Quinolizidine-Derived Lucanthone and Amitriptyline Analogues Endowed with Potent Antileishmanial Activity

 ,
,  ,
,  , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
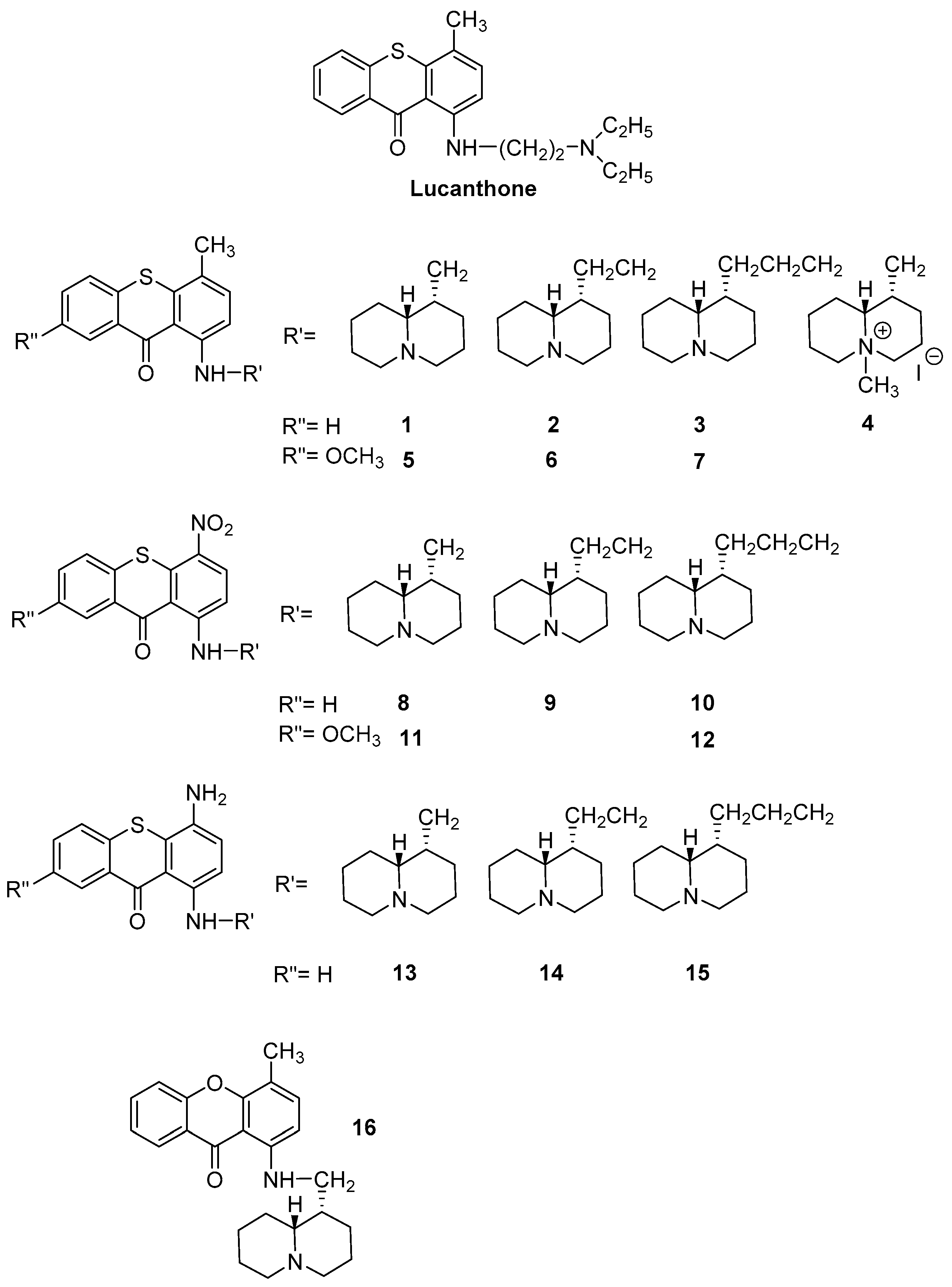
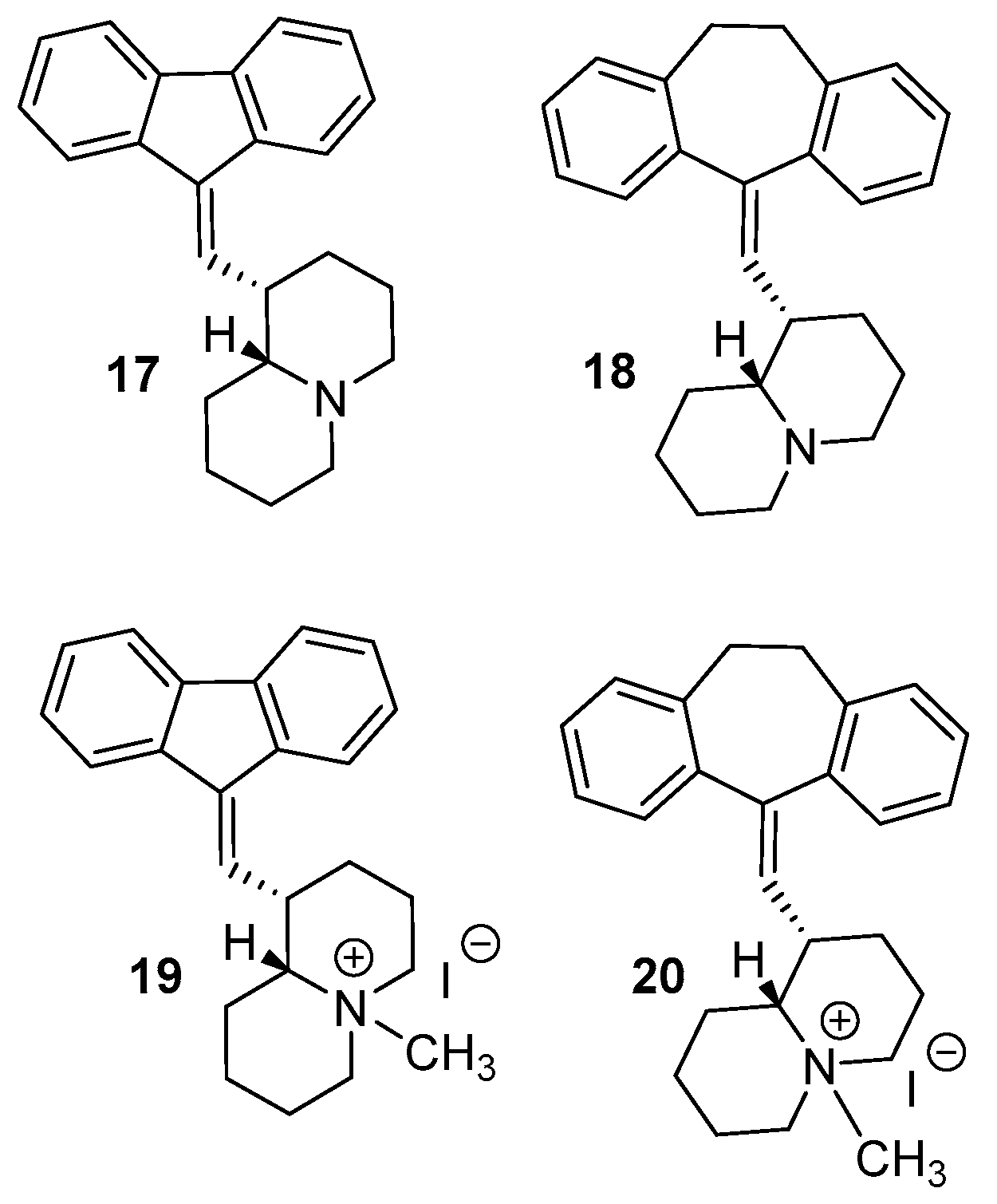
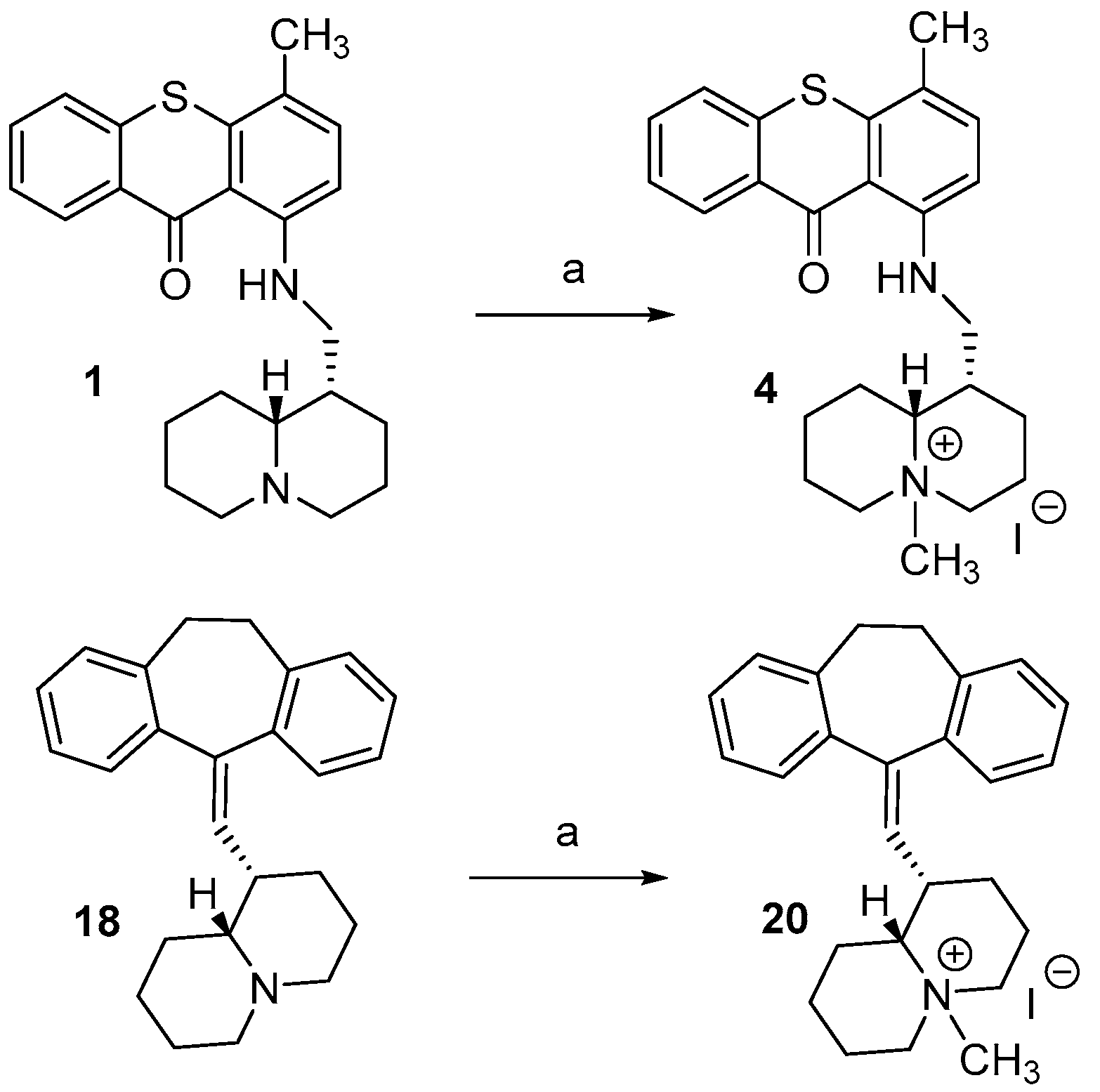
2.1. Chemistry
2.2. Biological Studies and SAR
