Synthesis and Glycosidase Inhibition Properties of Calix[8]arene-Based Iminosugar Click Clusters

 , ,
, ,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
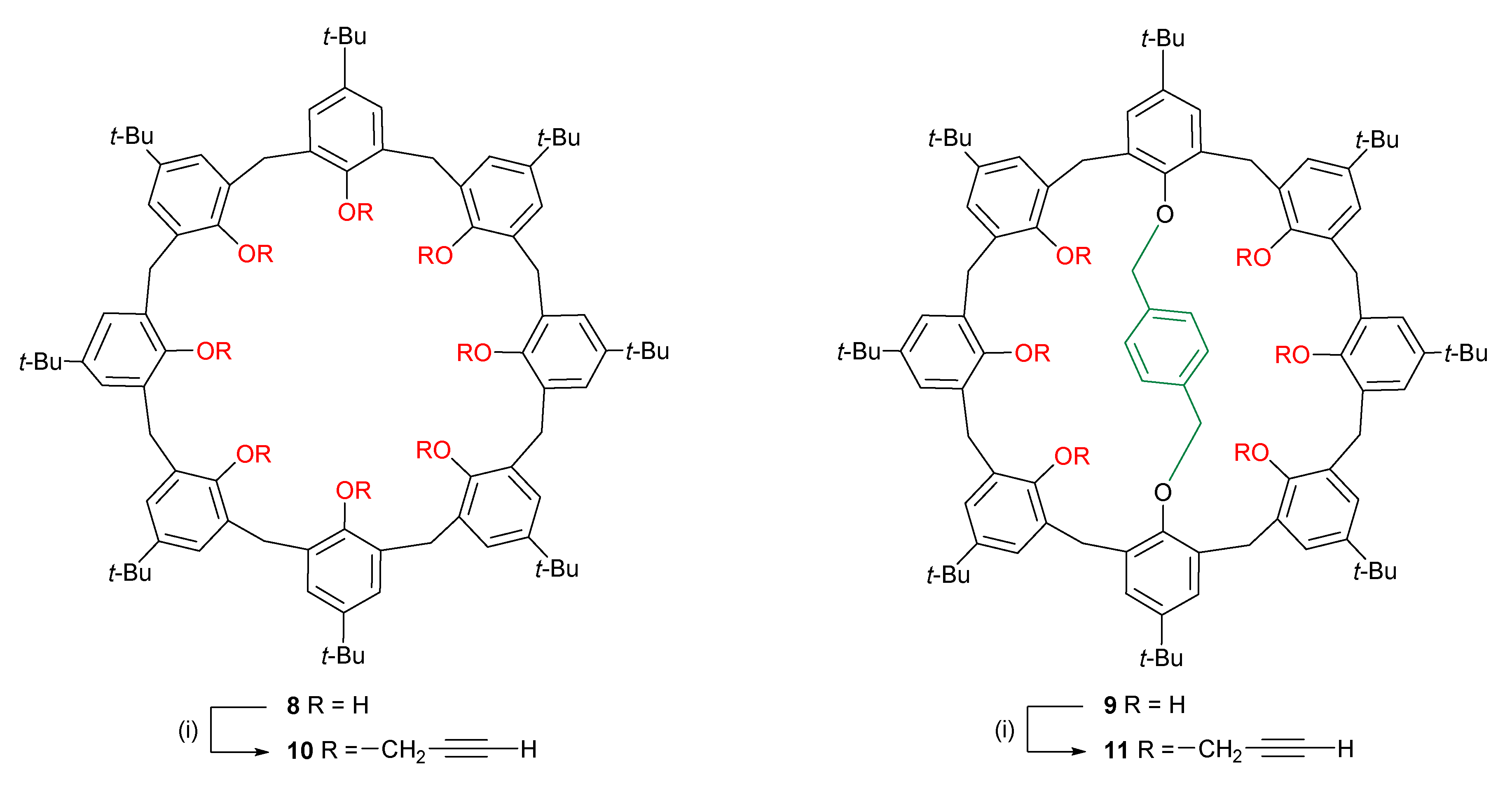
2.1. Chemistry
2.2. Biological Assays
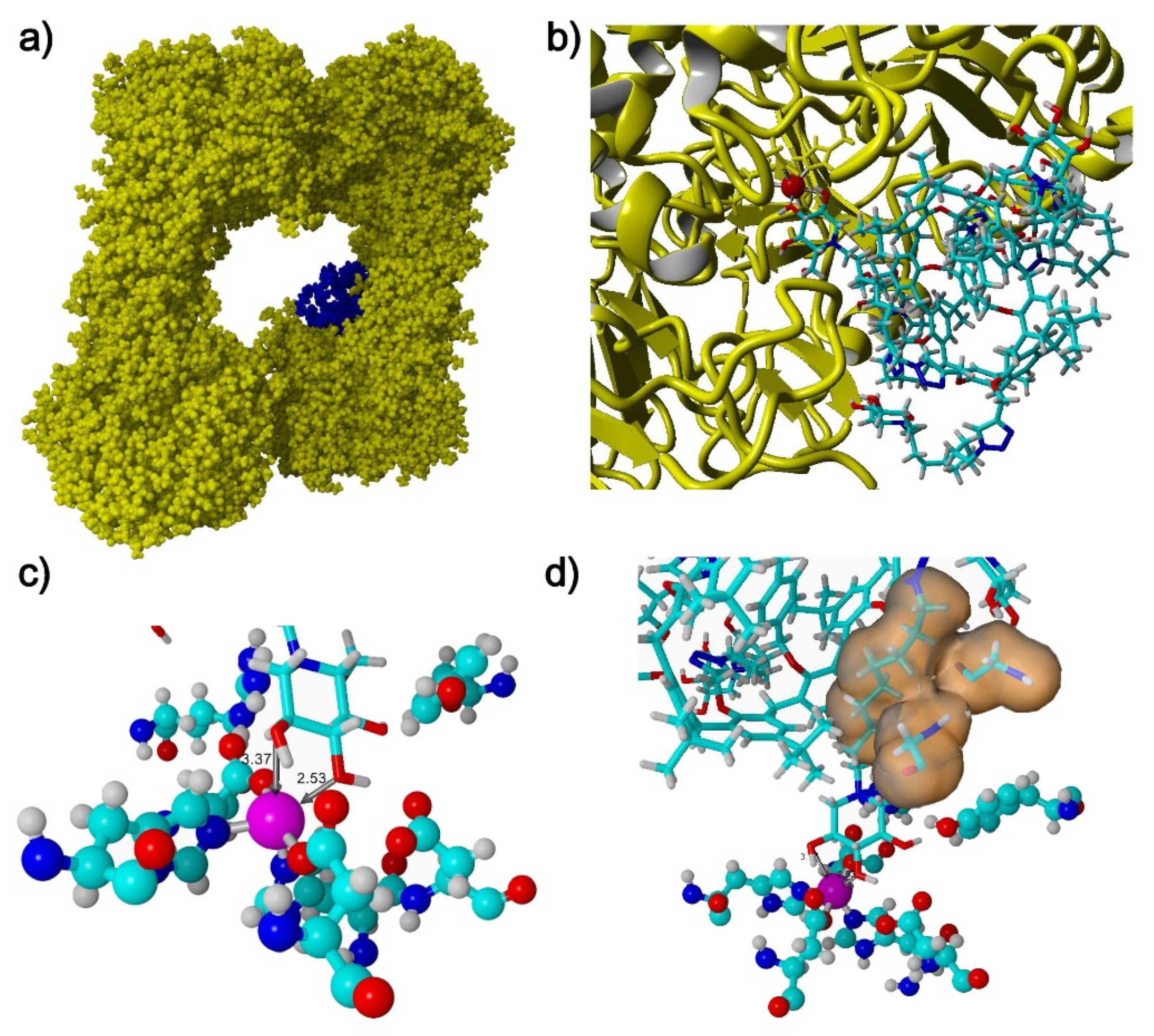
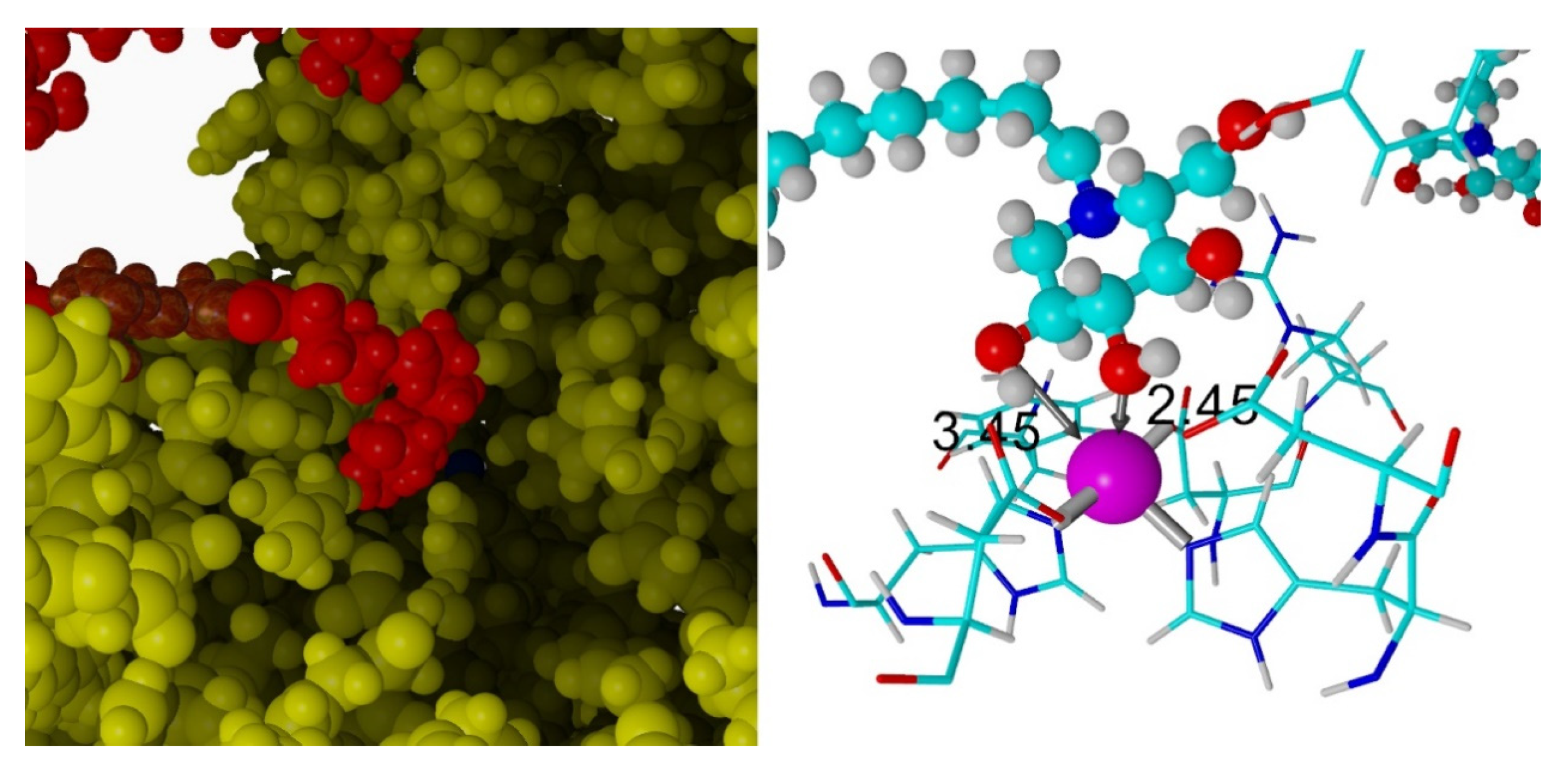
2.3. Docking Studies
3. Materials and Methods
3.1. Chemistry
3.2. Biological Assays
3.3. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peczuh, M.W.; Hamilton, A.D. Peptide and Protein Recognition by Designed Molecules. Chem. Rev. 2000, 100, 2479–2494. [Google Scholar] [CrossRef]
- Baldini, L.; Casnati, A.; Sansone, F.; Ungaro, R. Calixarene-based multivalent ligands. Chem. Soc. Rev. 2007, 36, 254–266. [Google Scholar] [CrossRef]
- Park, H.S.; Lin, Q.; Hamilton, A.D. Protein Surface Recognition by Synthetic Receptors: A Route to Novel Submicromolar Inhibitors for α-Chymotrypsin. J. Am. Chem. Soc. 1999, 121, 8–13. [Google Scholar] [CrossRef]
- Sansone, F.; Casnati, A. Multivalent glycocalixarenes for recognition of biological macromolecules: Glycocalyx mimics capable of multitasking. Chem. Soc. Rev. 2013, 42, 4623. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, M.; Morbioli, I.; Sansone, F.; Casnati, A. Moulding calixarenes for biomacromolecule targeting. Chem. Commun. 2015, 51, 14140–14159. [Google Scholar] [CrossRef]
- McGovern, R.E.; Fernandes, H.; Khan, A.R.; Power, N.P.; Crowley, P.B. Protein camouflage in cytochrome c-calixarene complexes. Nat. Chem. 2012, 4, 527–533. [Google Scholar] [CrossRef]
- Sansone, F.; Baldini, L.; Casnati, A.; Ungaro, R. Calixarenes: From biomimetic receptors to multivalent ligands for biomolecular recognition. New J. Chem. 2010, 34, 2715–2728. [Google Scholar] [CrossRef]
- Gutsche, C.D. Calixarenes: An Introduction, 2nd ed.; Royal Society of Chemistry: Cambridge, UK, 2008; ISBN 978-0-85404-258-6. [Google Scholar]
- Asfari, M.-Z.; Böhmer, V.; Harrowfield, J.; Vicens, J. Calixarenes 2001, 2001 ed.; Springer: Dordrecht, The Netherlands; Boston, MA, USA, 2001; ISBN 978-0-7923-6960-8. [Google Scholar]
- Neri, P.; Sessler, J.L.; Wang, M.-X. Calixarenes and Beyond; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; ISBN 978-3-319-31865-3. [Google Scholar]
