A Photocleavable Contrast Agent for Light-Responsive MRI

 and
and
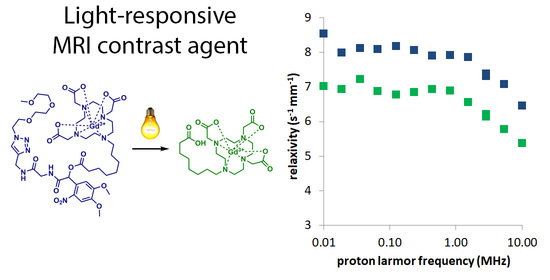
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Photochemical Analysis
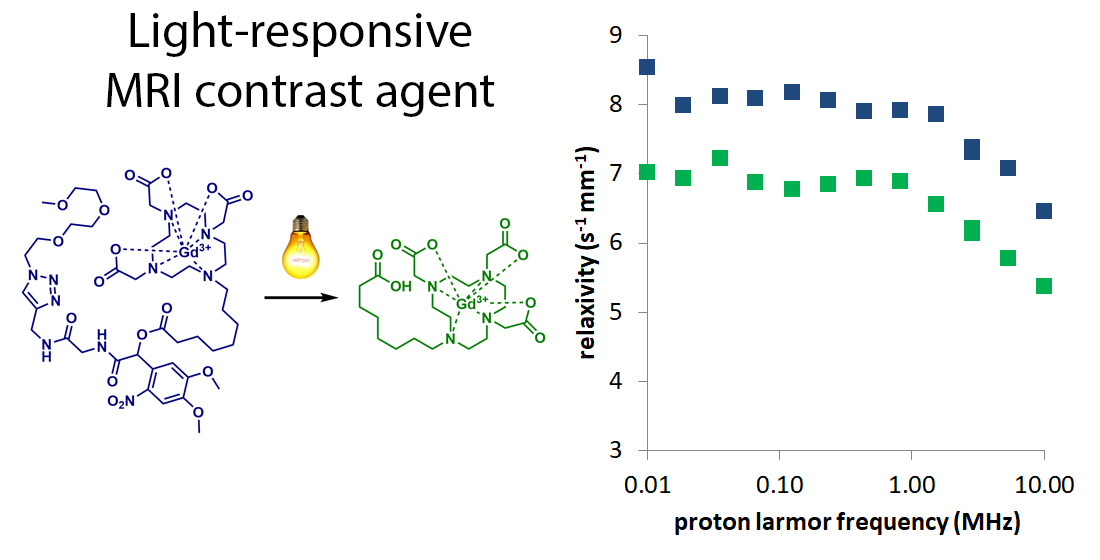
2.2. Fast Field Cycling Relaxometric Analysis
2.3. Relaxometric Measurements on Clinical 1.5T and 3.0T Systems
2.4. Assessment of Free Gadolinium(III) Ions
3. Discussion
4. Materials and Methods
4.1. General Information
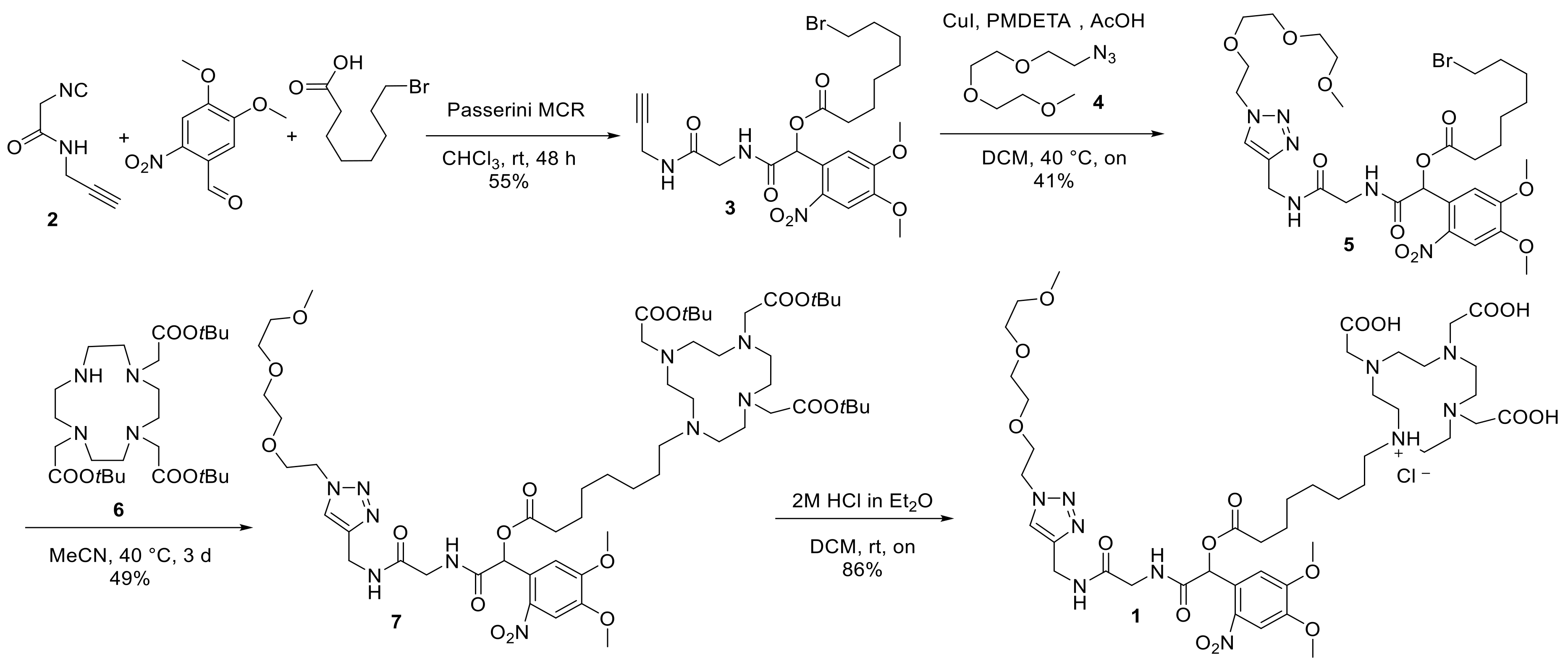
4.2. Synthetic Procedures and Spectroscopic Data
4.3. Quantum Yield Determination
4.4. FFC Relaxometry
4.5. Measurements on 1.5T and 3.0T Clinical MRI Systems
4.6. Determination of Free Gadolinium(III) Concentration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weissleder, R.; Ntziachristos, V. Shedding light onto live molecular targets. Nat. Med. 2003, 9, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.A.; Evans, D.H.; Abrahamse, H. Photodynamic therapy (PDT): A short review on cellular mechanisms and cancer research applications for PDT. J. Photochem. Photobiol. B 2009, 96, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one—Photosensitizers, photochemistry and cellular localization. Photodiagn. Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Woodgate, P.; Jardine, L.A. Neonatal jaundice: Phototherapy. BMJ Clin. Evid. 2015, 2015, 0319. [Google Scholar] [PubMed]
- Hüll, K.; Morstein, J.; Trauner, D. In Vivo Photopharmacology. Chem. Rev. 2018, 118, 10710–10747. [Google Scholar] [CrossRef] [PubMed]
- Velema, W.A.; Szymański, W.; Feringa, B.L. Photopharmacology: Beyond proof of principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenno, L.; Yizhar, O.; Deisseroth, K. The Development and Application of Optogenetics. Annu. Rev. Neurosci. 2011, 34, 389–412. [Google Scholar] [CrossRef]
- Reeßing, F.; Szymański, W. Beyond Photodynamic Therapy: Light-Activated Cancer Chemotherapy. Curr. Med. Chem. 2017, 24, 4905–4950. [Google Scholar] [CrossRef]
- Koch, M.; Symvoulidis, P.; Ntziachristos, V. Tackling standardization in fluorescence molecular imaging. Nat. Photonics 2018, 12, 505–515. [Google Scholar] [CrossRef]
- Steinberg, I.; Huland, D.M.; Vermesh, O.; Frostig, H.E.; Tummers, W.S.; Gambhir, S.S. Photoacoustic clinical imaging. Photoacoustics 2019, 14, 77–98. [Google Scholar] [CrossRef]