2.2.1. Antileishmanial Activity Against L. tropica and L. infantum Promastigotes
2.2.2. Cytotoxicity
2.2.3. Antileishmanial Activity against L. Infantum Amastigotes
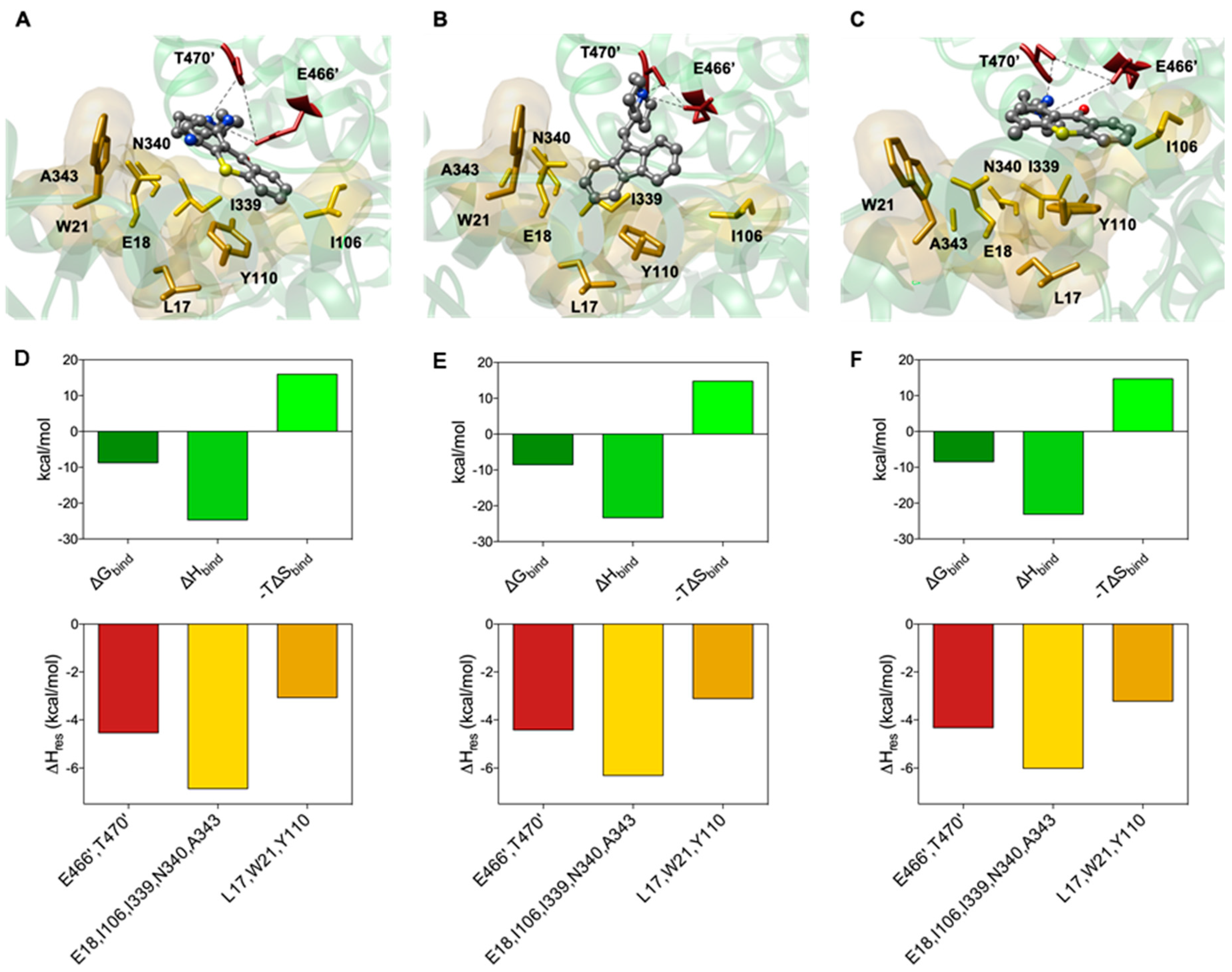
2.3. Molecular Modelling Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Procedure for the Synthesis of Quaternary Ammonium Iodides (4 and 20)
3.2. Biological Tests
3.2.1. Antileishmanial Activity
3.2.2. In Vitro Intracellular Amastigote Susceptibility Assays
3.2.3. Cell Cytotoxicity Assays
3.3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Factsheet. Available online: https://www.afro.who.int/health-topics/Leishmaniasis (accessed on 2 March 2020).