- Sebti, S.M.; Hamilton, A.D. Design of growth factor antagonists with antiangiogenic and antitumor properties. Oncogene 2000, 19, 6566–6573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaskovich, M.A.; Lin, Q.; Delarue, F.L.; Sun, J.; Park, H.S.; Coppola, D.; Hamilton, A.D.; Sebti, S.M. Design of GFB-111, a platelet-derived growth factor binding molecule with antiangiogenic and anticancer activity against human tumors in mice. Nat. Biotechnol. 2000, 18, 1065–1070. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, D.; Baldini, L.; Ennis, E.; Jain, R.; Carie, A.; Sebti, S.M.; Hamilton, A.D. Structure–activity studies on a library of potent calix[4]arene-based PDGF antagonists that inhibit PDGF-stimulated PDGFR tyrosine phosphorylation. Org. Biomol. Chem. 2006, 4, 2376–2386. [Google Scholar] [CrossRef]
- Tommasone, S.; Talotta, C.; Gaeta, C.; Margarucci, L.; Monti, M.C.; Casapullo, A.; Macchi, B.; Prete, S.P.; Ladeira De Araujo, A.; Neri, P. Biomolecular Fishing for Calixarene Partners by a Chemoproteomic Approach. Angew. Chem. Int. Ed. 2015, 54, 15405–15409. [Google Scholar] [CrossRef] [PubMed]
- Chini, M.G.; Terracciano, S.; Riccio, R.; Bifulco, G.; Ciao, R.; Gaeta, C.; Troisi, F.; Neri, P. Conformationally Locked Calixarene-Based Histone Deacetylase Inhibitors. Org. Lett. 2010, 12, 5382–5385. [Google Scholar] [CrossRef]
- Morbioli, I.; Porkolab, V.; Magini, A.; Casnati, A.; Fieschi, F.; Sansone, F. Mannosylcalix[n]arenes as multivalent ligands for DC-SIGN. Carbohydr. Res. 2017, 453–454, 36–43. [Google Scholar] [CrossRef]
- Geraci, C.; Consoli, G.M.L.; Granata, G.; Galante, E.; Palmigiano, A.; Pappalardo, M.; Di Puma, S.D.; Spadaro, A. First Self-Adjuvant Multicomponent Potential Vaccine Candidates by Tethering of Four or Eight MUC1 Antigenic Immunodominant PDTRP Units on a Calixarene Platform: Synthesis and Biological Evaluation. Bioconjugate Chem. 2013, 24, 1710–1720. [Google Scholar] [CrossRef]
- Hevey, R. Strategies for the Development of Glycomimetic Drug Candidates. Pharmaceuticals 2019, 12, 55. [Google Scholar] [CrossRef] [Green Version]
- Brás, N.F.; Cerqueira, N.M.; Ramos, M.J.; Fernandes, P.A. Glycosidase inhibitors: A patent review (2008–2013). Expert Opin. Ther. Patents 2014, 24, 857–874. [Google Scholar] [CrossRef]
- Wadood, A.; Ghufran, M.; Khan, A.; Azam, S.S.; Jelani, M.; Uddin, R. Selective glycosidase inhibitors: A patent review (2012–present). Int. J. Biol. Macromol. 2018, 111, 82–91. [Google Scholar] [CrossRef]
- Cipolla, L.; La Ferla, B.; Airoldi, C.; Zona, C.; Orsato, A.; Shaikh, N.; Russo, L.; Nicotra, F. Carbohydrate mimetics and scaffolds: Sweet spots in medicinal chemistry. Future Med. Chem. 2010, 2, 587–599. [Google Scholar] [CrossRef]
- Davies, G.J.; Gloster, T.M.; Henrissat, B. Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr. Opin. Struct. Biol. 2005, 15, 637–645. [Google Scholar] [CrossRef]