- Kobayashi, H.; Ogawa, M.; Alford, R.; Choyke, P.L.; Urano, Y. New strategies for fluorescent probe design in medical diagnostic imaging. Chem. Rev. 2010, 110, 2620–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogue, B.W. Perspective review of what is needed for molecular-specific fluorescence-guided surgery. J. Biomed. Opt. 2018, 23, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linssen, M.D.; ter Weele, E.J.; Allersma, D.P.; Lub-de Hooge, M.N.; van Dam, G.M.; Jorritsma-Smit, A.; Nagengast, W.B. Roadmap for the development and clinical translation of optical tracers cetuximab-800CW and trastuzumab-800CW. J. Nucl. Med. 2019, 60, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Antaris, A.L.; Chen, H.; Cheng, K.; Sun, Y.; Hong, G.; Qu, C.; Diao, S.; Deng, Z.; Hu, X.; Zhang, B.; et al. A small-molecule dye for NIR-II imaging. Nat. Mater. 2016, 15, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Gioux, S.; Choi, H.S.; Frangioni, J.V. Image-Guided Surgery Using Invisible Near-Infrared Light: Fundamentals of Clinical Translation. Mol. Imaging 2010, 9, 237–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badr, C.E.; Tannous, B.A. Bioluminescence imaging: Progress and applications. Trends Biotechnol. 2011, 29, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadikot, R.T.; Blackwell, T.S. Bioluminescence imaging. Proc. Am. Thorac. Soc. 2005, 2, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Barger, N.; Litovco, P.; Li, X.; Habib, M.; Daniel, R. Synthetic metabolic computation in a bioluminescence-sensing system. Nucleic Acids Res. 2019, 47, 10464–10474. [Google Scholar] [CrossRef]
- Close, D.M.; Patterson, S.S.; Ripp, S.; Baek, S.J.; Sanseverino, J.; Sayler, G.S. Autonomous Bioluminescent Expression of the Bacterial Luciferase Gene Cassette (lux) in a Mammalian Cell Line. PLoS ONE 2010, 5, e12441. [Google Scholar] [CrossRef]
- Xu, T.; Close, D.; Handagama, W.; Marr, E.; Sayler, G.; Ripp, S. The expanding toolbox of in vivo bioluminescent imaging. Front. Oncol. 2016, 6, 150. [Google Scholar] [CrossRef] [Green Version]
- Hao, D.; Ai, T.; Goerner, F.; Hu, X.; Runge, V.M.; Tweedle, M. MRI contrast agents: Basic chemistry and safety. J. Magn. Res. Imaging 2012, 36, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Dommaschk, M.; Gröbner, J.; Wellm, V.; Hövener, J.B.; Riedel, C.; Herges, R. Dendronised Ni(II) porphyrins as photoswitchable contrast agents for MRI. Phys. Chem. Chem. Phys. 2019, 21, 24296–24299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataramani, S.; Jana, U.; Dommaschk, M.; Sönnichsen, F.D.; Tuczek, F.; Herges, R. Magnetic bistability of molecules in homogeneous solution at room temperature. Science 2011, 331, 445–448. [Google Scholar] [CrossRef]