- Maslov, D.A.; Opperdoes, F.R.; Kostygov, A.Y.; Hashimi, H.; Lukeš, J.; Yurchenko, V. Recent advances in trypanosomatid research: Genome organization, expression, metabolism, taxonomy and evolution. Parasitology 2019, 146, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Avila-Levy, C.M.; Boucinha, C.; Kostygov, A.; Santos, H.L.; Morelli, K.A.; Grybchuk-Ieremenko, A.; Duval, L.; Votýpka, J.; Yurchenko, V.; Grellier, P.; et al. Exploring the environmental diversity of kinetoplastid flagellates in the high-throughput DNA sequencing era. Mem. Inst. Oswaldo Cruz 2015, 110, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Sangshetti, J.N.; Kalam Khan, F.A.; Kulkarni, A.A.; Arote, R.; Patilc, R.H. Antileishmanial drug discovery: Comprehensive review of the last 10 years. RSC Adv. 2015, 5, 32376–32415. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, M.S.; Hayat, F.; Shin, D. Advances in the discovery of novel antiprotozoal agents. Molecules 2019, 24, 3886. [Google Scholar] [CrossRef] [Green Version]
- Razzaghi-Asl, N.; Sepehri, S.; Ebadi, A.; Karami, P.; Nejatkhah, N.; Johari-Ahar, M. Insights into the current status of privileged N-heterocycles as antileishmanial agents. Mol. Divers. 2020, 24, 525–569. [Google Scholar] [CrossRef]
- Andrade-Neto, V.V.; Ferreira Cunha-Junior, E.; Dos Santos Faioes, V.; Pereira, T.M.; Silva, R.L.; Leon, L.L.; Torres-Santos, E.C. Leishmaniasis treatment: Update of possibilities for drug repurposing. Front. Biosci. 2018, 23, 967–999. [Google Scholar]
- Bekhit, A.A.; El-Agroudy, E.; Helmy, A.; Ibrahim, T.M.; Shavandi, A.; Bekhit, A.E.A. Leishmania treatment and prevention: Natural and synthesized drugs. Eur. J. Med. Chem. 2018, 160, 229–244. [Google Scholar] [CrossRef] [Green Version]
- Mowbray, C.E. Drug Discovery for Leishmaniasis; Rivas, L., Gil, C., Eds.; Royal Society of Chemistry: London, UK, 2018; pp. 26–36. [Google Scholar]
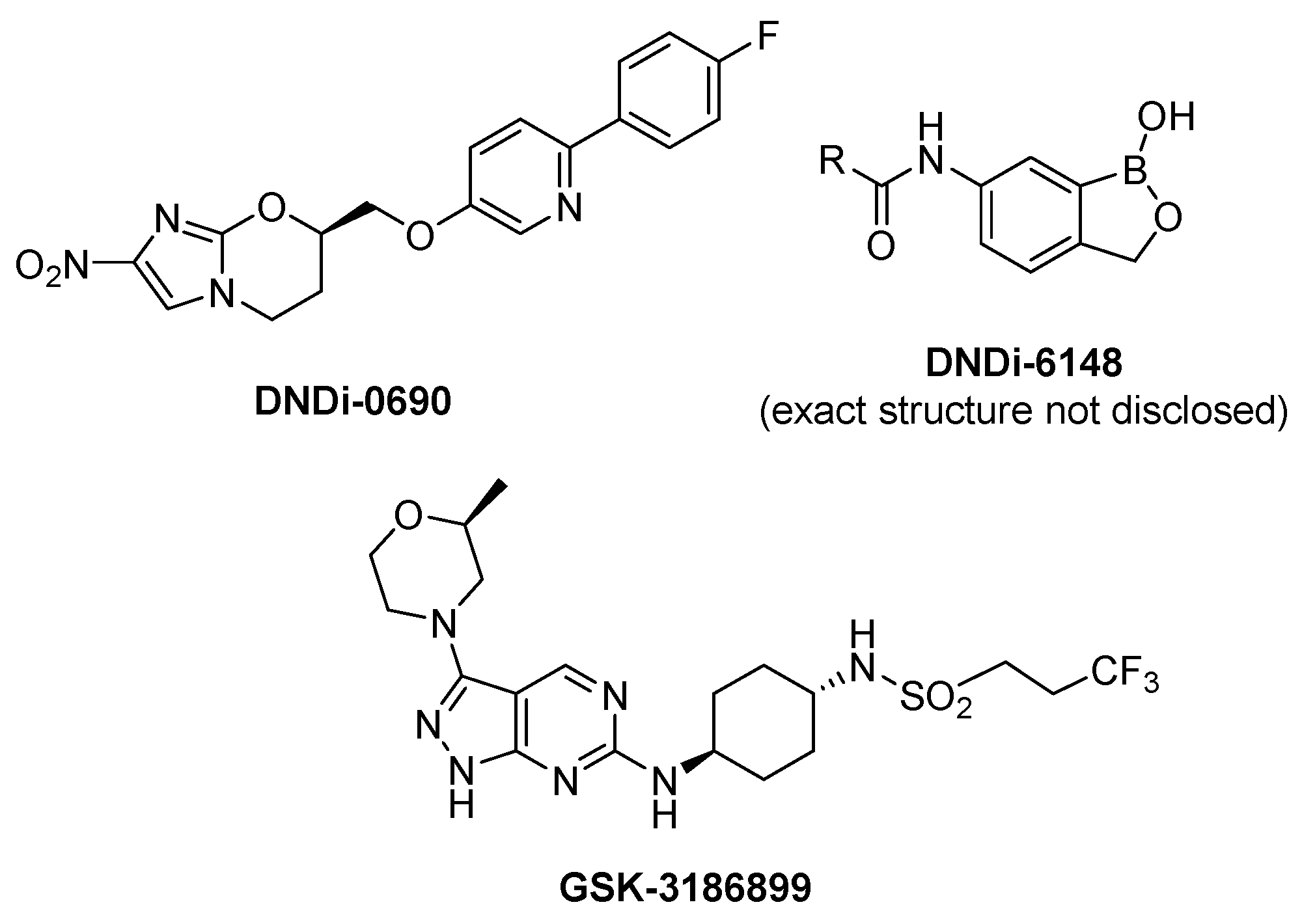
- Wijnant, G.-J.; Croft, S.L.; de la Flor, R.; Alavijeh, M.; Yardley, V.; Braillard, S.; Mowbray, C.; van Bocxlaer, K. Pharmacokinetics and pharmacodynamics of the nitroimidazole DNDI-0690 in mouse models of cutaneous Leishmaniasis. Antimicrob. Agents Chemother. 2019, 63, e00829-19. [Google Scholar] [CrossRef] [Green Version]
- van den Kerkhof, M.; Mabille, D.; Chatelain, E.; Mowbray, C.E.; Braillard, S.; Hendrickx, S.; Maes, L.; Caljon, G. In vitro and in vivo pharmacodynamics of three novel antileishmanial lead series. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 81–86. [Google Scholar] [CrossRef]
- Thomas, M.G.; De Rycker, M.; Ajakane, M.; Albrecht, S.; Álvarez-Pedraglio, A.I.; Boesche, M.; Brand, S.; Campbell, L.; Cantizani-Perez, J.; Cleghorn, L.A.T.; et al. Identification of GSK3186899/DDD853651 as a preclinical development candidate for the treatment of visceral Leishmaniasis. J. Med. Chem. 2019, 62, 1180–1202. [Google Scholar] [CrossRef] [Green Version]
- Zilberstein, D.; Dwyer, D.M. Antidepressants cause lethal disruption of membrane function in the human protozoan parasite Leishmania. Science 1984, 226, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.T.; Croft, S.L. Antileishmanial actions of tricyclic neuroleptics appear to lack structural specificity. Biochem. Pharmacol. 1994, 48, 613–616. [Google Scholar] [CrossRef]
- da Silva Rodrigues, J.H.; Miranda, N.; Volpato, H.; Ueda-Nakamura, T.; Nakamura, C.V. The antidepressant clomipramine induces programmed cell death in Leishmania amazonensis through a mitochondrial pathway. Parasitol. Res. 2019, 118, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Ferreira Cunha-Júnior, E.; Andrade-Neto, V.V.; Lima, M.L.; da Costa-Silva, T.A.; Galisteo Junior, A.J.; Abengózar, M.A.; Barbas, C.; Rivas, L.; Almeida-Amaral, E.E.; Tempone, A.G.; et al. Cyclobenzaprine raises ROS levels in Leishmania infantum and reduces parasite burden in infected mice. PLoS Negl. Trop. Dis. 2017, 11, e0005281. [Google Scholar] [CrossRef]
- Mattock, N.M.; Peters, W. The experimental chemotherapy of leishmaniasis, III Detection of antileishmanial activity in some new synthetic compounds in a tissue culture model. Ann. Trop. Med. Parasitol. 1975, 69, 449–462. [Google Scholar] [CrossRef]
