CrossRef]
- Schmitt, F.; Gosch, L.C.; Dittmer, A.; Rothemund, M.; Mueller, T.; Schobert, R.; Biersack, B.; Volkamer, A.; Höpfner, M. Oxazole-Bridged Combretastatin a-4 Derivatives with Tethered Hydroxamic Acids: Structure⁻Activity Relations of New Inhibitors of Hdac and/or Tubulin Function. Int. J. Mol. Sci. 2019, 20, 383. [Google Scholar] [CrossRef] [Green Version]
- Khelifi, I.; Naret, T.; Renko, D.; Hamze, A.; Bernadat, G.; Bignon, J.; Lenoir, C.; Dubois, J.; Brion, J.-D.; Provot, O.; et al. Design, Synthesis and Anticancer Properties of Isocombretaquinolines as Potent Tubulin Assembly Inhibitors. Eur. J. Med. Chem. 2017, 127, 1025–1034. [Google Scholar] [CrossRef]
- Patil, R.; Patil, S.A.; Beaman, K.D.; Patil, S.A. Indole Molecules as Inhibitors of Tubulin Polymerization: Potential New Anticancer Agents, an Update (2013–2015). Future Med. Chem. 2016, 8, 1291–1316. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.A.; Patil, R.; Miller, D.D. Indole Molecules as Inhibitors of Tubulin Polymerization: Potential New Anticancer Agents. Future Med. Chem. 2012, 4, 2085–2115. [Google Scholar] [CrossRef] [PubMed]
- Brancale, A.; Silvestri, R. Indole, a Core Nucleus for Potent Inhibitors of Tubulin Polymerization. Med. Res. Rev. 2007, 27, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shuai, W.; Sun, H.; Xu, F.; Bi, Y.; Xu, J.; Ma, C.; Yao, H.; Zhu, Z.; Xu, S. Design, Synthesis and Biological Evaluation of Quinoline-Indole Derivatives as Anti-Tubulin Agents Targeting the Colchicine Binding Site. Eur. J. Med. Chem. 2019, 163, 428–442. [Google Scholar] [CrossRef]
- Naret, T.; Khelifi, I.; Provot, O.; Bignon, J.; Levaique, H.; Dubois, J.; Souce, M.; Kasselouri, A.; Deroussent, A.; Paci, A.; et al. 1,1-Diheterocyclic Ethylenes Derived from Quinaldine and Carbazole as New Tubulin-Polymerization Inhibitors: Synthesis, Metabolism, and Biological Evaluation. J. Med. Chem. 2019, 62, 1902–1916. [Google Scholar] [CrossRef]
- Niu, H.; Strecker, T.E.; Gerberich, J.L.; Campbell, J.W.; Saha, D.; Mondal, D.; Hamel, E.; Chaplin, D.J.; Mason, R.P.; Trawick, M.L.; et al. Structure Guided Design, Synthesis, and Biological Evaluation of Novel Benzosuberene Analogues as Inhibitors of Tubulin Polymerization. J. Med. Chem. 2019, 62, 5594–5615. [Google Scholar] [CrossRef]
- Mustafa, M.; Anwar, S.; Elgamal, F.; Ahmed, E.R.; Aly, O.M. Potent Combretastatin a-4 Analogs Containing 1,2,4-Triazole: Synthesis, Antiproliferative, Anti-Tubulin Activity, and Docking Study. Eur. J. Med. Chem. 2019, 183, 111697. [Google Scholar] [CrossRef]
- King, M.L.S.; Sullivan, M.M. The Similarity of the Effect of Podophyllin and Colchicine and Their Use in the Treatment of Condylomata Acuminata. Science 1946, 104, 244–245. [Google Scholar] [CrossRef]
- Kern, A.B.; Fanger, H. Podophyllin in the Treatment of Cutaneous Carcinoma. JAMA Dermatol. 1950, 62, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Canel, C.; Moraes, R.M.; Dayan, F.E.; Ferreira, D. Podophyllotoxin. Phytochemistry 2000, 54, 115–120. [Google Scholar] [CrossRef]
- Stähelin, H.F.; von Wartburg, A. The Chemical and Biological Route from Podophyllotoxin Glucoside to Etoposide: Ninth Cain Memorial Award Lecture. Cancer Res. 1991, 51, 5–15. [Google Scholar] [PubMed]
- FDA Vumon® (Teniposide Injection). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020119s010s011lbl.pdf (accessed on 24 December 2019).