- Rempel, B.P.; Withers, S.G. Covalent inhibitors of glycosidases and their applications in biochemistry and biology. Glycobiology 2008, 18, 570–586. [Google Scholar] [CrossRef] [Green Version]
- Herscovics, A. Importance of glycosidases in mammalian glycoprotein biosynthesis. Biochim. Biophys. Acta (BBA)—Gener. Subj. 1999, 1473, 96–107. [Google Scholar] [CrossRef]
- Asano, N. Glycosidase inhibitors: Update and perspectives on practical use. Glycobiology 2003, 13, 93R–104R. [Google Scholar] [CrossRef]
- Ghani, U. Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: Finding needle in the haystack. Eur. J. Med. Chem. 2015, 103, 133–162. [Google Scholar] [CrossRef]
- Simone, M.I.; Mares, L.J.; Eveleens, C.A.; McCluskey, A.; Pappin, B.B.; Kiefel, M.J.; Houston, T.A. Back to (non-)Basics: An Update on Neutral and Charge-Balanced Glycosidase Inhibitors. MRMC 2018, 18, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.J.; Goddard-Borger, E.D. α-glucosidase inhibitors as host-directed antiviral agents with potential for the treatment of COVID-19. Biochem. Soc. Trans. 2020, BST20200505. [Google Scholar] [CrossRef]
- Pérez-García, L.A.; Martínez-Duncker, I.; Mora Montes, H.M. The Endoplasmic Reticulum Alpha-Glycosidases as Potential Targets for Virus Control. CPPS 2017, 18, 1090–1097. [Google Scholar] [CrossRef]
- Robina, I.; Moreno-Vargas, A.; Carmona, A.; Vogel, P. Glycosidase Inhibitors as Potential HIV Entry Inhibitors? CDM 2004, 5, 329–361. [Google Scholar] [CrossRef]
- Ikeda, K. Sialic Acid Derivative Synthesis and Inhibitory Activities against Human Parainfluenza Virus Type 1. Trends Glycosci. Glycotechnol. 2011, 23, 14–32. [Google Scholar] [CrossRef]
- Hossain, F.; Andreana, P.R. Developments in Carbohydrate-Based Cancer Therapeutics. Pharmaceuticals 2019, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Kolarich, D. The promise of protein glycosylation for personalised medicine. Biochim. Biophys. Acta (BBA)—Gener. Subj. 2016, 1860, 1583–1595. [Google Scholar] [CrossRef]
- Wu, L.; Armstrong, Z.; Schröder, S.P.; de Boer, C.; Artola, M.; Aerts, J.M.; Overkleeft, H.S.; Davies, G.J. An overview of activity-based probes for glycosidases. Curr. Opin. Chem. Biol. 2019, 53, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Decroocq, C.; Iehl, J.; Holler, M.; Hazelard, D.; Mena Barragán, T.; Ortiz Mellet, C.; Nierengarten, J.-F. Glycosidase Inhibition with Fullerene Iminosugar Balls: A Dramatic Multivalent Effect. Angew. Chem. Int. Ed. 2010, 49, 5753–5756. [Google Scholar] [CrossRef] [PubMed]
