Assembly of Peptidoglycan Fragments—A Synthetic Challenge



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
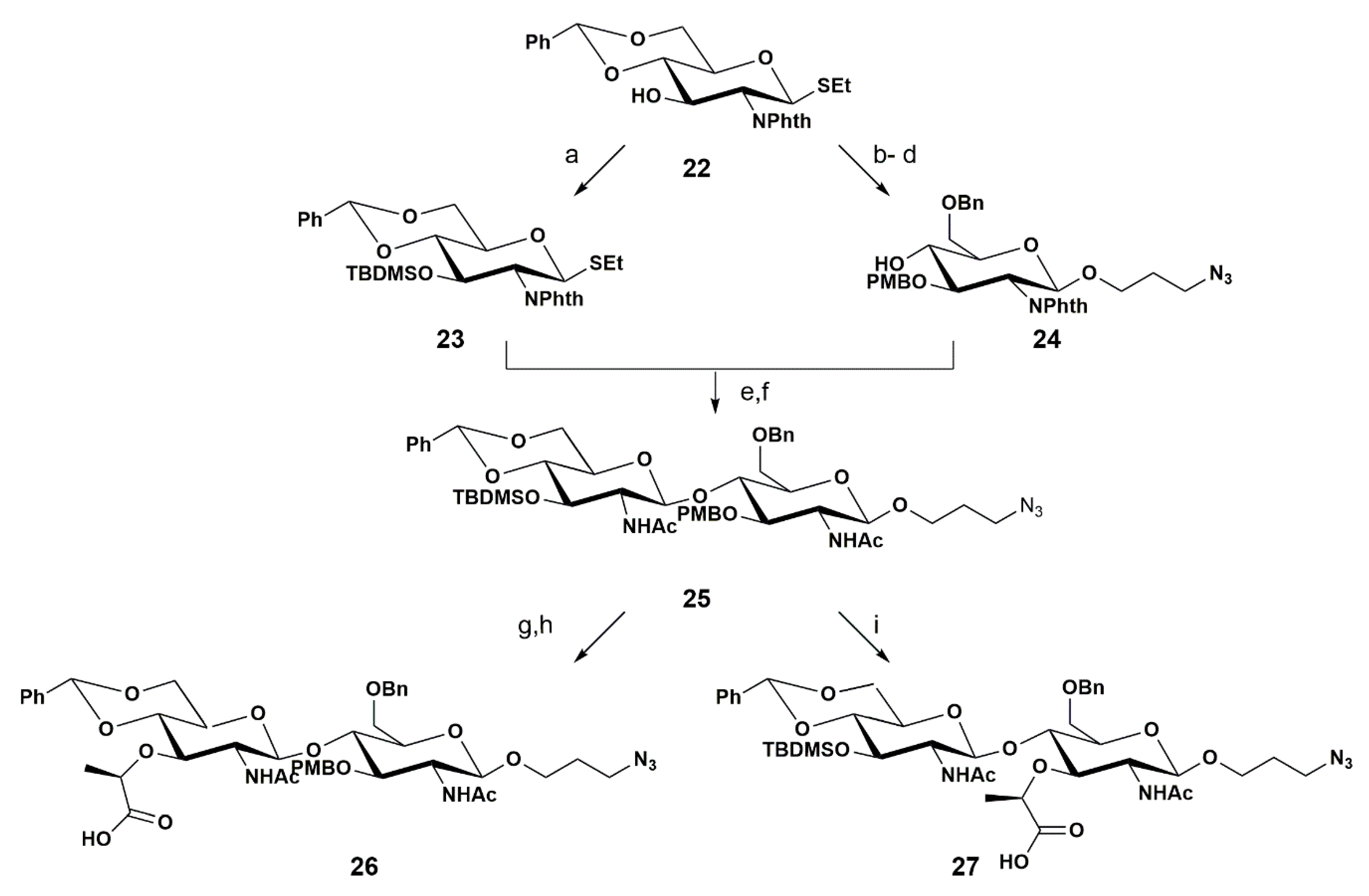{kind=link}
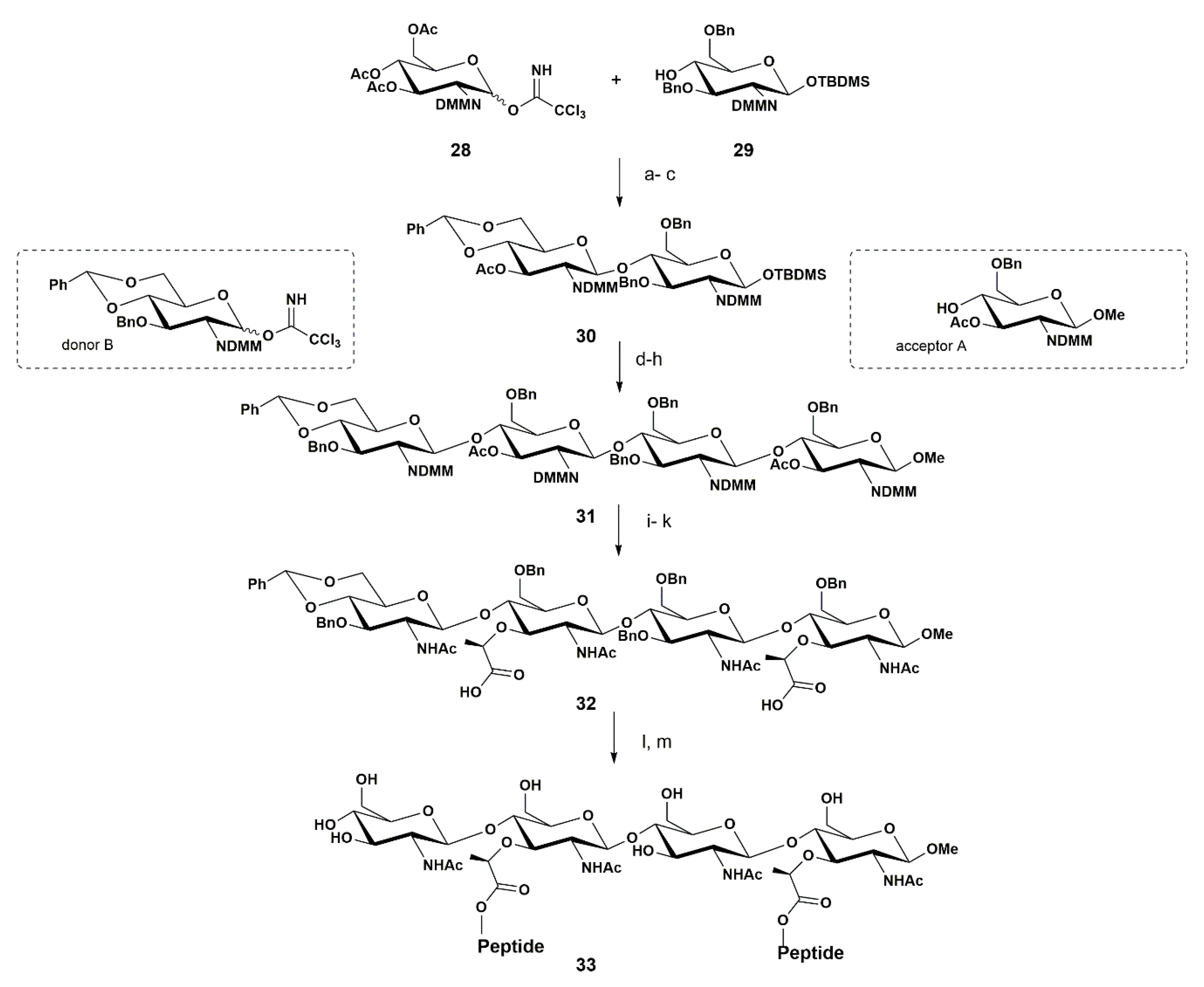{kind=link}
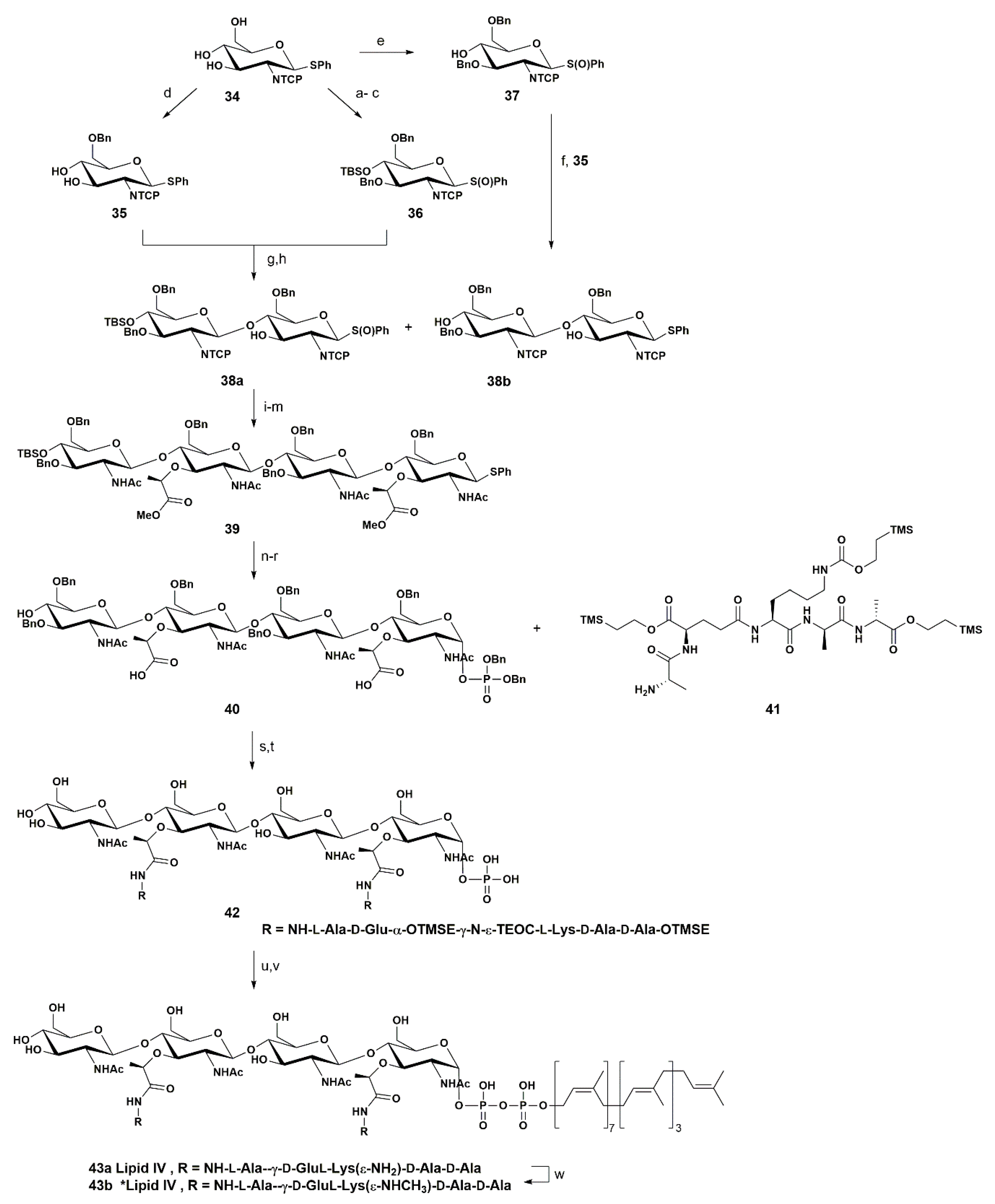{kind=link}
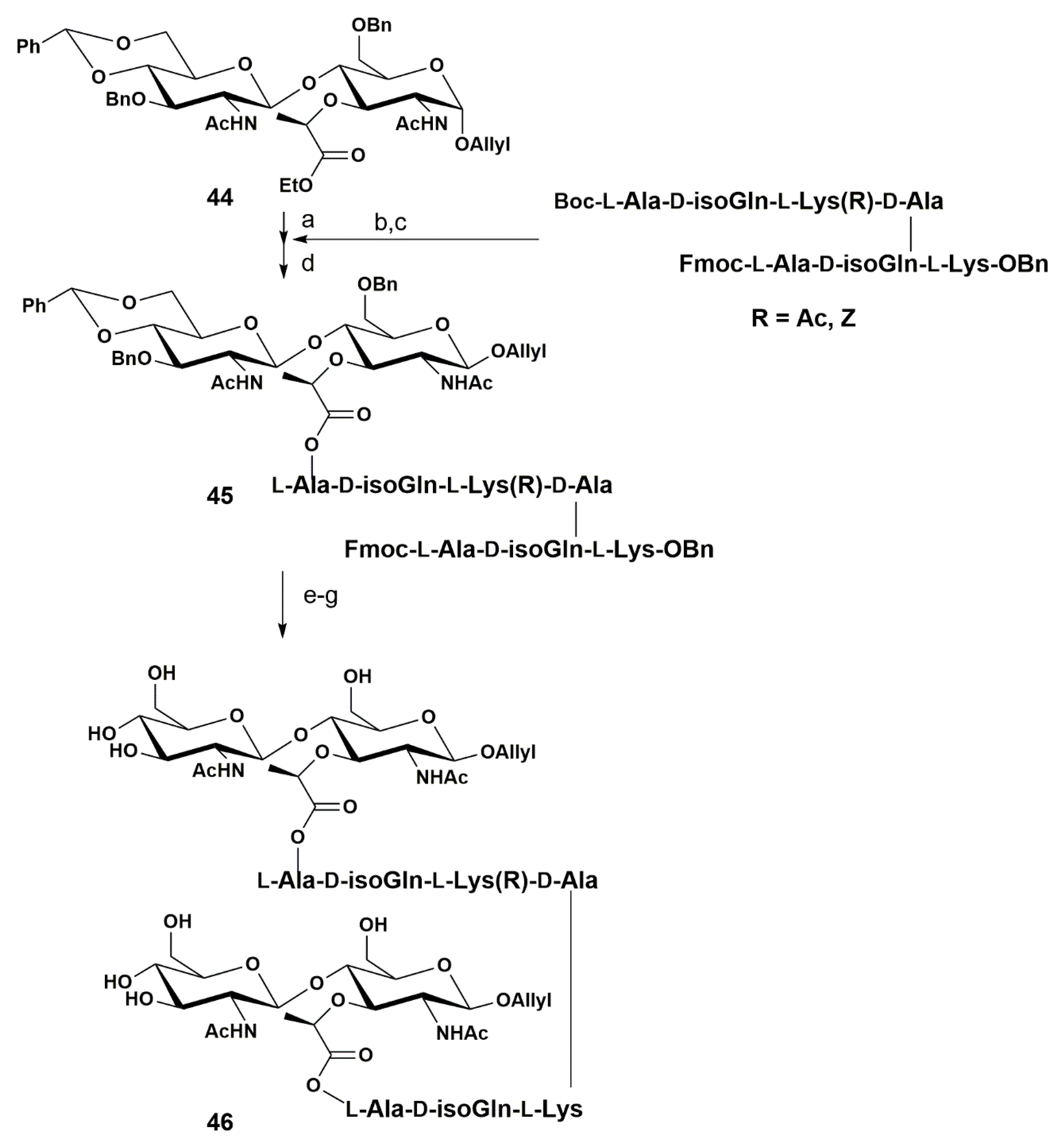{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
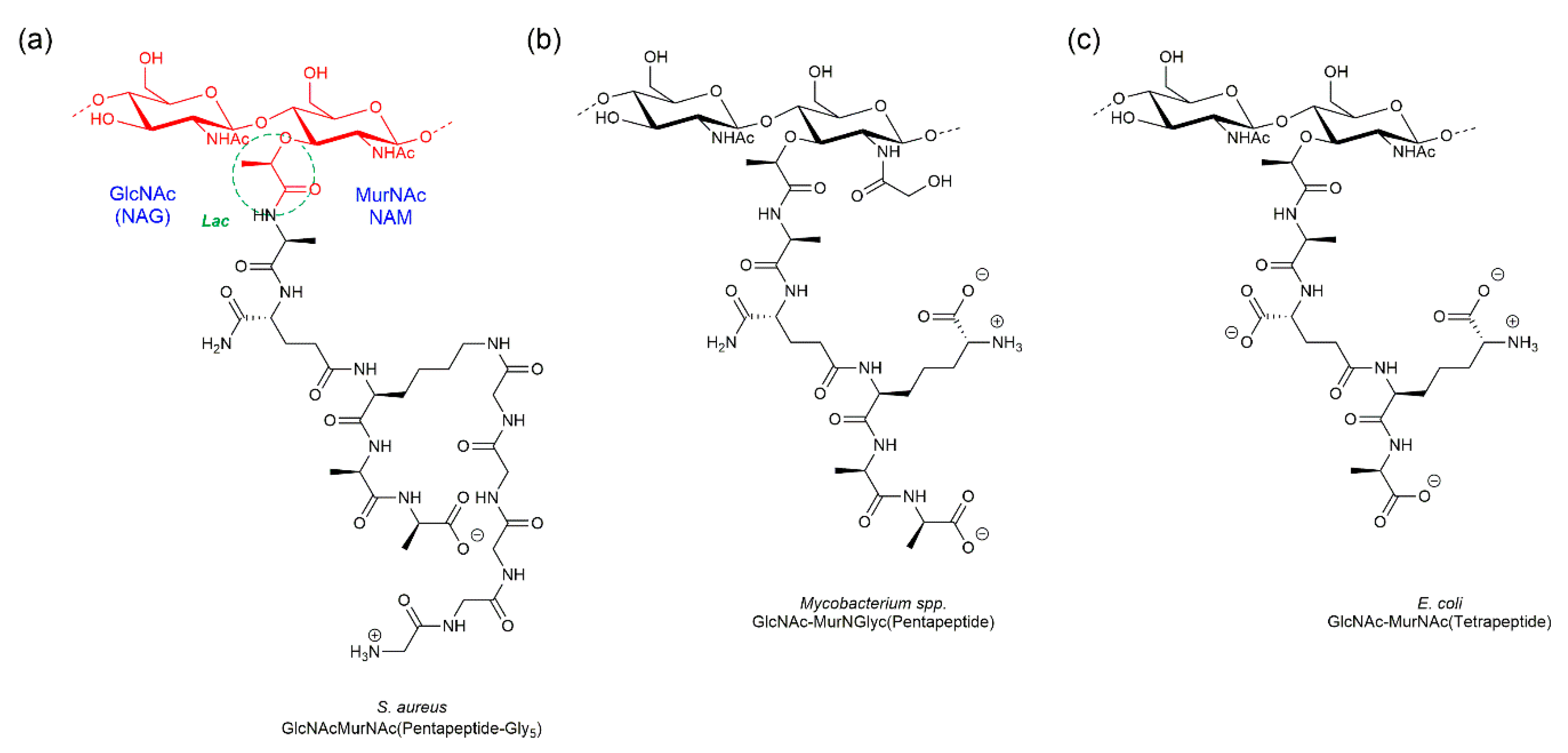
:1. Introduction
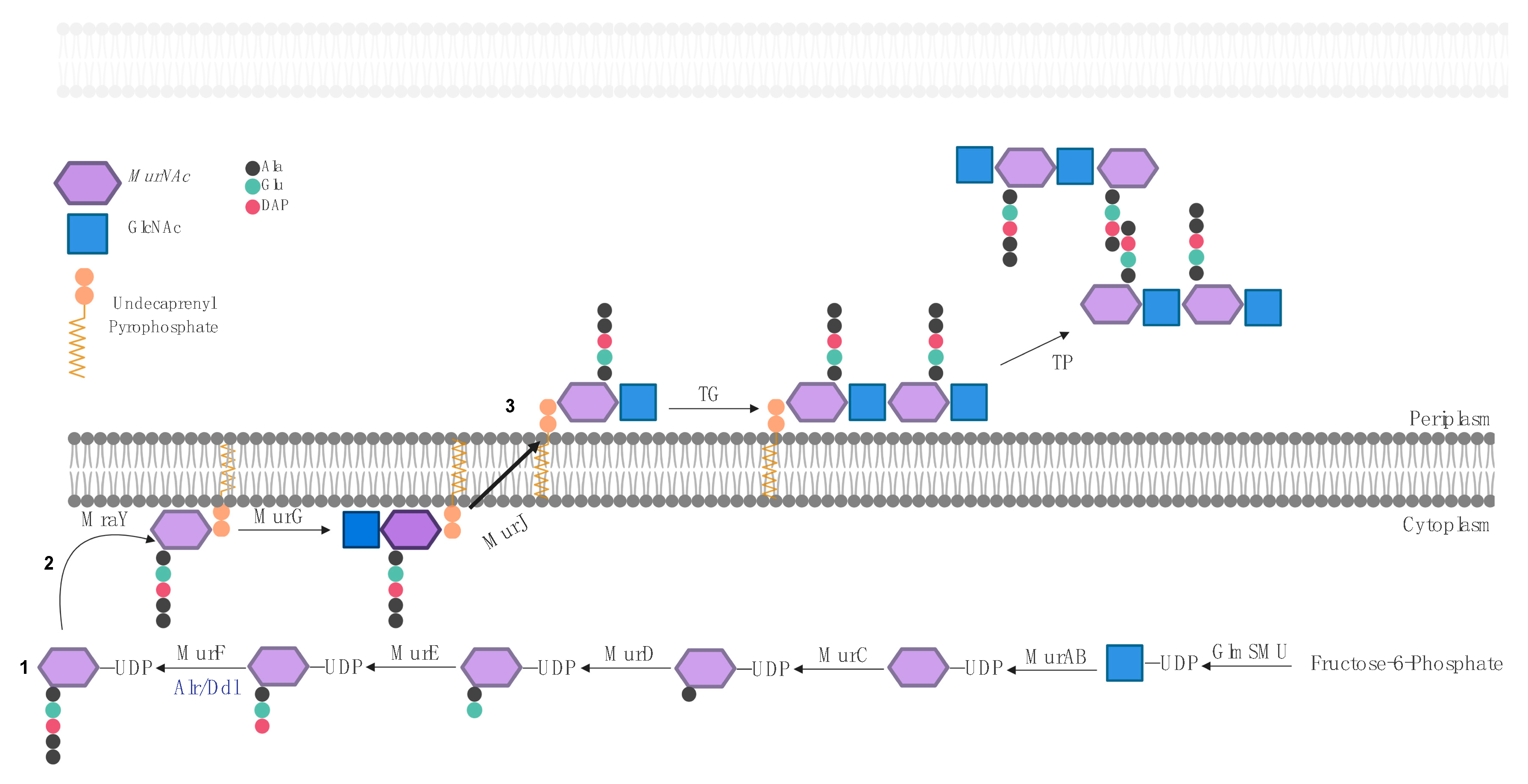
2. Biosynthesis of the Bacterial Peptidoglycan
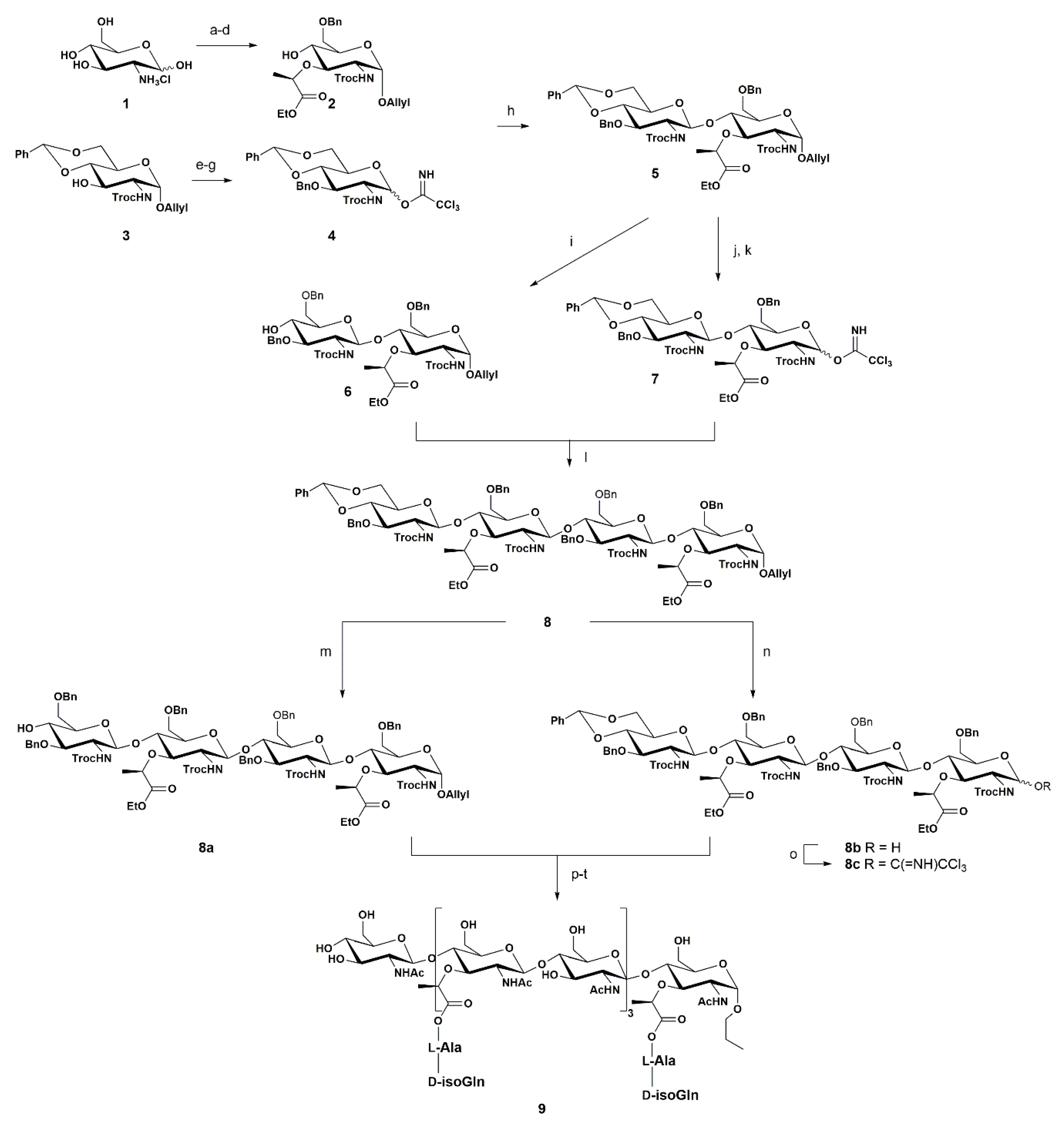
3. Synthetic Approaches towards PGN Fragments from Glucosamine I
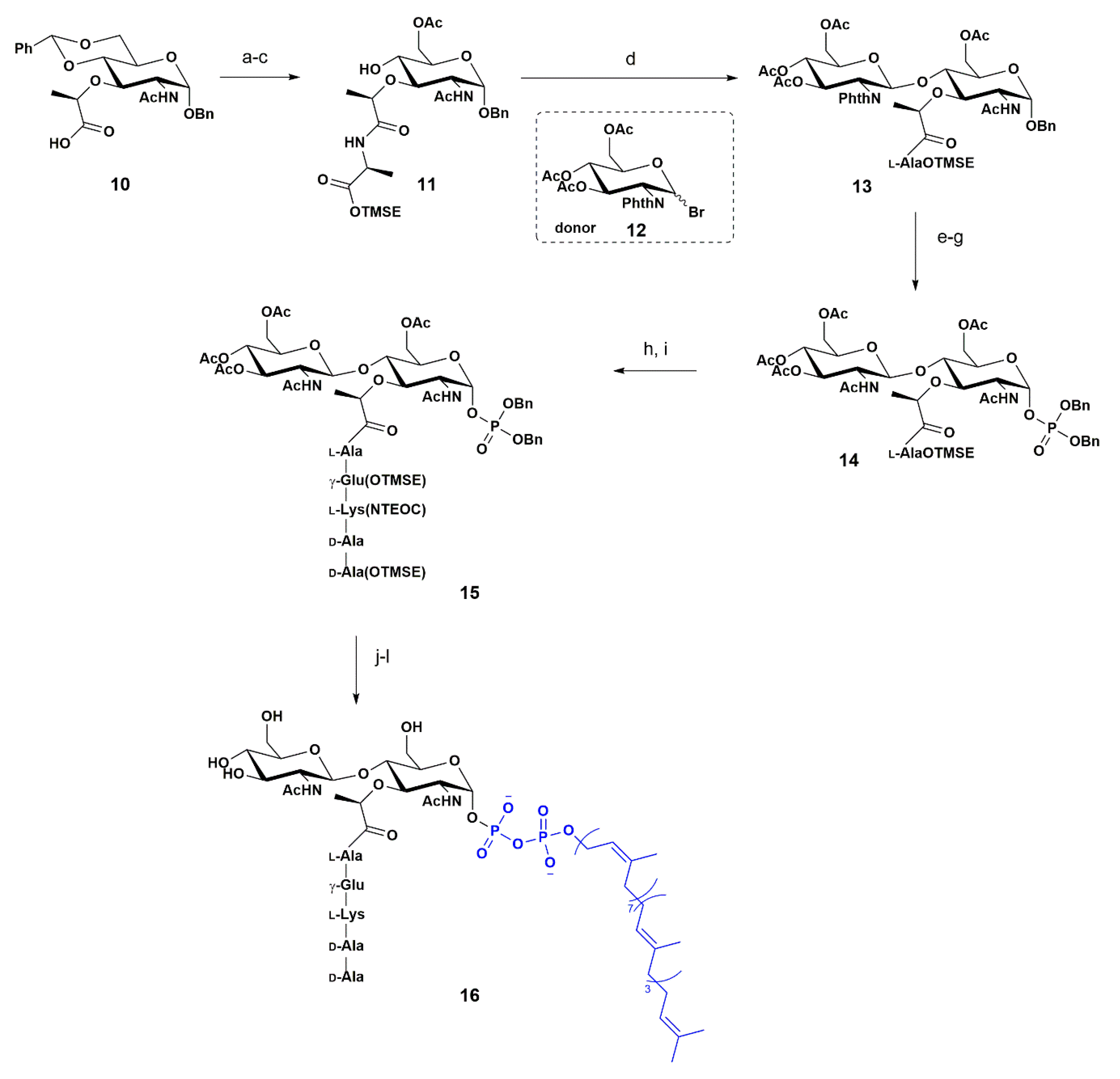
4. Synthetic Approaches towards the Carbohydrate Backbone of PGN from Biopolymers
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell-walls and their taxonomic implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtje, J.V. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 1998, 62, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roemer, T.; Schneider, T.; Pinho, M.G. Auxiliary factors: A chink in the armor of MRSA resistance to beta-lactam antibiotics. Curr. Opin. Microbiol. 2013, 16, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Courvalin, P. Vancomycin resistance in gram-positive cocci. Clin. Infect. Dis. 2006, 42, S25–S34. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, W.; Joris, B.; Charlier, P.; Foster, S. Bacterial peptidoglycan (murein) hydrolases. Fems Microbiol. Rev. 2008, 32, 259–286. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Uehara, T.; Bernhardt, T.G. Beta-Lactam Antibiotics Induce a Lethal Malfunctioning of the Bacterial Cell Wall Synthesis Machinery. Cell 2014, 159, 1300–1311. [Google Scholar] [CrossRef] [Green Version]
- Catalao, M.J.; Gil, F.; Moniz-Pereira, J.; Sao-Jose, C.; Pimentel, M. Diversity in bacterial lysis systems: Bacteriophages show the way. Fems Microbiol. Rev. 2013, 37, 554–571. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.H.; Weber, A.N.; Atilano, M.L.; Filipe, S.R.; Gay, N.J.; Ligoxygakis, P. Sensing of Gram-positive bacteria in Drosophila: GNBP1 is needed to process and present peptidoglycan to PGRP-SA. Embo J. 2006, 25, 5005–5014. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yu, B. Recent advances in the synthesis of chitooligosaccharides and congeners. Tetrahedron 2014, 70, 1023–1046. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, T.A.; Yang, Y.L.; Wu, Q.Y.; Yang, Q.; Yu, B.A. Synthesis, Evaluation, and Mechanism of N,N,N-Trimethyl-D-glucosamine-(1 -> 4)-chitooligosaccharides as Selective Inhibitors of Glycosyl Hydrolase Family 20 beta-N-Acetyl-D-hexosaminidases. Chembiochem 2011, 12, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.W.; Chen, K.T.; Cheng, T.J.R.; Wong, C.H.; Cheng, W.C. A New Synthetic Approach toward Bacterial Transglycosylase Substrates, Lipid II and Lipid IV. Org. Lett. 2011, 13, 4600–4603. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, N.; Brossay, A.; Gasciolli, V.; Bono, J.J.; Baron, A.; Beau, J.M.; Urban, D.; Boyer, F.D.; Vauzeilles, B. Synthesis of lipo-chitooligosaccharide analogues and their interaction with LYR3, a high affinity binding protein for Nod factors and Myc-LCOs. Org. Biomol. Chem. 2017, 15, 7802–7812. [Google Scholar] [CrossRef] [PubMed]
- Enugala, R.; Carvalho, L.C.R.; Pires, M.J.D.; Marques, M.M.B. Stereoselective Glycosylation of Glucosamine: The Role of the N-Protecting Group. Chem. Asian J. 2012, 7, 2482–2501. [Google Scholar] [CrossRef] [PubMed]
- Bongat, A.F.G.; Demchenko, A.V. Recent trends in the synthesis of O-glycosides of 2-amino-2-deoxysugars. Carbohydr. Res. 2007, 342, 374–406. [Google Scholar] [CrossRef] [PubMed]
- Arihara, R.; Nakamura, S.; Hashimoto, S. Direct and stereoselective synthesis of 2-acetamido-2-deoxy-beta-D-glycopyranosides by using the phosphite method. Angew. Chem. Int. Ed. 2005, 44, 2245–2249. [Google Scholar] [CrossRef] [PubMed]
- Stevenin, A.; Boyer, F.D.; Beau, J.M. Highly Selective Formation of beta-Glycosides of N-Acetylglucosamine Using Catalytic Iron(III) Triflate. Eur. J. Org. Chem. 2012, 2012, 1699–1702. [Google Scholar] [CrossRef]
- Crich, D.; Dudkin, V. Why are the hydroxy groups of partially protected N-acetylglucosamine derivatives such poor glycosyl accepters, and what can be done about it? A comparative study of the reactivity of N-acetyl-, N-phthalimido-, and 2-azido-2-deoxy-glucosamine derivatives in glycosylation. 2-picolinyl ethers as reactivity-enhancing replacements for benzyl ethers. J. Am. Chem. Soc. 2001, 123, 6819–6825. [Google Scholar] [CrossRef]
- Badet, B.; Vermoote, P.; Legoffic, F. Glucosamine synthetase from escherichia-coli kinetic mechanism and inhibition by n3-fumaroyl-l-2,3-diaminopropionic derivatives. Biochemistry 1988, 27, 2282–2287. [Google Scholar] [CrossRef]
- Badetdenisot, M.A.; Badet, B. Chemical modification of glucosamine-6-phosphate synthase by diethyl pyrocarbonate—Evidence of histidine requirement for enzymatic-activity. Arch. Biochem. Biophys. 1992, 292, 475–478. [Google Scholar] [CrossRef]
- Gehring, A.M.; Lees, W.J.; Mindiola, D.J.; Walsh, C.T.; Brown, E.D. Acetyltransfer precedes uridylyltransfer in the formation of UDP-N-acetylglucosamine in separable active sites of the bifunctional GlmU protein of Escherichia coli. Biochemistry 1996, 35, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Jolly, L.; Ferrari, P.; Blanot, D.; van Heijenoort, J.; Fassy, F.; Mengin-Lecreulx, D. Reaction mechanism of phosphoglucosamine mutase from Escherichia coli. Eur. J. Biochem. 