- Duca, M.; Guianvarc’h, D.; Meresse, P.; Bertounesque, E.; Dauzonne, D.; Kraus-Berthier, L.; Thirot, S.; Léonce, S.; Pierré, A.; Pfeiffer, B.; et al. Synthesis and Biological Study of a New Series of 4‘-Demethylepipodophyllotoxin Derivatives. J. Med. Chem. 2005, 48, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Rituximab: A Review of Its Use in Chronic Lymphocytic Leukaemia, Low-Grade or Follicular Lymphoma and Diffuse Large B-Cell Lymphoma. Drugs 2010, 70, 1445–1476. [Google Scholar] [CrossRef] [PubMed]
- Saulnier, M.G.; Langley, D.R.; Kadow, J.F.; Senter, P.D.; Knipe, J.O.; Tun, M.M.; Vyas, D.M.; Doyle, T.W. Synthesis of Etoposide Phosphate, Bmy-40481: A Water-Soluble Clinically Active Prodrug of Etoposide. Bioorg. Med. Chem. Lett. 1994, 4, 2567–2572. [Google Scholar] [CrossRef]
- Hande, K.R. Etoposide: Four Decades of Development of a Topoisomerase Ii Inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Doré, J.; Viel, C. Antitumor Chemotherapy. Ix. Cytotoxic Activity in Cultured Tumor Cells of Chalcone Substituants and Related Compounds. Journal de Pharmacie de Belgique 1974, 29, 341. [Google Scholar]
- Peyrot, V.; Leynadier, D.; Sarrazin, M.; Briand, C.; Menendez, M.; Laynez, J.; Andreu, J.M. Mechanism of Binding of the New Antimitotic Drug Mdl 27048 to the Colchicine Site of Tubulin: Equilibrium Studies. Biochemistry 1992, 31, 11125–11132. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, W.; Wang, J.; Liu, L.; Li, L.; Yang, J.; Wang, G.; Cao, D.; Zhang, R.; Tang, M.; et al. Synthesis and Biological Evaluation of Novel Millepachine Derivatives as a New Class of Tubulin Polymerization Inhibitors. J. Med. Chem. 2014, 57, 7977–7989. [Google Scholar] [CrossRef]
- Cao, D.; Han, X.; Wang, G.; Yang, Z.; Peng, F.; Ma, L.; Zhang, R.; Ye, H.; Tang, M.; Wu, W.; et al. Synthesis and Biological Evaluation of Novel Pyranochalcone Derivatives as a New Class of Microtubule Stabilizing Agents. Eur. J. Med. Chem. 2013, 62, 579–589. [Google Scholar] [CrossRef]
- Yang, J.; Yan, W.; Yu, Y.; Wang, Y.; Yang, T.; Xue, L.; Yuan, X.; Long, C.; Liu, Z.; Chen, X.; et al. The Compound Millepachine and Its Derivatives Inhibit Tubulin Polymerization by Irreversibly Binding to the Colchicine-Binding Site in Β-Tubulin. J. Biol. Chem. 2018, 293, 9461–9472. [Google Scholar] [CrossRef] [Green Version]