- Diot, J.; García-Moreno, M.I.; Gouin, S.G.; Ortiz Mellet, C.; Haupt, K.; Kovensky, J. Multivalent iminosugars to modulate affinity and selectivity for glycosidases. Org. Biomol. Chem. 2009, 7, 357. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Bodlenner, A. The Multivalent Effect in Glycosidase Inhibition: A New, Rapidly Emerging Topic in Glycoscience. ChemBioChem 2014, 15, 1239–1251. [Google Scholar] [CrossRef]
- Gouin, S.G. Multivalent Inhibitors for Carbohydrate-Processing Enzymes: Beyond the “Lock-and-Key” Concept. Chem. Eur. J. 2014, 20, 11616–11628. [Google Scholar] [CrossRef]
- Zelli, R.; Longevial, J.-F.; Dumy, P.; Marra, A. Synthesis and biological properties of multivalent iminosugars. New J. Chem. 2015, 39, 5050–5074. [Google Scholar] [CrossRef]
- Matassini, C.; Parmeggiani, C.; Cardona, F.; Goti, A. Are enzymes sensitive to the multivalent effect? Emerging evidence with glycosidases. Tetrahedron Lett. 2016, 57, 5407–5415. [Google Scholar] [CrossRef]
- Mellet, C.O.; Nierengarten, J.-F.; Fernández, J.M.G. Multivalency as an action principle in multimodal lectin recognition and glycosidase inhibition: A paradigm shift driven by carbon-based glyconanomaterials. J. Mater. Chem. B 2017, 5, 6428–6436. [Google Scholar] [CrossRef]
- Compain, P. Multivalent Effect in Glycosidase Inhibition: The End of the Beginning. Chem. Rec. 2020, 20, 10–22. [Google Scholar] [CrossRef]
- González-Cuesta, M.; Ortiz Mellet, C.; García Fernández, J.M. Carbohydrate supramolecular chemistry: Beyond the multivalent effect. Chem. Commun. 2020, 56, 5207–5222. [Google Scholar] [CrossRef] [Green Version]
- Lepage, M.L.; Schneider, J.P.; Bodlenner, A.; Meli, A.; De Riccardis, F.; Schmitt, M.; Tarnus, C.; Nguyen-Huynh, N.-T.; Francois, Y.-N.; Leize-Wagner, E.; et al. Iminosugar-Cyclopeptoid Conjugates Raise Multivalent Effect in Glycosidase Inhibition at Unprecedented High Levels. Chem. A Eur. J. 2016, 22, 5151–5155. [Google Scholar] [CrossRef] [PubMed]
- Decroocq, C.; Joosten, A.; Sergent, R.; Mena Barragán, T.; Ortiz Mellet, C.; Compain, P. The Multivalent Effect in Glycosidase Inhibition: Probing the Influence of Valency, Peripheral Ligand Structure, and Topology with Cyclodextrin-Based Iminosugar Click Clusters. ChemBioChem 2013, 14, 2038–2049. [Google Scholar] [CrossRef]
- Rísquez-Cuadro, R.; García Fernández, J.M.; Nierengarten, J.-F.; Ortiz Mellet, C. Fullerene-sp2-Iminosugar Balls as Multimodal Ligands for Lectins and Glycosidases: A Mechanistic Hypothesis for the Inhibitory Multivalent Effect. Chem. Eur. J. 2013, 19, 16791–16803. [Google Scholar] [CrossRef] [Green Version]