- Peters, W.; Trotter, E.R.; Robinson, B.L. The experimental chemotherapy of leishmaniasis, VII. Ann. Trop. Med. Parasitol. 1980, 74, 321–335. [Google Scholar] [CrossRef]
- Berman, J.D.; Lee, L.S. Activity of oral drugs against Leishmania tropica in human macrophages in vitro. Am. J. Trop. Med. Hyg. 1983, 32, 947–951. [Google Scholar] [CrossRef]
- Osorio, Y.; Travi, B.L.; Renslo, A.R.; Peniche, A.G.; Melby, P.C. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl. Trop. Dis. 2011, 5, e962. [Google Scholar] [CrossRef] [Green Version]
- Cioli, D.; Pica-Mattoccia, L.; Archer, S. Antischistosomal drugs: Past, present... and future? Pharmacol. Ther. 1995, 68, 35–85. [Google Scholar] [CrossRef]
- Russell, W.L. Results of tests for possible transmitted genetic effects of hycanthone in mammals. J. Toxicol. Environ. Health 1975, 1, 301–304. [Google Scholar] [CrossRef]
- Hartman, P.E.; Hulbert, P.B.; Bueding, E.; Taylor, D.D. Microsomal activation to mutagens of antischistosomal methyl thioxanthenones and initial tests on a possibly non-mutagenic analogue. Mutat. Res. 1975, 31, 87–95. [Google Scholar] [CrossRef]
- Hartman, P.E.; Hulbert, P.B. Genetic activity spectra of some antischistosomal compounds, with particular emphasis on thioxanthenones and benzothiopyranoindazoles. J. Toxicol. Environ. Health 1975, 1, 243–270. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Vasconcelos, M.H.; Paiva, A.; Fernandes, M.X.; Pinto, M.; Sousa, E. Dual inhibitors of P-glycoprotein and tumor cell growth: (Re)discovering thioxanthones. Biochem. Pharmacol. 2012, 83, 57–68. [Google Scholar] [CrossRef]
- Barbosa, J.; Lima, R.T.; Sousa, D.; Gomes, A.S.; Palmeira, A.; Seca, H.; Choosang, K.; Pakkong, P.; Bousbaa, H.; Pinto, M.M.; et al. Screening a small library of xanthones for antitumor activity and identification of a hit compound which induces apoptosis. Molecules 2016, 21, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasso, B.; Novelli, F.; Tonelli, M.; Barteselli, A.; Basilico, N.; Parapini, S.; Taramelli, D.; Sparatore, A.; Sparatore, F. Synthesis and antiplasmodial activity of novel chloroquine analogues with bulky basic side chains. ChemMedChem 2015, 10, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Sparatore, A.; Basilico, N.; Casagrande, M.; Parapini, S.; Taramelli, D.; Brun, R.; Wittlin, S.; Sparatore, F. Antimalarial activity of novel pyrrolizidinyl derivatives of 4-aminoquinoline. Bioorg. Med. Chem. Lett. 2008, 18, 3737–3740. [Google Scholar] [CrossRef] [PubMed]
- Sparatore, A.; Basilico, N.; Parapini, S.; Romeo, S.; Novelli, F.; Sparatore, F.; Taramelli, D. 4-Aminoquinoline quinolizidinyl- and quinolizidinylalkyl-derivatives with antimalarial activity. Bioorg. Med. Chem. 2005, 13, 5338–5345. [Google Scholar] [CrossRef] [PubMed]
- Barteselli, A.; Casagrande, M.; Basilico, N.; Parapini, S.; Rusconi, C.M.; Tonelli, M.; Boido, V.; Taramelli, D.; Sparatore, F.; Sparatore, A. Clofazimine analogs with antileishmanial and antiplasmodial activity. Bioorg. Med. Chem. 2015, 23, 55–65. [Google Scholar] [CrossRef]
- Tonelli, M.; Gabriele, E.; Piazza, F.; Basilico, N.; Parapini, S.; Tasso, B.; Loddo, R.; Sparatore, F.; Sparatore, A. Benzimidazole derivatives endowed with potent antileishmanial activity. J. Enzyme Inhib. Med. Chem. 2018, 33, 210–226. [Google Scholar] [CrossRef] [Green Version]
- Archer, S.; Rej, R. Nitro and amino derivatives of lucanthone as antitumor agents. J. Med. Chem. 1982, 25, 328–331. [Google Scholar] [CrossRef]
- Sparatore, A.; Veronese, M.; Sparatore, F. Quinolizidine derivatives with antimicrobial activity. Farmaco Ed. Sci. 1987, 42, 159–174. [Google Scholar]
- Boido Canu, C.; Iusco, G.; Boido, V.; Sparatore, F.; Sparatore, A. Synthesis and antileukemic activity of 1-[(quinolizidinylalkyl)amino]4/7-R-thioxanthen-9-ones. Farmaco 1989, 44, 1069–1082. [Google Scholar] [PubMed]
- Sparatore, A.; Marchi, M.; Maura, G.; Paudice, P.; Raiteri, M. Effects of some rigid analogues of imipramine and amitriptyline on the uptake of noradrenaline, serotonin and choline in rat brain synaptosomes. Pharmacol. Res. Commun. 1982, 14, 257–265. [Google Scholar] [CrossRef]