- Engel, J.; Lategahn, J.; Rauh, D. Hope and Disappointment: Covalent Inhibitors to Overcome Drug Resistance in Non-Small Cell Lung Cancer. ACS Med. Chem. Lett. 2015, 7, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.-J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of Covalent Inhibitors That Can Overcome Resistance to First-Generation Fgfr Kinase Inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 4869–4877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindamulage, I.K.; Vu, H.-Y.; Karthikeyan, C.; Knockleby, J.; Lee, Y.-F.; Trivedi, P.; Lee, H. Novel Quinolone Chalcones Targeting Colchicine-Binding Pocket Kill Multidrug-Resistant Cancer Cells by Inhibiting Tubulin Activity and Mrp1 Function. Sci. Rep. 2017, 7, 10298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Greene, L.M.; Keely, N.O.; Wang, S.; Cotter, T.S.; Zisterer, D.M.; Meegan, M.J. Synthesis and Biochemical Activities of Antiproliferative Amino Acid and Phosphate Derivatives of Microtubule-Disrupting Β-Lactam Combretastatins. Eur. J. Med. Chem. 2013, 62, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, T.; Follis, A.V.; Kriwacki, R.W.; Green, D.R. Many Players in Bcl-2 Family Affairs. Trends Biochem. Sci. 2014, 39, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Xu, F.; Shuai, W.; Sun, H.; Yao, H.; Ma, C.; Xu, S.; Yao, H.; Zhu, Z.; Yang, D.-H.; et al. Discovery of Novel Quinoline–Chalcone Derivatives as Potent Antitumor Agents with Microtubule Polymerization Inhibitory Activity. J. Med. Chem. 2019, 62, 993–1013. [Google Scholar] [CrossRef]
- Hamel, E. Antimitotic Natural Products and Their Interactions with Tubulin. Med. Res. Rev. 1996, 16, 207–231. [Google Scholar] [CrossRef]
- Wipf, P.; Reeves, J.T.; Day, B.W. Chemistry and Biology of Curacin, A. Curr. Pharm. Des. 2004, 10, 1417–1437. [Google Scholar] [CrossRef]
- Lu, Y.; Li, C.-M.; Wang, Z.; Chen, J.; Mohler, M.L.; Li, W.; Dalton, J.T.; Miller, D.D. Design, Synthesis, and Sar Studies of 4-Substituted Methoxylbenzoyl-Aryl-Thiazoles Analogues as Potent and Orally Bioavailable Anticancer Agents. J. Med. Chem. 2011, 54, 4678–4693. [Google Scholar] [CrossRef] [Green Version]
- RCSB 6NNG Tubulin-Rb3_Sld-Ttl in Complex with Compound DJ95. Available online: https://www.rcsb.org/structure/6NNG (accessed on 10 November 2019).
- Arnst, K.E.; Wang, Y.; Lei, Z.-N.; Hwang, D.-J.; Kumar, G.; Ma, D.; Parke, D.N.; Chen, Q.; Yang, J.; White, S.W.; et al. Colchicine Binding Site Agent Dj95 Overcomes Drug Resistance and Exhibits Antitumor Efficacy. Mol. Pharmacol. 2019, 96, 73–89. [Google Scholar] [CrossRef]
- Matei, D.; Schilder, J.; Sutton, G.; Perkins, S.; Breen, T.; Quon, C.; Sidor, C. Activity of 2 Methoxyestradiol (Panzem® Ncd) in Advanced, Platinum-Resistant Ovarian Cancer and Primary Peritoneal Carcinomatosis: A Hoosier Oncology Group Trial. Gynecol. Oncol. 2009, 115, 90–96. [Google Scholar] [CrossRef] [PubMed]