- Mirabella, S.; D’Adamio, G.; Matassini, C.; Goti, A.; Delgado, S.; Gimeno, A.; Robina, I.; Moreno-Vargas, A.J.; Šesták, S.; Jiménez-Barbero, J.; et al. Mechanistic Insight into the Binding of Multivalent Pyrrolidines to α-Mannosidases. Chem. A Eur. J. 2017, 23, 14585–14596. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Clavijo, E.; Carmona, A.T.; Moreno-Vargas, A.J.; Molina, L.; Wright, D.W.; Davies, G.J.; Robina, I. Exploring a Multivalent Approach to α-L-Fucosidase Inhibition. Eur. J. Organ. Chem. 2013, 2013, 7328–7336. [Google Scholar] [CrossRef]
- Martínez-Bailén, M.; Jiménez-Ortega, E.; Carmona, A.T.; Robina, I.; Sanz-Aparicio, J.; Talens-Perales, D.; Polaina, J.; Matassini, C.; Cardona, F.; Moreno-Vargas, A.J. Structural basis of the inhibition of GH1 β-glucosidases by multivalent pyrrolidine iminosugars. Bioorgan. Chem. 2019, 89, 103026. [Google Scholar] [CrossRef]
- Howard, E.; Cousido-Siah, A.; Lepage, M.L.; Schneider, J.P.; Bodlenner, A.; Mitschler, A.; Meli, A.; Izzo, I.; Alvarez, H.A.; Podjarny, A.; et al. Structural Basis of Outstanding Multivalent Effects in Jack Bean α-Mannosidase Inhibition. Angew. Chem. Int. Ed. 2018, 57, 8002–8006. [Google Scholar] [CrossRef] [Green Version]
- Hottin, A.; Wright, D.W.; Moreno-Clavijo, E.; Moreno-Vargas, A.J.; Davies, G.J.; Behr, J.-B. Exploring the divalent effect in fucosidase inhibition with stereoisomeric pyrrolidine dimers. Org. Biomol. Chem. 2016, 14, 4718–4727. [Google Scholar] [CrossRef]
- Joosten, A.; Decroocq, C.; de Sousa, J.; Schneider, J.P.; Etamé, E.; Bodlenner, A.; Butters, T.D.; Compain, P. A Systematic Investigation of Iminosugar Click Clusters as Pharmacological Chaperones for the Treatment of Gaucher Disease. ChemBioChem 2014, 15, 309–319. [Google Scholar] [CrossRef]
- Compain, P.; Decroocq, C.; Joosten, A.; de Sousa, J.; Rodríguez-Lucena, D.; Butters, T.D.; Bertrand, J.; Clément, R.; Boinot, C.; Becq, F.; et al. Rescue of Functional CFTR Channels in Cystic Fibrosis: A Dramatic Multivalent Effect Using Iminosugar Cluster-Based Correctors. ChemBioChem 2013, 14, 2050–2058. [Google Scholar] [CrossRef]
- Li, J.-J.; Wang, K.-R.; Li, R.-F.; Yang, J.-X.; Li, M.; Zhang, H.-X.; Cao, Z.-R.; Li, X.-L. Synthesis, self-assembly behaviours and multivalent glycosidase inhibition effects of a deoxynojirimycin modified perylene bisimide derivative. J. Mater. Chem. B 2019, 7, 1270–1275. [Google Scholar] [CrossRef]
- Pichon, M.M.; Stauffert, F.; Bodlenner, A.; Compain, P. Tight-binding inhibition of Jack bean α-mannosidase by glycoimidazole clusters. Org. Biomol. Chem. 2019, 17, 5801–5817. [Google Scholar] [CrossRef]