- Sparatore, A.; Marchi, M.; Maura, G.; Raiteri, M. Effect of some quinolizidine derivatives on the release of serotonin, noradrenaline, dopamine and acetylcholine from rat brain synaptosomes. Farmaco 1989, 44, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Boido, V.; Sparatore, F. Derivatives of natural amino alcohols and diamines of pharmacologic interest. IV. Action of lupinylmagnesium chloride on aromatic and heterocyclic ketones. Ann. Chim. 1966, 56, 1603–1613. [Google Scholar]
- Tonelli, M.; Catto, M.; Tasso, B.; Novelli, F.; Canu, C.; Iusco, G.; Pisani, L.; De Stradis, A.; Denora, N.; Sparatore, A.; et al. Multitarget therapeutic leads for Alzheimer’s disease: Quinolizidinyl derivatives of bi- and tricyclic systems as dual inhibitors of cholinesterases and β-amyloid (Aβ) aggregation. ChemMedChem 2015, 10, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Tasso, B.; Catto, M.; Nicolotti, O.; Novelli, F.; Tonelli, M.; Giangreco, I.; Pisani, L.; Sparatore, A.; Boido, V.; Carotti, A.; et al. Quinolizidinyl derivatives of bi- and tricyclic systems as potent inhibitors of acetyl- and butyrylcholinesterase with potential in Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 2170–2184. [Google Scholar] [CrossRef]
- Blanz, E.J., Jr.; French, F.A. A systematic investigation of thioxanthen-9-ones and analogs as potential antitumor agents. J. Med. Chem. 1963, 6, 185–191. [Google Scholar] [CrossRef]
- Naidu, M.D.; Agarwal, R.; Pena, L.A.; Cunha, L.; Mezei, M.; Shen, M.; Wilson, D.M.; Liu, Y.; Sanchez, Z.; Chaudhary, P.; et al. Lucanthone and its derivative hycanthone inhibit apurinic endonuclease-1 (APE1) by direct protein binding. PLoS ONE 2011, 6, e23679. [Google Scholar] [CrossRef] [Green Version]
- Carew, J.S.; Espitia, C.M.; Esquivel, J.A., 2nd; Mahalingam, D.; Kelly, K.R.; Reddy, G.; Giles, F.J.; Nawrocki, S.T. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J. Biol. Chem. 2011, 286, 6602–6613. [Google Scholar] [CrossRef] [Green Version]
- Lima, R.; Sousa, D.; Gomes, A.; Mendes, N.; Matthiesen, R.; Pedro, M.; Marques, F.; Pinto, M.M.; Sousa, E.; Vasconcelo, M.H. The antitumor activity of a lead thioxanthone is associated with alterations in cholesterol localization. Molecules 2018, 23, 3301. [Google Scholar] [CrossRef] [Green Version]
- Tischer, M.; Pradel, G.; Ohlsen, K.; Holzgrabe, U. Quaternary ammonium salts and their antimicrobial potential: Targets or nonspecific interactions? ChemMedChem 2012, 7, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Mamoun, C.B. Choline transport in Leishmania major promastigotes and its inhibition by choline and phosphocholine analogs. Mol. Biochem. Parasitol. 2002, 125, 127–134. [Google Scholar] [CrossRef]
- Duque-Benítez, S.M.; Ríos-Vásquez, L.A.; Ocampo-Cardona, R.; Cedeño, D.L.; Jones, M.A.; Vélez, I.D.; Robledo, S.M. Synthesis of novel quaternary ammonium salts and their in vitro antileishmanial activity and U-937 cell cytotoxicity. Molecules 2016, 21, 381. [Google Scholar] [CrossRef]
- Papanastasiou, I.; Prousis, K.C.; Georgikopoulou, K.; Pavlidis, T.; Scoulica, E.; Kolocouris, N.; Calogeropoulou, T. Design and synthesis of new adamantyl-substituted antileishmanial ether phospholipids. Bioorg. Med. Chem. Lett. 2010, 20, 5484–5487. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Chernoff, R.; Finkelstein, I.; Hirschberg, E. Miracil D: An inhibitor of ribonucleic acid synthesis in Bacillus subtilis. Mol. Pharmacol. 1965, 1, 297–305. [Google Scholar] [PubMed]
- Archer, S.; Yarinsky, A. Recent developments in the chemotherapy of schistosomiasis. Prog. Drug Res. 1972, 16, 11–66. [Google Scholar] [PubMed]
- Krauth-Siegel, R.L.; Meiering, S.K.; Schmidt, H. The parasite-specific trypanothione metabolism of Trypanosoma and Leishmania. Biol. Chem. 2003, 384, 539–549. [Google Scholar] [CrossRef]
- Kumar, S.; Ali, M.R.; Bawa, S. Mini review on tricyclic compounds as an inhibitor of trypanothione reductase. J. Pharm. Bioallied Sci. 2014, 6, 222–228. [Google Scholar]
- Khan, M.O. Trypanothione reductase: A viable chemotherapeutic target for antitrypanosomal and antileishmanial drug design. Drug Targ. Insights 2007, 2, 129–146. [Google Scholar] [CrossRef]