- LaVallee, T.M.; Burke, P.A.; Swartz, G.M.; Hamel, E.; Agoston, G.E.; Shah, J.; Suwandi, L.; Hanson, A.D.; Fogler, W.E.; Sidor, C.F.; et al. Significant Antitumor Activity In Vivo Following Treatment with the Microtubule Agent Enmd-1198. Mol. Cancer Ther. 2008, 7, 1472–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquier, E.; Sinnappan, S.; Munoz, M.A.; Kavallaris, M. Enmd-1198, a New Analogue of 2-Methoxyestradiol, Displays Both Antiangiogenic and Vascular-Disrupting Properties. Mol. Cancer Ther. 2010, 9, 1408–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteve, M.-A.; Honore, S.; McKay, N.; Bachmann, F.; Lane, H.; Braguer, D. Abstract 1977: Bal27862: A Unique Microtubule-Targeted Drug That Suppresses Microtubule Dynamics, Severs Microtubules, and Overcomes Bcl-2- and Tubulin Subtype-Related Drug Resistance. Cancer Res. 2010, 70, 1977. [Google Scholar]
- Zheng, S.; Zhong, Q.; Mottamal, M.; Zhang, Q.; Zhang, C.; LeMelle, E.; McFerrin, H.; Wang, G. Design, Synthesis, and Biological Evaluation of Novel Pyridine-Bridged Analogues of Combretastatin-A4 as Anticancer Agents. J. Med. Chem. 2014, 57, 3369–3381. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-B.; Gong, J.-H.; Liu, X.-J.; Wu, S.-Y.; Li, Y.; Xu, X.-D.; Shang, B.-Y.; Zhou, J.-M.; Zhu, Z.-L.; Si, S.-Y.; et al. A Novel Nitrobenzoate Microtubule Inhibitor That Overcomes Multidrug Resistance Exhibits Antitumor Activity. Sci. Rep. 2016, 6, 31472. [Google Scholar] [CrossRef] [Green Version]
- Bai, Z.; Gao, M.; Zhang, H.; Guan, Q.; Xu, J.; Li, Y.; Qi, H.; Li, Z.; Zuo, D.; Zhang, W.; et al. BZML, a Novel Colchicine Binding Site Inhibitor, Overcomes Multidrug Resistance in A549/Taxol Cells by Inhibiting P-Gp Function and Inducing Mitotic Catastrophe. Cancer Lett. 2017, 402, 81–92. [Google Scholar] [CrossRef]
- Gourdeau, H.; Leblond, L.; Hamelin, B.; Desputeau, C.; Dong, K.; Kianicka, I.; Custeau, D.; Boudreau, C.; Geerts, L.; Cai, S.-X.; et al. Antivascular and Antitumor Evaluation of 2-Amino-4-(3-Bromo-4,5-Dimethoxy-Phenyl)-3-Cyano-4-Chromenes, a Novel Series of Anticancer Agents. Mol. Cancer Ther. 2004, 3, 1375–1384. [Google Scholar]
- Liu, Y.; Yang, D.; Hong, Z.; Guo, S.; Liu, M.; Zuo, D.; Ge, D.; Qin, M.; Sun, D. Synthesis and Biological Evaluation of 4,6-Diphenyl-2-(1h-Pyrrol-1-Yl)Nicotinonitrile Analogues of Crolibulin and Combretastatin a-4. Eur. J. Med. Chem. 2018, 146, 185–193. [Google Scholar] [CrossRef]