- Brissonnet, Y.; Ortiz Mellet, C.; Morandat, S.; Garcia Moreno, M.I.; Deniaud, D.; Matthews, S.E.; Vidal, S.; Šesták, S.; El Kirat, K.; Gouin, S.G. Topological Effects and Binding Modes Operating with Multivalent Iminosugar-Based Glycoclusters and Mannosidases. J. Am. Chem. Soc. 2013, 135, 18427–18435. [Google Scholar] [CrossRef]
- Marra, A.; Zelli, R.; D’Orazio, G.; La Ferla, B.; Dondoni, A. Synthesis and glycosidase inhibition properties of triazole-linked calixarene–iminosugar clusters. Tetrahedron 2014, 70, 9387–9393. [Google Scholar] [CrossRef]
- Lepage, M.L.; Meli, A.; Bodlenner, A.; Tarnus, C.; De Riccardis, F.; Izzo, I.; Compain, P. Synthesis of the first examples of iminosugar clusters based on cyclopeptoid cores. Beilstein J. Org. Chem. 2014, 10, 1406–1412. [Google Scholar] [CrossRef] [Green Version]
- Gaeta, C.; Gregoli, L.; Martino, M.; Neri, P. Convenient regioselective functionalization at the upper-rim of p-tert-butylcalix[8]arene through a protection–deprotection procedure. Tetrahedron Lett. 2002, 43, 8875–8878. [Google Scholar] [CrossRef]
- Ho, W.-L.; Hsu, W.-M.; Huang, M.-C.; Kadomatsu, K.; Nakagawara, A. Protein glycosylation in cancers and its potential therapeutic applications in neuroblastoma. J. Hematol. Oncol. 2016, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Marradi, M.; Cicchi, S.; Sansone, F.; Casnati, A.; Goti, A. Low-generation dendrimers with a calixarene core and based on a chiral C2 -symmetric pyrrolidine as iminosugar mimics. Beilstein J. Org. Chem. 2012, 8, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardona, F.; Isoldi, G.; Sansone, F.; Casnati, A.; Goti, A. Building Multivalent Iminosugar-Based Ligands on Calixarene Cores via Nitrone Cycloadditions. J. Org. Chem. 2012, 77, 6980–6988. [Google Scholar] [CrossRef]
- Zelli, R.; Dumy, P.; Marra, A. Metal-free synthesis of imino-disaccharides and calix-iminosugars by photoinduced radical thiol–ene coupling (TEC). Org. Biomol. Chem. 2020, 18, 2392–2397. [Google Scholar] [CrossRef]
- Decroocq, C.; Rodríguez-Lucena, D.; Russo, V.; Mena Barragán, T.; Ortiz Mellet, C.; Compain, P. The Multivalent Effect in Glycosidase Inhibition: Probing the Influence of Architectural Parameters with Cyclodextrin-based Iminosugar Click Clusters. Chem. Eur. J. 2011, 17, 13825–13831. [Google Scholar] [CrossRef]
- Joosten, A.; Schneider, J.P.; Lepage, M.L.; Tarnus, C.; Bodlenner, A.; Compain, P. A Convergent Strategy for the Synthesis of Second-Generation Iminosugar Clusters Using “Clickable” Trivalent Dendrons: Synthesis of Second-Generation Iminosugar Clusters. Eur. J. Organ. Chem. 2014, 2014, 1866–1872. [Google Scholar] [CrossRef]