- Verdier, J.S.; Wolfe, A.D. Butyrylcholinesterase inhibition by miracil D and other compounds. Biochem. Pharmacol. 1986, 35, 1605–1608. [Google Scholar] [CrossRef]
- Pulido, S.A.; Nguyen, V.H.; Alzate, J.F.; Cedeño, D.L.; Makurath, M.A.; Ríos-Vásquez, A.; Duque-Benítez, S.M.; Jones, M.A.; Robledo, S.M.; Friesen, J.A. Insights into the phosphatidylcholine and phosphatidylethanolamine biosynthetic pathways in Leishmania parasites and characterization of a choline kinase from Leishmania infantum. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2017, 213, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Wassef, M.K.; Fioretti, T.B.; Dwyer, D.M. Lipid analyses of isolated surface membranes of Leishmania donovani promastigotes. Lipids 1985, 20, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Pezzementi, L.; Chatonnet, A. Evolution of cholinesterases in the animal kingdom. Chem. Biol. Interact. 2010, 187, 27–33. [Google Scholar] [CrossRef]
- Karczmar, A.G. Cholinesterases (ChEs) and the cholinergic system in ontogenesis and phylogenesis, and non-classical roles of cholinesterases-A review. Chem. Biol. Interact. 2010, 187, 34–43. [Google Scholar] [CrossRef]
- Halliday, A.C.; Greenfield, S.A. From protein to peptides: A spectrum of non-hydrolytic functions of acetylcholinesterase. Protein Pept. Lett. 2012, 19, 165–172. [Google Scholar] [CrossRef]
- Lenta, B.N.; Vonthron-Sénécheau, C.; Weniger, B.; Devkota, K.P.; Ngoupayo, J.; Kaiser, M.; Naz, Q.; Choudhary, M.I.; Tsamo, E.; Sewald, N. Leishmanicidal and cholinesterase inhibiting activities of phenolic compounds from Allanblackia monticola and Symphonia globulifera. Molecules 2007, 12, 1548–1557. [Google Scholar] [CrossRef]
- Vila-Nova, N.S.; Morais, S.M.; Falcão, M.J.C.; Bevilaqua, C.M.L.; Rondon, F.C.M.; Wilson, M.E.; Vieira, I.G.P.; Andrade, H.F. Leishmanicidal and cholinesterase inhibiting activities of phenolic compounds of Dimorphandra gardneriana and Platymiscium floribundum, native plants from Caatinga biome. Pesq. Vet. Bras. 2012, 32, 1164–1168. [Google Scholar] [CrossRef] [Green Version]
- Mogana, R.; Adhikari, A.; Debnath, S.; Hazra, S.; Hazra, B.; Teng-Jin, K.; Wiart, C. The antiacetylcholinesterase and antileishmanial activities of Canarium patentinervium Miq. Biomed Res. Int. 2014, 2014, 903529. [Google Scholar] [CrossRef] [Green Version]
- Benson, T.J.; McKie, J.H.; Garforth, J.; Borges, A.; Fairlamb, A.H.; Douglas, K.T. Rationally designed selective inhibitors of trypanothione reductase. Phenothiazines and related tricyclics as lead structures. Biochem. J. 1992, 286, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, S.K.; Shukla, A.K.; Dubey, V.K. Molecular docking studies of selected tricyclic and quinone derivatives on trypanothione reductase of Leishmania infantum. J. Comput. Chem. 2010, 31, 2463–2475. [Google Scholar] [PubMed]
- Briguglio, I.; Loddo, R.; Laurini, E.; Fermeglia, M.; Piras, S.; Corona, P.; Giunchedi, P.; Gavini, E.; Sanna, G.; Giliberti, G.; et al. Synthesis, cytotoxicity and antiviral evaluation of new series of imidazo[4,5-g]quinoline and pyrido[2,3-g]quinoxalinone derivatives. Eur. J. Med. Chem. 2015, 105, 63–79. [Google Scholar] [CrossRef]
- Carta, A.; Sanna, G.; Briguglio, I.; Madeddu, S.; Vitale, G.; Piras, S.; Corona, P.; Peana, A.T.; Laurini, E.; Fermeglia, M.; et al. Quinoxaline derivatives as new inhibitors of coxsackievirus B5. Eur. J. Med. Chem. 2018, 145, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Loddo, R.; Francesconi, V.; Laurini, E.; Boccardo, S.; Aulic, S.; Fermeglia, M.; Pricl, S.; Tonelli, M. 9-Aminoacridine-based agents impair the bovine viral diarrhea virus (BVDV) replication targeting the RNA-dependent RNA polymerase (RdRp). Bioorg. Med. Chem. 2018, 26, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Baiocco, P.; Ilari, A.; Ceci, P.; Orsini, S.; Gramiccia, M.; Di Muccio, T.; Colotti, G. Inhibitory effect of silver nanoparticles on trypanothione reductase activity and Leishmania infantum proliferation. ACS Med. Chem. Lett. 2010, 2, 230–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinović, J.; Videnović, M.; Orsini, S.; Bogojević, K.; D’Alessandro, S.; Scaccabarozzi, D.; Terzić Jovanović, N.; Gradoni, L.; Basilico, N.; Šolaja, B.A. Novel aminoquinoline derivatives significantly reduce parasite load in Leishmania infantum infected mice. ACS Med. Chem. Lett. 2018, 9, 629–634. [Google Scholar] [CrossRef]