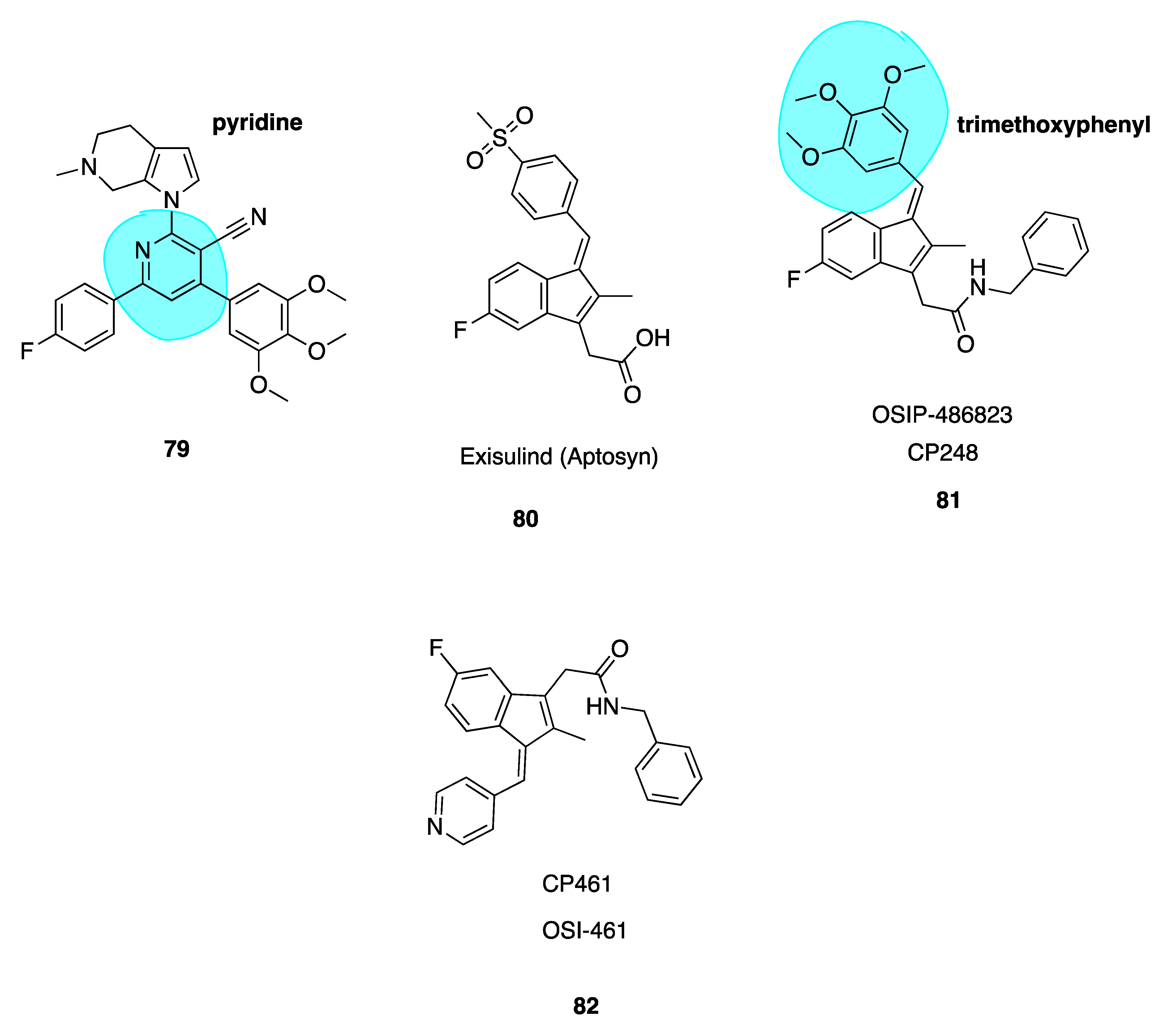
- Xiao, D.; Deguchi, A.; Gundersen, G.G.; Oehlen, B.; Arnold, L.; Weinstein, I.B. The Sulindac Derivatives Osi-461, Osip486823, and Osip487703 Arrest Colon Cancer Cells in Mitosis by Causing Microtubule Depolymerization. Mol. Cancer Ther. 2006, 5, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Haanen, C. Sulindac and Its Derivatives: A Novel Class of Anticancer Agents. Curr. Opin. Investig. Drugs 2001, 2, 677–683. [Google Scholar] [PubMed]
- Clinicaltrials Gov. 6 Studies Found For: CP 461/Osi-461. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=CP+461&cntry=&state=&city=&dist= (accessed on 19 September 2019).
- Pettit, G.R.; Lippert, J.W. Antineoplastic Agents 429. Syntheses of the Combretastatin a-1 and Combretastatin B-1 Prodrugs. Anti-Cancer Drug Des. 2000, 15, 203–216. [Google Scholar]
- Tozer, G.M.; Kanthou, C.; Parkins, C.S.; Hill, S.A. The Biology of the Combretastatins as Tumour Vascular Targeting Agents. Int. J. Exp. Pathol. 2002, 83, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, I.G.; Loadman, P.M.; Swaine, D.J.; Anthoney, D.A.; Pettit, G.R.; Lippert, J.W.; Shnyder, S.D.; Cooper, P.A.; Bibby, M.C. Comparative Preclinical Pharmacokinetic and Metabolic Studies of the Combretastatin Prodrugs Combretastatin A4 Phosphate and A1 Phosphate. Clin. Cancer Res. 2004, 10, 1446–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, C.J.; Nagaiah, G.; Fu, P.; Wasman, J.K.; Cooney, M.M.; Savvides, P.S.; Bokar, J.A.; Dowlati, A.; Wang, D.; Agarwala, S.S.; et al. A Phase Ii Trial of Fosbretabulin in Advanced Anaplastic Thyroid Carcinoma and Correlation of Baseline Serum-Soluble Intracellular Adhesion Molecule-1 with Outcome. Thyroid 2009, 19, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griggs, M.J.; Hesketh, R. Targeting Tumour Vasculature: The Development of Combretastatin A4. Lancet Oncol. 2001, 2, 82–87. [Google Scholar] [CrossRef]
- EMA. Public Summary of Opinion on Orphan Designation; Fosbretabulin Tromethamine for the Treatment of Gastro-Entero-Pancreatic Neuroendocrine Tumours; EMA, Ed.; EMA: London, UK, 2016; pp. 1–5. [Google Scholar]
- EMA. Public Summary of Opinion on Orphan Designation; Fosbretabulin Tromethamine for the Treatment of Ovarian Cancer; EMA, Ed.; EMA: London, UK, 2013; pp. 1–4. [Google Scholar]
- Therepeutics, M. Developing Innovative Therapeutic Approaches to Treat Cancer; Oxi4503. Available online: http://www.mateon.com/product-development/taboxi4503/ (accessed on 28 October 2019).