- Munch, J.H.; Gutsche, C.D. p-tert-Butylcalix[8]Arene. Org. Synth. 1990, 68, 243. [Google Scholar] [CrossRef]
- Cunsolo, F.; Consoli, G.M.L.; Piattelli, M.; Neri, P. Methylation of p-tert-Butylcalix[8]arene. Products Obtained in the Presence of Strong Bases. J. Org. Chem. 1998, 63, 6852–6858. [Google Scholar] [CrossRef]
- Consoli, G.M.L.; Cunsolo, F.; Piattelli, M.; Neri, P. Study on the Esterification of p-tert-Butylcalix[8]arene. J. Org. Chem. 1996, 61, 2195–2198. [Google Scholar] [CrossRef]
- Lapenta, R.; De Simone, N.A.; Buonerba, A.; Talotta, C.; Gaeta, C.; Neri, P.; Grassi, A.; Milione, S. Dinuclear zirconium complex bearing a 1,5-bridged-calix[8]arene ligand as an effective catalyst for the synthesis of macrolactones. Catal. Sci. Technol. 2018, 8, 2716–2727. [Google Scholar] [CrossRef]
- Consoli, G.M.L.; Cunsolo, F.; Geraci, C.; Gavuzzo, E.; Neri, P. Atropisomerism in 1,5-Bridged Calix[8]arenes. Org. Lett. 2002, 4, 2649–2652. [Google Scholar] [CrossRef]
- Segel, I.H. Enzyme Kinetics; Wiley: New York, NY, USA, 1975; p. 185. [Google Scholar]
- Baici, A. The Specific Velocity Plot. Eur. J. Biochem. 1981, 119, 9–14. [Google Scholar] [CrossRef]
- Morrison, J.F. Kinetics of the Reversible Inhibition of Enzyme-Catalyzed Reactions by Tight-Binding Inhibitors. Biochim. Biophys. Acta 1969, 185, 269–286. [Google Scholar] [CrossRef]
- Cha, S. Tight-binding inhibitors—I: Kinetic behavior. Biochem. Pharmacol. 1975, 24, 2177–2185. [Google Scholar] [CrossRef]
- Vovk, A.I.; Kalchenko, V.I.; Cherenok, S.A.; Kukhar, V.P.; Muzychka, V.M.; Lozynsky, M.O. Calix[4]arene methylenebisphosphonic acids as calf intestine alkaline phosphatase inhibitors. Org. Biomol. Chem. 2004, 2, 3162–3166. [Google Scholar] [CrossRef]
- Español, E.S.; Villamil, M.M. Calixarenes: Generalities and Their Role in Improving the Solubility, Biocompatibility, Stability, Bioavailability, Detection, and Transport of Biomolecules. Biomolecules 2019, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Khairutdinov, B.; Ermakova, E.; Sitnitsky, A.; Stoikov, I.; Zuev, Y. Supramolecular complex formed by DNA oligonucleotide and thiacalix[4]arene. NMR-spectroscopy and molecular docking. J. Mol. Struct. 2014, 1074, 126–133. [Google Scholar] [CrossRef]
- Jang, Y.M.; Yu, C.J.; Kim, J.S.; Kim, S.U. Ab initio design of drug carriers for zoledronate guest molecule using phosphonated and sulfonated calix[4]arene and calix[4]resorcinarene host molecules. J. Mater. Sci. 2018, 53, 5125–5139. [Google Scholar] [CrossRef] [Green Version]