1999, 262, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Menginlecreulx, D.; Vanheijenoort, J. Copurification of glucosamine-1-phosphate acetyltransferase and n-acetylglucosamine-1-phosphate uridyltransferase activities of escherichia-coli-characterization of the glmu gene-product as a bifunctional enzyme catalyzing 2 subsequent steps in the pathway for udp-n-acetylglucosamine synthesis. J. Bacteriol. 1994, 176, 5788–5795. [Google Scholar] [CrossRef] [Green Version]
- MenginLecreulx, D.; vanHeijenoort, J. Characterization of the essential gene glmM encoding phosphoglucosamine mutase in Escherichia coli. J. Biol. Chem. 1996, 271, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Benson, T.E.; Marquardt, J.L.; Marquardt, A.C.; Etzkorn, F.A.; Walsh, C.T. Overexpression, purification, and mechanistic study of udp-n-acetylenolpyruvylglucosamine reductase. Biochemistry 1993, 32, 2024–2030. [Google Scholar] [CrossRef] [PubMed]
- Benson, T.E.; Walsh, C.T.; Massey, V. Kinetic characterization of wild-type and S229A mutant MurB: Evidence for the role of ser 229 as a general acid. Biochemistry 1997, 36, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Krekel, F.; Samland, A.K.; Macheroux, P.; Amrhein, N.; Evans, J.N.S. Determination of the pK(a) value of C115 in MurA (UDP-N-acetylglucosamine enolpyruvyltransferase) from Enterobacter cloacae. Biochemistry 2000, 39, 12671–12677. [Google Scholar] [CrossRef]
- Doublet, P.; Vanheijenoort, J.; Bohin, J.P.; Menginlecreulx, D. The muri gene of escherichia-coli is an essential gene that encodes a glutamate racemase activity. J. Bacteriol. 1993, 175, 2970–2979. [Google Scholar] [CrossRef] [Green Version]
- Doublet, P.; Vanheijenoort, J.; Menginlecreulx, D. The glutamate racemase activity from escherichia-coli is regulated by peptidoglycan precursor udp-n-acetylmuramoyl-l-alanine. Biochemistry 1994, 33, 5285–5290. [Google Scholar] [CrossRef]
- Scaletti, E.R.; Luckner, S.R.; Krause, K.L. Structural features and kinetic characterization of alanine racemase from Staphylococcus aureus (Mu50). Acta Crystallogr. Sect. D Struct. Biol. 2012, 68, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Kouidmi, I.; Levesque, R.C.; Paradis-Bleau, C. The biology of Mur ligases as an antibacterial target. Mol. Microbiol. 