- Baiocco, P.; Colotti, G.; Franceschini, S.; Ilari, A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. The origin of layer structure artifacts in simulations of liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the generalized born model suitable for macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro III, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.F. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
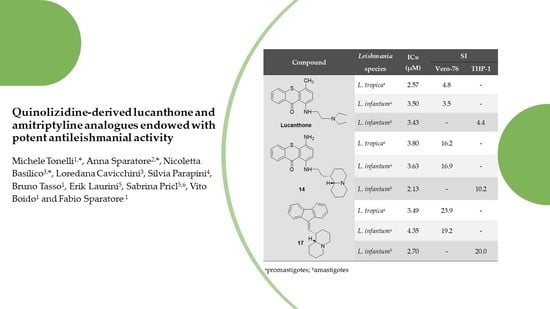
| Compd. | IC50 (µM) a L. tropica | Ratio b IC50 Miltef./IC50 Compd. | IC50 (µM) a L. infantum | Ratio b IC50 Miltef./IC50 Compd. |
|---|---|---|---|---|
| Lucanthone | 2.57 ± 1.03 | 16.8 | 3.50 ± 1.17 | 8.9 |
| 1 | 3.3 6 ± 1.53 | 12.9 | 3.87 ± 1.53 | 8.1 |
| 2 | 6.27 ± 2.75 | 6.9 | 10.60 ± 3.78 | 2.95 |
| 3 | 8.89 ± 3.02 | 4.87 | 17.19 ± 1.31 | 1.8 |
| 4 | 17.83 ± 5.61 | 2.4 | 13.00 ± 2.80 | 2.4 |
| 5 | 8.16 ± 5.23 | 5.3 | 8.99 ± 3.86 | 3.5 |
| 6 | 28.35 ± 14.34 | 1.5 | >46 | <0.7 |
| 7 | 12.00 ± 5.24 | 3.6 | 19.66 ± 5.79 | 1.6 |
| 8 | 19.24 ± 9.33 | 2.25 | 38.72 ± 7.67 | 0.8 |
| 9 | 4.64 ± 1.12 | 9.3 | 9.39 ± 4.34 | 3.3 |
| 10 | 11.05 ± 3.30 | 3.9 | 22.27 ± 3.30 | 1.4 |
| 11 | 6.35 ± 1.46 | 6.8 | 13.01 ± 0.97 | 2.4 |
| 12 | 7.39 ± 2.31 | 5.85 | 7.89 ± 2.41 | 3.95 |
| 13 | 2.87 ± 0.43 | 15.1 | 5.23 ± 1.83 | 6.0 |
| 14 | 3.80 ± 1.82 | 11.4 | 3.63 ± 0.78 | 8.6 |
| 15 | 3.72 ± 1.66 | 11.6 | 4.22 ± 1.95 | 7.4 |
| 16 | 6.56 ± 3.08 | 6.6 | 8.23 ± 1.51 | 3.8 |
| 17 | 3.49 ± 1.02 | 12.4 | 4.35 ± 0.37 | 7.2 |
| 18 | 24.25 ± 3.58 | 1.8 | 25.21 ± 1.92 | 1.24 |
| 19 | 10.52 ± 2.69 | 4.1 | 16.42 ± 0.59 | 1.90 |
| 20 | 7.21 ± 3.52 | 6.0 | 8.71 ± 0.68 | 3.58 |
| Miltefosine | 43.26 ± 11.36 | 1.0 | 31.26 ± 10.45 | 1.0 |
| Compd. | CC50 a (µM) Vero-76 | IC50 b (µM) | S.I. c | ||
|---|---|---|---|---|---|
| L. tropica | L. infantum | L. tropica | L. infantum | ||
| Lucanthone | 12.4 ± 2.1 | 2.57 ± 1.03 | 3.50 ± 1.17 | 4.8 | 3.5 |
| 1 | 23.5 ±1.8 | 3.36 ± 1.53 | 3.87 ± 1.53 | 7.0 | 6.6 |
| 2 | 22.9 ± 2.3 | 6.27 ± 2.75 | 10.60 ± 3.78 | 3.7 | 2.2 |
| 14 | 61.4 ± 2.6 | 3.80 ± 1.82 | 3.63 ± 0.78 | 16.2 | 16.9 |
| 17 | 83.4 ± 3.0 | 3.48 ± 1.02 | 4.35 ± 0.37 | 23.9 | 19.2 |
| 19 | 82.7 ± 4.7 | 10.52 ± 2.69 | 16.42 ± 0.59 | 7.9 | 5.0 |
| 20 | 89.9 ± 3.7 | 7.21 ± 3.52 | 8.71 ± 0.68 | 12.5 | 10.3 |
| Compd. | L. infantum Amastigotes IC50a (µM) | THP-1 CC50 a,b (µM) | SI c |
|---|---|---|---|
| 1 | 3.49 ± 0.18 | 27.97 ± 8.00 | 8.0 |
| 14 | 2.13 ± 1.35 | 21.66 ± 8.83 | 10.2 |
| 17 | 2.70 ± 0.68 | 53.90 ± 3.98 | 20.0 |
| Lucanthone | 3.43 ± 0.93 | 15.17 ± 0.27 | 4.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonelli, M.; Sparatore, A.; Basilico, N.; Cavicchini, L.; Parapini, S.; Tasso, B.; Laurini, E.; Pricl, S.; Boido, V.; Sparatore, F. Quinolizidine-Derived Lucanthone and Amitriptyline Analogues Endowed with Potent Antileishmanial Activity. Pharmaceuticals 2020, 13, 339. https://doi.org/10.3390/ph13110339
Tonelli M, Sparatore A, Basilico N, Cavicchini L, Parapini S, Tasso B, Laurini E, Pricl S, Boido V, Sparatore F. Quinolizidine-Derived Lucanthone and Amitriptyline Analogues Endowed with Potent Antileishmanial Activity. Pharmaceuticals. 2020; 13(11):339. https://doi.org/10.3390/ph13110339
Chicago/Turabian StyleTonelli, Michele, Anna Sparatore, Nicoletta Basilico, Loredana Cavicchini, Silvia Parapini, Bruno Tasso, Erik Laurini, Sabrina Pricl, Vito Boido, and Fabio Sparatore. 2020. "Quinolizidine-Derived Lucanthone and Amitriptyline Analogues Endowed with Potent Antileishmanial Activity" Pharmaceuticals 13, no. 11: 339. https://doi.org/10.3390/ph13110339
APA StyleTonelli, M., Sparatore, A., Basilico, N., Cavicchini, L., Parapini, S., Tasso, B., Laurini, E., Pricl, S., Boido, V., & Sparatore, F. (2020). Quinolizidine-Derived Lucanthone and Amitriptyline Analogues Endowed with Potent Antileishmanial Activity. Pharmaceuticals, 13(11), 339. https://doi.org/10.3390/ph13110339