- Ang, T.-F.; Salleh, A.B.; Normi, Y.M.; Leow, T.C. For a recent report regarding docking calculations using YASARA. In silico design of potentially functional artificial metallo-haloalkane dehalogenase containing catalytic zinc. Biotech 2018, 8, 314. [Google Scholar]
- Maier, J.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.J. A point-charge force field for molecular mechanics simulations of proteins. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charge. AM1-BCC model: II. Parameterization and validation. J. Comp. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Koilman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comp. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock VINA: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]








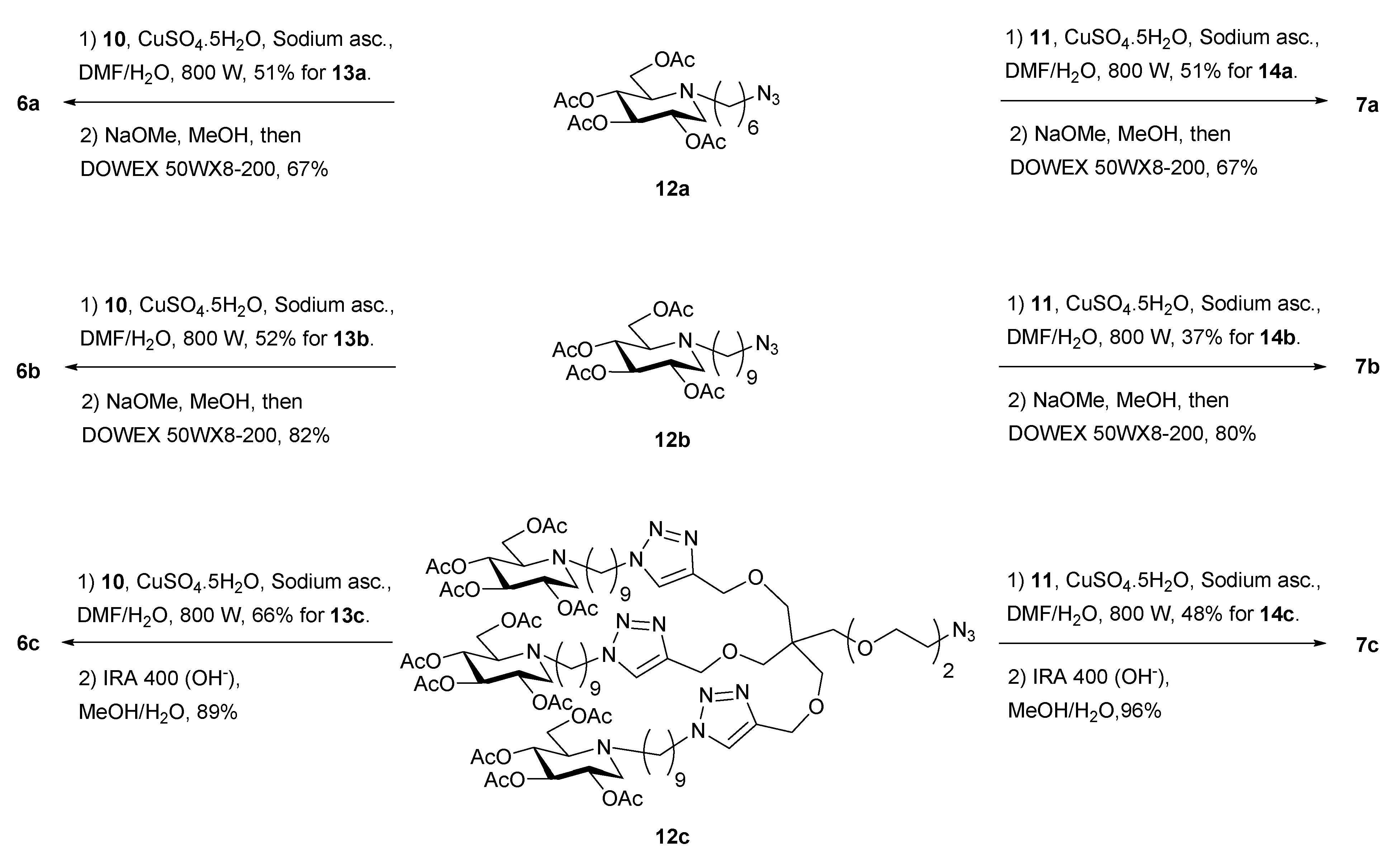
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
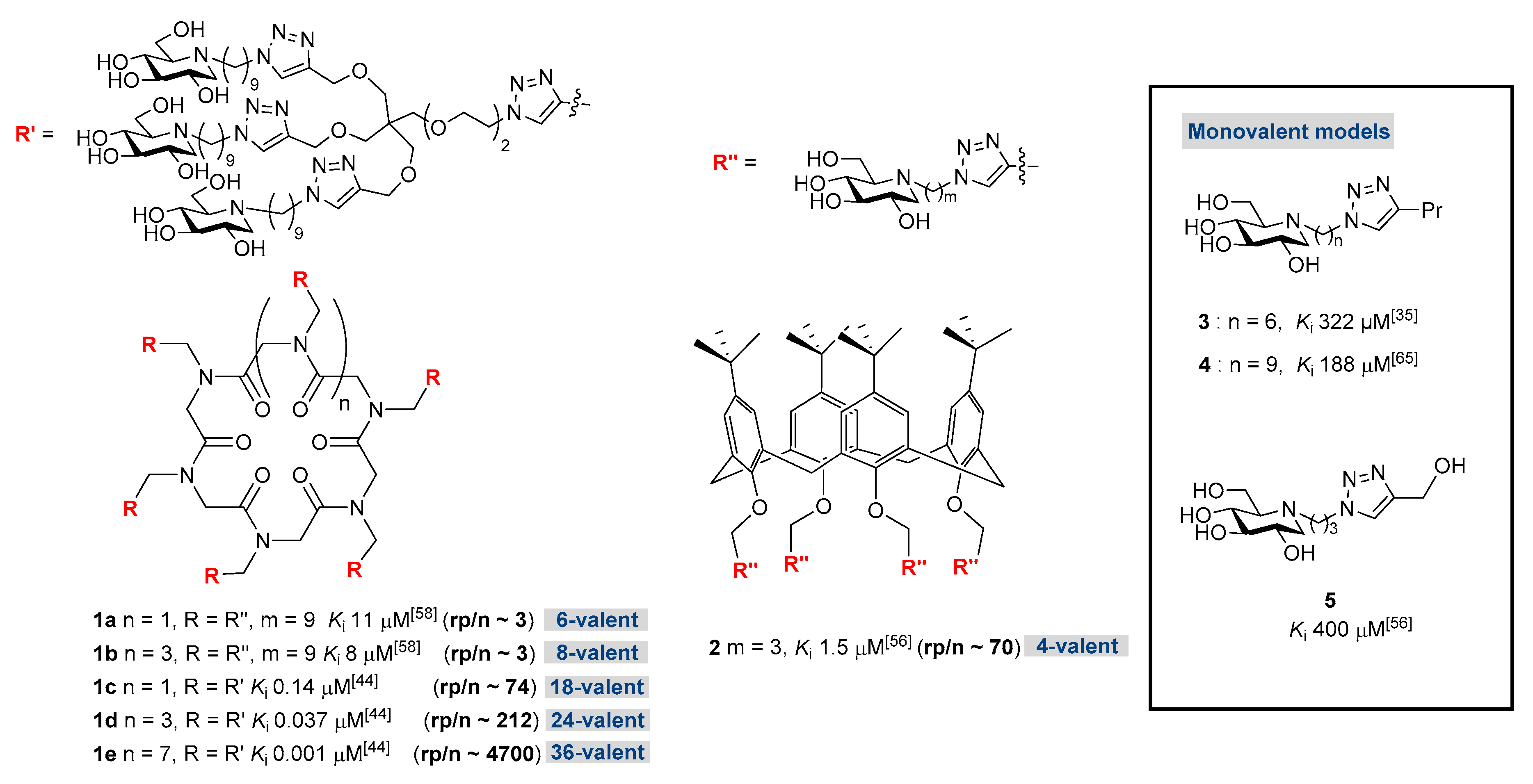
| Entry | Compound | Alkyl Chain Length | DNJ Unit | Kia (µM) | rpb | rp/nc |
|---|---|---|---|---|---|---|
| 1 | 3 [35] | C6 | 1 | 322 [35] | - | - |
| 2 | 4 [65] | C9 | 1 | 188 [65] | - | - |
| 3 | 7a | C6 | 6 | 7.7 ± 0.7 | 42 | 7 |
| 4 | 7b | C9 | 6 | 0.38 ± 0.01 | 495 | 82 |
| 5 | 6a | C6 | 8 | 80 ± 24 | 4 | 0.5 |
| 6 | 6b | C9 | 8 | 0.32 ± 0.05 | 588 | 73 |
| 7 | 7c | C9 | 18 | 0.092 ± 0.008 d 0.213 ± 0.042 | 2043 | 113 |
| 8 | 6c | C9 | 24 | 0.050±0.012 e | 3760 | 157 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, J.P.; Tommasone, S.; Della Sala, P.; Gaeta, C.; Talotta, C.; Tarnus, C.; Neri, P.; Bodlenner, A.; Compain, P. Synthesis and Glycosidase Inhibition Properties of Calix[8]arene-Based Iminosugar Click Clusters. Pharmaceuticals 2020, 13, 366. https://doi.org/10.3390/ph13110366
Schneider JP, Tommasone S, Della Sala P, Gaeta C, Talotta C, Tarnus C, Neri P, Bodlenner A, Compain P. Synthesis and Glycosidase Inhibition Properties of Calix[8]arene-Based Iminosugar Click Clusters. Pharmaceuticals. 2020; 13(11):366. https://doi.org/10.3390/ph13110366
Chicago/Turabian StyleSchneider, Jérémy P., Stefano Tommasone, Paolo Della Sala, Carmine Gaeta, Carmen Talotta, Céline Tarnus, Placido Neri, Anne Bodlenner, and Philippe Compain. 2020. "Synthesis and Glycosidase Inhibition Properties of Calix[8]arene-Based Iminosugar Click Clusters" Pharmaceuticals 13, no. 11: 366. https://doi.org/10.3390/ph13110366
APA StyleSchneider, J. P., Tommasone, S., Della Sala, P., Gaeta, C., Talotta, C., Tarnus, C., Neri, P., Bodlenner, A., & Compain, P. (2020). Synthesis and Glycosidase Inhibition Properties of Calix[8]arene-Based Iminosugar Click Clusters. Pharmaceuticals, 13(11), 366. https://doi.org/10.3390/ph13110366