2014, 94, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Ruane, K.M.; Lloyd, A.J.; Fulop, V.; Dowson, C.G.; Barreteau, H.; Boniface, A.; Dementin, S.; Blanot, D.; Mengin-Lecreulx, D.; Gobec, S.; et al. Specificity Determinants for Lysine Incorporation in Staphylococcus aureus Peptidoglycan as Revealed by the Structure of a MurE Enzyme Ternary Complex. J. Biol. Chem. 2013, 288, 33439–33448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, J.A.; Auger, G.; Martin, L.; Fanchon, E.; Blanot, D.; La Beller, D.; van Heijenoort, J.; Dideberg, O. Determination of the MurD mechanism through crystallographic analysis of enzyme complexes. J. Mol. Biol. 1999, 289, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, J.J.; Jin, H.Y.; Jacobson, B.L.; Chang, C.Y.Y.; Einspahr, H.M.; Villafranca, J.J. Kinetic and crystallographic studies of Escherichia coli UDP-N-acetylmuramate: L-alanine ligase. Protein Sci. 1996, 5, 2566–2574. [Google Scholar] [CrossRef] [Green Version]
- Zawadzke, L.E.; Bugg, T.D.H.; Walsh, C.T. Existence of 2 d-alanine-d-alanine ligases in escherichia-coli-cloning and sequencing of the ddla gene and purification and characterization of the ddla and ddlb enzymes. Biochemistry 1991, 30, 1673–1682. [Google Scholar] [CrossRef]
- Anderson, M.S.; Eveland, S.S.; Onishi, H.R.; Pompliano, D.L. Kinetic mechanism of the Escherichia coli UDPMurNAc-tripeptide D-alanyl-D-alanine-adding enzyme: Use of a glutathione S-transferase fusion. Biochemistry 1996, 35, 16264–16269. [Google Scholar] [CrossRef]
- Albar, O.A.M.; Oconnor, C.D.; Giles, I.G.; Akhtar, M. D-alanine-d-alanine ligase of escherichia-coli-expression, purification and inhibitory studies on the cloned enzyme. Biochem. J. 1992, 282, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Al-Dabbagh, B.; Henry, X.; El Ghachi, M.; Auger, G.; Blanot, D.; Parquet, C.; Mengin-Lecreulx, D.; Bouhss, A. Active site mapping of MraY, a member of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily, catalyzing the first membrane step of peptidoglycan biosynthesis. Biochemistry 2008, 47, 8919–8928. [Google Scholar] [CrossRef]
- Chung, B.C.; Zhao, J.S.; Gillespie, R.A.; Kwon, D.Y.; Guan, Z.Q.; Hong, J.Y.; Zhou, P.; Lee, S.Y. Crystal Structure of MraY, an Essential Membrane Enzyme for Bacterial Cell Wall Synthesis. Science 2013, 341, 1012–1016. [Google Scholar] [CrossRef] [Green Version]
- Bouhss, A.; Crouvoisier, M.; Blanot, D.; Mengin-Lecreulx, D. Purification and characterization of the bacterial MraY translocase catalyzing the first membrane step of peptidoglycan biosynthesis. J. Biol. Chem. 2004, 279, 29974–29980. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.S.; Matsuhashi, M.; Haskin, M.A.; Strominger, J.L. Lipid-phosphoacetylmuramyl-pentapeptide and lipid-phosphodisaccharide-pentapeptide–presumed membrane transport intermediates in cell wall synthesis. Proc. Natl. Acad. Sci. USA 1965, 53, 881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Heijenoort, J. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol. Mol. Biol. Rev. 2007, 71, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menginlecreulx, D.; Texier, L.; Rousseau, M.; Vanheijenoort, J. The murg gene of escherichia-coli codes for the udp-n-acetylglucosamine-n-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol n-acetylglucosamine transferase involved in the membrane steps of peptidoglycan synthesis. J. Bacteriol. 1991, 173, 4625–4636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, T.; van Dam, V.; Sijbrandi, R.; Vernet, T.; Zapun, A.; Bouhss, A.; Diepeveen-de Bruin, M.; Nguyen-Disteche, M.; de Kruijff, B.; Breukink, E. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. Embo J. 2011, 30, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 15553–15557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sham, L.T.; Butler, E.K.; Lebar, M.D.; Kahne, D.; Bernhardt, T.G.; Ruiz, N. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 2014, 345, 220–222. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, N. Lipid Flippases for Bacterial Peptidoglycan Biosynthesis. Lipid Insights 2015, 8, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Teo, A.C.K.; Roper, D.I. Core Steps of Membrane-Bound Peptidoglycan Biosynthesis: Recent Advances, Insight and Opportunities. Antibiot. Basel 2015, 4, 495–520. [Google Scholar] [CrossRef]
- Meeske, A.J.; Sham, L.T.; Kimsey, H.; Koo, B.M.; Gross, C.A.; Bernhardt, T.G.; Rudner, D.Z. MurJ and a novel lipid II flippase are required for cell wall biogenesis in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2015, 112, 6437–6442. [Google Scholar] [CrossRef] [Green Version]
- Egan, A.J.F.; Errington, J.; Vollmer, W. Regulation of peptidoglycan synthesis and remodelling. Nat. Rev. Microbiol. 2020, 18, 446–460. [Google Scholar] [CrossRef]
- Manat, G.; Roure, S.; Auger, R.; Bouhss, A.; Barreteau, H.; Mengin-Lecreulx, D.; Touzé, T. Deciphering the metabolism of undecaprenyl-phosphate: The bacterial cell-wall unit carrier at the membrane frontier. Microb. Drug Resist. 2014, 20, 199–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebar, M.D.; May, J.M.; Meeske, A.J.; Leiman, S.A.; Lupoli, T.J.; Tsukamoto, H.; Losick, R.; Rudner, D.Z.; Walker, S.; Kahne, D. Reconstitution of Peptidoglycan Cross-Linking Leads to Improved Fluorescent Probes of Cell Wall Synthesis. J. Am. Chem. Soc. 2014, 136, 10874–10877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travassos, L.H.; Girardin, S.E.; Philpott, D.J.; Blanot, D.; Nahori, M.A.; Werts, C.; Boneca, I.G. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 2004, 5, 1000–1006. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.J.; Underhill, D.M. Peptidoglycan recognition by the innate immune system. Nat. Rev. Immunol. 2018, 18, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Inamura, S.; Fukase, K.; Kusumoto, S. Synthetic study of peptidoglycan partial structures. Synthesis of tetrasaccharide and octasaccharide fragments. Tetrahedron Lett. 2001, 42, 7613–7616. [Google Scholar] [CrossRef]