- Heitmann, G.; Schütt, C.; Gröbner, J.; Huber, L.; Herges, R. Azoimidazole functionalized Ni-porphyrins for molecular spin switching and light responsive MRI contrast agents. Dalton Trans. 2016, 45, 11407–11412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruttwig, K.; Yankelevich, D.R.; Brueggemann, C.; Tu, C.; L’Etoile, N.; Knoesen, A.; Louie, A.Y. Reversible Low-Light Induced Photoswitching of Crowned Spiropyran-DO3A Complexed with Gadolinium(III) Ions. Molecules 2012, 17, 6605–6624. [Google Scholar] [CrossRef] [Green Version]
- Reeßing, F.; Stuart, M.C.A.; Samplonius, D.F.; Dierckx, R.A.J.O.; Feringa, B.L.; Helfrich, W.; Szymański, W. A light-responsive liposomal agent for MRI contrast enhancement and monitoring of cargo delivery. Chem. Commun. 2019, 55, 10784–10787. [Google Scholar] [CrossRef] [PubMed]
- Szymański, W.; Velema, W.A.; Feringa, B.L. Photocaging of Carboxylic Acids: A Modular Approach. Angew. Chem. Int. Ed. 2014, 53, 8682–8686. [Google Scholar] [CrossRef]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable protecting groups in chemistry and biology: Reaction mechanisms and efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef]
- Šolomek, T.; Mercier, S.; Bally, T.; Bochet, C.G. Photolysis of ortho-nitrobenzylic derivatives: The importance of the leaving group. Photochem. Photobiol. Sci. 2012, 11, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Gautier, A.; Nguyen, D.P.; Lusic, H.; An, W.; Deiters, A.; Chin, J.W. Genetically encoded photocontrol of protein localization in mammalian cells. J. Am. Chem. Soc. 2010, 132, 4086–4088. [Google Scholar] [CrossRef]
- Schelkle, K.M.; Griesbaum, T.; Ollech, D.; Becht, S.; Buckup, T.; Hamburger, M.; Wombacher, R. Light-Induced Protein Dimerization by One- and Two-Photon Activation of Gibberellic Acid Derivatives in Living Cells. Angew. Chem. Int. Ed. 2015, 54, 2825–2829. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, B.; Horbert, R.; Döbber, A.; Kuhl, L.; Peifer, C. Photoactivatable Caged Prodrugs of VEGFR-2 Kinase Inhibitors. Molecules 2016, 21, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, C.L.; Grøtli, M.; Andréasson, J. On-Command Regulation of Kinase Activity using Photonic Stimuli. ChemPhotoChem 2019, 3, 318–326. [Google Scholar] [CrossRef] [Green Version]
- Kolarski, D.; Sugiyama, A.; Breton, G.; Rakers, C.; Ono, D.; Schulte, A.; Tama, F.; Itami, K.; Szymański, W.; Hirota, T.; et al. Controlling the Circadian Clock with High Temporal Resolution through Photodosing. J. Am. Chem. Soc. 2019, 141, 15784–15791. [Google Scholar] [CrossRef] [Green Version]