- Kim, T.J.; Ravoori, M.; Landen, C.N.; Kamat, A.A.; Han, L.Y.; Lu, C.; Lin, Y.G.; Merritt, W.M.; Jennings, N.; Spannuth, W.A.; et al. Antitumor and Antivascular Effects of Ave8062 in Ovarian Carcinoma. Cancer Res. 2007, 67, 9337–9345. [Google Scholar] [CrossRef] [Green Version]
- Morinaga, Y.; Suga, Y.; Ehara, S.; Harada, K.; Nihei, Y.; Suzuki, M. Combination Effect of Ac-7700, a Novel Combretastatin a-4 Derivative, and Cisplatin against Murine and Human Tumors in Vivo. Cancer Sci. 2003, 94, 200–204. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Pápai, Z.; Tolcher, A.W.; Italiano, A.; Cupissol, D.; López-Pousa, A.; Chawla, S.P.; Bompas, E.; Babovic, N.; Penel, N.; et al. Ombrabulin Plus Cisplatin Versus Placebo Plus Cisplatin in Patients with Advanced Soft-Tissue Sarcomas after Failure of Anthracycline and Ifosfamide Chemotherapy: A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2015, 16, 531–540. [Google Scholar] [CrossRef]
- Munshi, N.; Jeay, S.; Li, Y.; Chen, C.-R.; France, D.S.; Ashwell, M.A.; Hill, J.; Moussa, M.M.; Leggett, D.S.; Li, C.J. Arq 197, a Novel and Selective Inhibitor of the Human C-Met Receptor Tyrosine Kinase with Antitumor Activity. Mol. Cancer Ther. 2010, 9, 1544–1553. [Google Scholar] [CrossRef] [Green Version]
- Graveel, C.R.; Tolbert, D.; Vande Woude, G.F. Met: A Critical Player in Tumorigenesis and Therapeutic Target. Cold Spring Harb. Perspect. Biol. 2013, 5, a009209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, A.; Katayama, R.; Oh-hara, T.; Sato, S.; Okuno, Y.; Fujita, N. Tivantinib (Arq 197) Exhibits Antitumor Activity by Directly Interacting with Tubulin and Overcomes Abc Transporter–Mediated Drug Resistance. Mol. Cancer Ther. 2014, 13, 2978–2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clincialtrials Gov. Tivantinib: Phase 3. Available online: https://clinicaltrials.gov/ct2/results?term=tivantinib&age_v=&gndr=&type=&rslt=&phase=2&Search=Apply (accessed on 25 November 2019).
- Cabibbo, G.; Enea, M.; Attanasio, M.; Bruix, J.; Craxi, A.; Camma, C. A Meta-Analysis of Survival Rates of Untreated Patients in Randomized Clinical Trials of Hepatocellular Carcinoma. Hepatology 2010, 51, 1274–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (Checkmate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Rota Caremoli, E.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for Second-Line Treatment of Met-High, Advanced Hepatocellular Carcinoma (Metiv-Hcc): A Final Analysis of a Phase 3, Randomised, Placebo-Controlled Study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- KyowaKirin Kyowa Hakko Kirin Announces Discontinuation for Developing Arq 197 (Tivantinib). Available online: https://www.kyowakirin.com/media_center/news_releases/2017/e20171006_01.html (accessed on 25 November 2019).
- Prior, I.; Lewis, P.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Beyondspring’s Novel Study 103 Phase 3 Design in Nsclc Presented at 2019 Iaslc World Conference on Lung Cancer. Available online: https://www.globenewswire.com/ (accessed on 25 November 2019).