- Kusumoto, S.; Yamamoto, K.; Imoto, M.; Inage, M.; Tsujimoto, M.; Kotani, S.; Shiba, T. Chemical synthesis and biological-activities of 2 disaccharide dipeptides corresponding to the repeating units of bacterial peptidoglycan. Bull. Chem. Soc. Jpn. 1986, 59, 1411–1417. [Google Scholar] [CrossRef]
- Schwartz, B.; Markwalder, J.A.; Wang, Y. Lipid II: Total synthesis of the bacterial cell wall precursor and utilization as a substrate for glycosyltransfer and transpeptidation by penicillin binding protein (PBP) 1b of Eschericia coli. J. Am. Chem. Soc. 2001, 123, 11638–11643. [Google Scholar] [CrossRef]
- Saha, S.L.; Van Nieuwenhze, M.S.; Hornback, W.J.; Aikins, J.A.; Blaszczak, L.C. Synthesis of an orthogonally protected precursor to the glycan repeating unit of the bacterial cell wall. Org. Lett. 2001, 3, 3575–3577. [Google Scholar] [CrossRef]
- VanNieuwenhze, M.S.; Mauldin, S.C.; Zia-Ebrahimi, M.; Winger, B.E.; Hornback, W.J.; Saha, S.L.; Aikins, J.A.; Blaszczak, L.C. The first total synthesis of lipid II: The final monomeric intermediate in bacterial cell wall biosynthesis. J. Am. Chem. Soc. 2002, 124, 3656–3660. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Siriwardena, A.; Boons, G.J. A highly convergent approach for the synthesis of disaccharide repeating units of peptidoglycan. Tetrahedron Lett. 2002, 43, 7805–7807. [Google Scholar] [CrossRef]
- Hesek, D.; Lee, M.J.; Morio, K.I.; Mobashery, S. Synthesis of a fragment of bacterial cell wall. J. Org. Chem. 2004, 69, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Inamura, S.; Fujimoto, Y.; Kawasaki, A.; Shiokawa, Z.; Woelk, E.; Heine, H.; Lindner, B.; Inohara, N.; Kusumoto, S.; Fukase, K. Synthesis of peptidoglycan fragments and evaluation of their biological activity. Org. Biomol. Chem. 2006, 4, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fechter, E.J.; Wang, T.S.A.; Barrett, D.; Walker, S.; Kahne, D.E. Synthesis of heptaprenyl-lipid IV to analyze peptidoglycan glycosyltransferases. J. Am. Chem. Soc. 2007, 129, 3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.Y.; Lo, M.C.; Brunner, L.; Walker, D.; Kahne, D.; Walker, S. Better substrates for bacterial transglycosylases. J. Am. Chem. Soc. 2001, 123, 3155–3156. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Konishi, Y.; Kubo, O.; Hasegawa, M.; Inohara, N.; Fukase, K. Synthesis of crosslinked peptidoglycan fragments for investigation of their immunobiological functions. Tetrahedron Lett. 2009, 50, 3631–3634. [Google Scholar] [CrossRef]