- Dcona, M.M.; Mitra, D.; Goehe, R.W.; Gewirtz, D.A.; Lebman, D.A.; Hartman, M.C.T. Photocaged permeability: A new strategy for controlled drug release. Chem. Commun. 2012, 48, 4755–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, M.; Kiefer, G.E.; Bott, S.; Castillo-Muzquiz, A.; Eshelbrenner, C.; Michaudet, L.; McMillan, K.; Mudigunda, S.D.K.; Ogrin, D.; Tircsó, G.; et al. Synthesis, relaxometric and photophysical properties of a new pH-responsive MRI contrast agent: The effect of other ligating groups on dissociation of a p-nitrophenolic pendant arm. J. Am. Chem. Soc. 2004, 126, 9248–9256. [Google Scholar] [CrossRef] [Green Version]
- Major, J.L.; Parigi, G.; Luchinat, C.; Meade, T.J. The synthesis and in vitro testing of a zinc-activated MRI contrast agent. Proc. Natl. Acad. Sci. USA 2007, 104, 13881–13886. [Google Scholar] [CrossRef] [Green Version]
- Moats, R.A.; Fraser, S.E.; Meade, T.J. A “Smart” Magnetic Resonance Imaging Agent That Reports on Specific Enzymatic Activity. Angew. Chem. Int. Ed. 1997, 36, 726–728. [Google Scholar] [CrossRef]
- Oukhatar, F.; Même, S.; Même, W.; Szeremeta, F.; Logothetis, N.K.; Angelovski, G.; Tóth, É. MRI sensing of neurotransmitters with a crown ether appended Gd3+ complex. ACS Chem. Neurosci. 2015, 6, 219–225. [Google Scholar] [CrossRef]
- Botta, M. Second Coordination Sphere Water Molecules and Relaxivity of Gadolinium(III) Complexes: Implications for MRI Contrast Agents. Eur. J. Inorg. Chem. 2000, 2000, 399–407. [Google Scholar] [CrossRef]
- Li, H.; Meade, T.J. Molecular Magnetic Resonance Imaging with Gd(III)-Based Contrast Agents: Challenges and Key Advances. J. Am. Chem. Soc. 2019, 141, 17025–17041. [Google Scholar] [CrossRef] [PubMed]
- Aime, S.; Botta, M.; Esteban-Gómez, D.; Platas-Iglesias, C. Characterisation of magnetic resonance imaging (MRI) contrast agents using NMR relaxometry. Mol. Phys. 2019, 117, 898–909. [Google Scholar] [CrossRef]
- Dömling, A.; Beck, B.; Fuchs, T.; Yazbak, A. Parallel synthesis of arrays of amino-acid-derived isocyanoamides useful as starting materials in IMCR. J. Comb. Chem. 2006, 8, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Wang, X.; Zhang, Q.; Luo, S.; Zhao, J.; Hu, Y. Acid-base jointly promoted copper(I)-catalyzed azide-alkyne cycloaddition. J. Org. Chem. 2011, 76, 6832–6836. [Google Scholar] [CrossRef]
- Jagadish, B.; Brickert-Albrecht, G.L.; Nichol, G.S.; Mash, E.A.; Raghunand, N. On the synthesis of 1,4,7-tris(tert-butoxycarbonylmethyl)-1,4,7,10- tetraazacyclododecane. Tetrahedron Lett. 2011, 52, 2058–2061. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.; Balabanova, A.; Frakes, S.A.; Bargmann, A.; Grimm, J.; Koch, T.H.; Yin, H. Photoactivatable Prodrug of Doxazolidine Targeting Exosomes. J. Med. Chem. 2019, 62, 1959–1970. [Google Scholar] [CrossRef]