- An Open-Label Study of Intravenous Bal101553 in Adult Patients with Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01397929 (accessed on 20 November 2019).
- Calvert, A.H.; Gonzalez, M.; Ganguli, S.; Ng, M.; Benafif, S.; Capelan, M.; Goldstein, R.; Shah, K.; Jarvis, C.; Flynn, M.; et al. A First-in-Human (Fih) Dose-Escalation Study of the Safety, Pharmacokinetics (Pk), and Pharmacodynamics (Pd) of Intravenous Bal101553, a Novel Microtubule Inhibitor, in Adult Patients with Advanced Solid Tumors. J. Clin. Oncol. 2013, 31, 2566. [Google Scholar]
- Kolb, E.A.; Gorlick, R.; Keir, S.T.; Maris, J.M.; Kang, M.H.; Reynolds, C.P.; Lock, R.B.; Carol, H.; Wu, J.; Kurmasheva, R.T.; et al. Initial Testing (Stage 1) of Bal101553, a Novel Tubulin Binding Agent, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2015, 62, 1106–1109. [Google Scholar] [CrossRef] [Green Version]
- Lisavanbulin and Radiation Therapy in Treating Patients with Newly Diagnosed Glioblastoma. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCI-2016-01847&r=1 (accessed on 18 December 2019).
- Clincialtrials Gov. A Phase 1/11 Trial of Crolibulin (Epc2407) Plus Cisplatin in Adults with Solid Tumors with a Focus on Anaplastic Thyroid Cancer (Atc). Available online: https://clinicaltrials.gov/ct2/show/NCT01240590 (accessed on 18 December 2019).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Section | Structural Features | Stage in Clinical Trials and Disease Treated | Company Developing Drug |
|---|---|---|---|---|
| ENMD1198 (74) | Section 5.5 | 2-Methoxyestradiol derivative | Phase I refractory solid tumours | Casi pharmaceuticals (no data published in recent years) |
| CP461/OSI-461 (82) | Section 5.11 | Derivative of exisulind | Phase II
| Astellas |
| Tivantinib (86) | Section 6.2.1 | Heterocyclic fused ring system | Phase II
| Kyowa Hakko Kirin Co. Ltd. (development discontinued in 2018 following multiple poor clinical trial outcomes) |
| Plinabulin (75b) | Section 6.2.2 | Piperazinedione structure | Phase III stage IIIb/IV NSCLC (in combination with docetaxel) | Beyond Spring Inc. |
| Lisavanbulin (BAL101553) (87) | Section 6.2.3 | Lysine prodrug of Avanbulin | Phase I/IIa advanced solid tumours, refractory to standard therapy | Basilea Pharmaceutica |
| Crolibulin (78) | Section 6.2.4 | Chromene derivative | Phase I/II Clinical trials Anaplastic Thyroid Cancer | Immune Pharmaceuticals Inc; National Cancer Institute (USA) (clinical trial progression limited due to recruitment issues) |
| Drug Name | Section | Structural Features | Stage in Clinical Trials and Disease Treated | Company Developing Drug |
|---|---|---|---|---|
| Fosbretabulin (83) | Section 6.1.1 | Phosphate prodrug of CA-4 | Phase I/II/III
| Diamond Biopharm Limited |
| Oxi4503 (84) | Section 6.1.2 | DiPhosphate prodrug of CA-1 | Phase Ib/II
| Mateon Therapeutics (previously Oxigene) |
| Ombrabulin/AVE8062 (85) | Section 6.1.3 | Serine prodrug of CA-4 | Phase II
| Sanofi Aventis |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. https://doi.org/10.3390/ph13010008
McLoughlin EC, O’Boyle NM. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals. 2020; 13(1):8. https://doi.org/10.3390/ph13010008
Chicago/Turabian StyleMcLoughlin, Eavan C., and Niamh M. O’Boyle. 2020. "Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review" Pharmaceuticals 13, no. 1: 8. https://doi.org/10.3390/ph13010008
APA StyleMcLoughlin, E. C., & O’Boyle, N. M. (2020). Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals, 13(1), 8. https://doi.org/10.3390/ph13010008