- Uehara, A.; Yang, S.; Fujimoto, Y.; Fukase, K.; Kusumoto, S.; Shibata, K.; Sugawara, S.; Takada, H. Muramyldipeptide and diaminopimelic acid-containing desmuramylpeptides in combination with chemically synthesized Toll-like receptor agonists synergistically induced production of interleukin-8 in a NOD2- and NOD1-dependent manner, respectively, in human monocytic cells in culture. Cell. Microbiol. 2005, 7, 53–61. [Google Scholar] [CrossRef]
- Wang, N.; Huang, C.; Hasegawa, M.; Inohara, N.; Fujimoto, Y.; Fukase, K. Glycan Sequence-Dependent Nod2 Activation Investigated by Using a Chemically Synthesized Bacterial Peptidoglycan Fragment Library. Chembiochem 2013, 14, 482–488. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Pradipta, A.R.; Inohara, N.; Fukase, K. Peptidoglycan as Nod1 ligand; fragment structures in the environment, chemical synthesis, and their innate immunostimulation. Nat. Prod. Rep. 2012, 29, 568–579. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Inamura, S.; Kawasaki, A.; Shiokawa, Z.; Shimoyama, A.; Hashimoto, T.; Kusumoto, S.; Fukase, K. Chemical synthesis of peptidoglycan fragments for elucidation of the immunostimulating mechanism. J. Endotoxin Res. 2007, 13, 189–196. [Google Scholar] [CrossRef]
- Enugala, R.; Pires, M.J.D.; Marques, M.M.B. Synthesis of the NAG-NAM disaccharide via a versatile intermediate. Carbohydr. Res. 2014, 384, 112–118. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Matsuo, Y.; Pradipta, A.R.; Inohara, N.; Fujimoto, Y.; Fukase, K. Synthesis of characteristic Mycobacterium peptidoglycan (PGN) fragments utilizing with chemoenzymatic preparation of meso-diaminopimelic acid (DAP), and their modulation of innate immune responses. Org. Biomol. Chem. 2016, 14, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Enugala, R.; Marques, M.M.B. Synthesis of a 3-hydroxyl-free N-acetyl glucosamine disaccharide. Arkivoc 2012, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Marqvorsen, M.H.S.; Pedersen, M.J.; Rasmussen, M.R.; Kristensen, S.K.; Dahl-Lassen, R.; Jensen, H.H. Why Is Direct Glycosylation with N-Acetylglucosamine Donors Such a Poor Reaction and What Can Be Done about It? J. Org. Chem. 2017, 82, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Xolin, A.; Stevenin, A.; Pucheault, M.; Norsikian, S.; Boyer, F.D.; Beau, J.M. Glycosylation with N-acetyl glycosamine donors using catalytic iron(III) triflate: From microwave batch chemistry to a scalable continuous-flow process. Org. Chem. Front. 2014, 1, 992–1000. [Google Scholar] [CrossRef]
- Enugala, R.; Carvalho, L.C.R.; Marques, M.M.B. Towards Glucosamine Building Blocks: Regioselective One-Pot Protection and Deallylation Procedures. Synlett 2010, 2711–2716. [Google Scholar] [CrossRef]