- Berroy, P.; Viriot, M.L.; Carré, M.C. Photolabile group for 5′-OH protection of nucleosides: Synthesis and photodeprotection rate. Sens. Actuators B 2001, 74, 186–189. [Google Scholar] [CrossRef]
- Aujard, I.; Benbrahim, C.; Gouget, M.; Ruel, O.; Baudin, J.-B.; Neveu, P.; Jullien, L. o-Nitrobenzyl Photolabile Protecting Groups with Red-Shifted Absorption: Syntheses and Uncaging Cross-Sections for One- and Two-Photon Excitation. Chem. Eur. J. 2006, 12, 6865–6879. [Google Scholar] [CrossRef]
- Roher, M.; Bauer, H.; Mintorovitch, J.; Requard, M.; Weinmann, H.-J. Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Investig. Radiol. 2005, 40, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Barge, A.; Cravotto, G.; Gianolio, E.; Fedeli, F. How to determine free Gd and free ligand in solution of Gd chelates. A technical note. Contrast Media Mol. Imaging 2006, 1, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Slanina, T.; Shrestha, P.; Palao, E.; Kand, D.; Peterson, J.A.; Dutton, A.S.; Rubinstein, N.; Weinstain, R.; Winter, A.H.; Klán, P. In Search of the Perfect Photocage: Structure-Reactivity Relationships in meso-Methyl BODIPY Photoremovable Protecting Groups. J. Am. Chem. Soc. 2017, 139, 15168–15175. [Google Scholar] [CrossRef] [PubMed]
- Sitkowska, K.; Feringa, B.L.; Szymański, W. Green-Light-Sensitive BODIPY Photoprotecting Groups for Amines. J. Org. Chem. 2018, 83, 1819–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, H.J.; Braslavsky, S.E.; Schmidt, R. Chemical actinometry (IUPAC technical report). Pure Appl. Chem. 2004, 76, 2105–2146. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reeßing, F.; Huijsse, S.E.M.; Dierckx, R.A.J.O.; Feringa, B.L.; Borra, R.J.H.; Szymański, W. A Photocleavable Contrast Agent for Light-Responsive MRI. Pharmaceuticals 2020, 13, 296. https://doi.org/10.3390/ph13100296
Reeßing F, Huijsse SEM, Dierckx RAJO, Feringa BL, Borra RJH, Szymański W. A Photocleavable Contrast Agent for Light-Responsive MRI. Pharmaceuticals. 2020; 13(10):296. https://doi.org/10.3390/ph13100296
Chicago/Turabian StyleReeßing, Friederike, Sèvrin E. M. Huijsse, Rudi A. J. O. Dierckx, Ben L. Feringa, Ronald J.H. Borra, and Wiktor Szymański. 2020. "A Photocleavable Contrast Agent for Light-Responsive MRI" Pharmaceuticals 13, no. 10: 296. https://doi.org/10.3390/ph13100296
APA StyleReeßing, F., Huijsse, S. E. M., Dierckx, R. A. J. O., Feringa, B. L., Borra, R. J. H., & Szymański, W. (2020). A Photocleavable Contrast Agent for Light-Responsive MRI. Pharmaceuticals, 13(10), 296. https://doi.org/10.3390/ph13100296