- Carvalho, L.R.; Corvo, M.C.; Enugala, R.; Marques, M.M.B.; Cabrita, E.J. Application of HR-MAS NMR in the solid-phase synthesis of a glycopeptide using Sieber amide resin. Magn. Reson. Chem. 2010, 48, 323–330. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Wang, C.C.; Sabbavarapu, N.M.; Podilapu, A.R.; Liao, P.H.; Hung, S.C. “One-Pot” Protection, Glycosylation, and Protection-Glycosylation Strategies of Carbohydrates. Chem. Rev. 2018, 118, 8025–8104. [Google Scholar] [CrossRef]
- Ito, Y.; Kanie, O.; Ogawa, T. Orthogonal glycosylation strategy for rapid assembly of oligosaccharides on a polymer support. Angew. Chem. Int. Ed. Engl. 1996, 35, 2510–2512. [Google Scholar] [CrossRef]
- Carvalho, L.C.R.; Queda, F.; Almeida, C.V.; Filipe, S.R.; Marques, M.M.B. From a Natural Polymer to Relevant NAG-NAM Precursors. Asian J. Org. Chem. 2018, 7, 2544–2551. [Google Scholar] [CrossRef]
- Nishimura, S.I.; Kuzuhara, H.; Takiguchi, Y.; Shimahara, K. Peracetylated chitobiose-preparation by specific degradations of chitin, and chemical manipulations. Carbohydr. Res. 1989, 194, 223–231. [Google Scholar] [CrossRef]
- Chang, R.; Yeager, A.R.; Finney, N.S. Probing the mechanism of a fungal glycosyltransferase essential for cell wall biosynthesis. UDP-chitobiose is not a substrate for chitin synthase. Org. Biomol. Chem. 2003, 1, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Despras, G.; Alix, A.; Urban, D.; Vauzeilles, B.; Beau, J.M. From Chitin to Bioactive Chitooligosaccharides and Conjugates: Access to Lipochitooligosaccharides and the TMG-chitotriomycin. Angew. Chem. Int. Ed. 2014, 53, 11912–11916. [Google Scholar] [CrossRef] [PubMed]
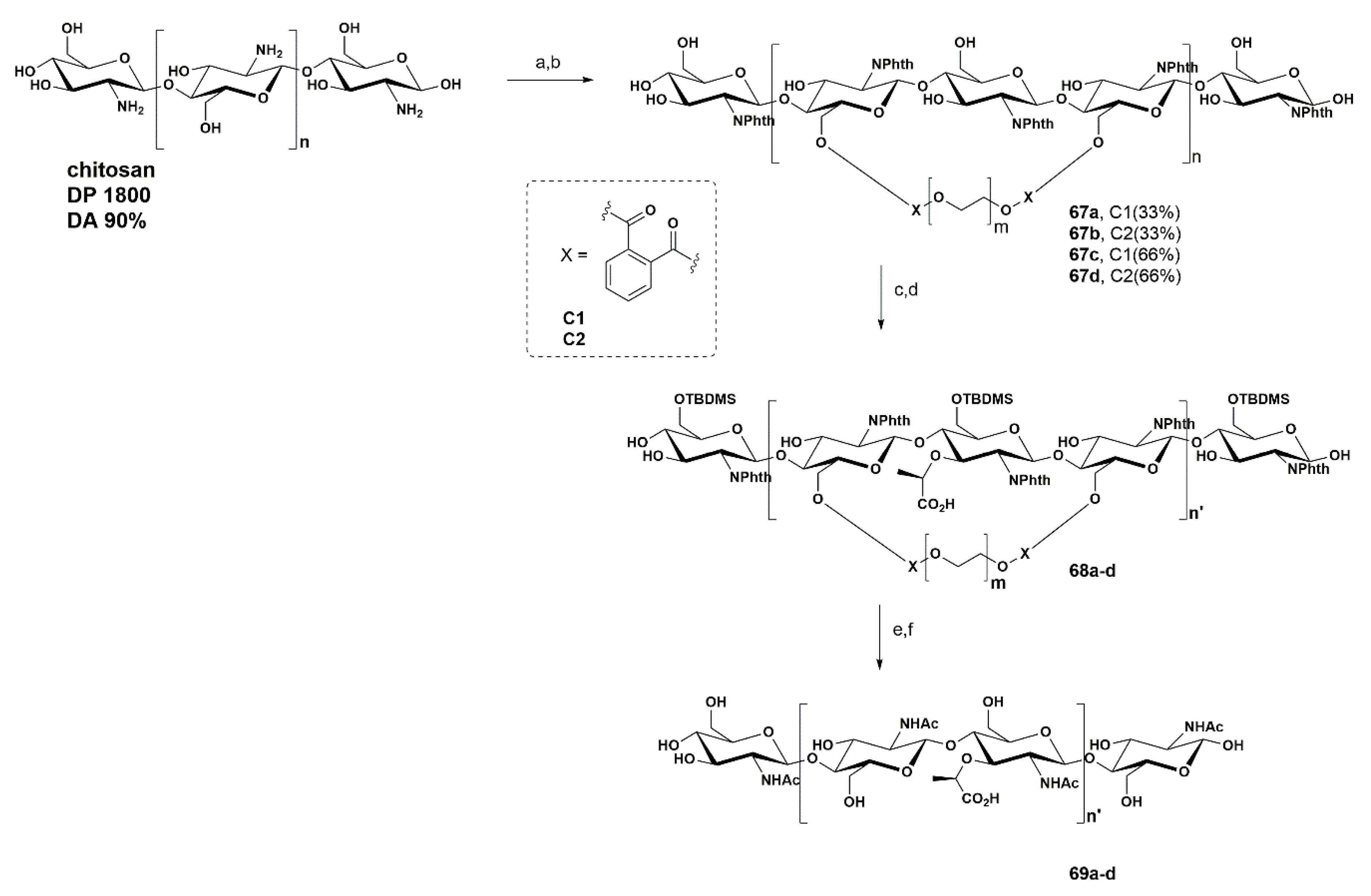
- Queda, F.; Covas, G.; Silva, T.; Santos, C.A.; Bronze, M.R.; Canada, F.J.; Corvo, M.C.; Filipe, S.R.; Marques, M.M.B. A top-down chemo-enzymatic approach towards N-acetylglucosamine-N-acetylmuramic oligosaccharides: Chitosan as a reliable template. Carbohydr. Polym. 2019, 224. [Google Scholar] [CrossRef] [PubMed]
- Goodman, H.; Pollock, J.J.; Iacono, V.J.; Wong, W.; Shockman, G.D. Peptidoglycan loss during hen egg-white lysozyme-inorganic salt lysis of streptococcus-mutans. J. Bacteriol. 1981, 146, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Kurita, K.; Ikeda, H.; Yoshida, Y.; Shimojoh, M.; Harata, M. Chemoselective protection of the amino groups of chitosan by controlled phthaloylation: Facile preparation of a precursor useful for chemical modifications. Biomacromolecules 2002, 3, 1–4. [Google Scholar] [CrossRef]
- Binette, A.; Gagnon, J. Regioselective silylation of N-phthaloylchitosan with TBDMS and TBDPS groups. Biomacromolecules 2007, 8, 1812–1815. [Google Scholar] [CrossRef]
- Lee, M.; Artola-Recolons, C.; Carrasco-Lopez, C.; Martinez-Caballero, S.; Hesek, D.; Spink, E.; Lastochkin, E.; Zhang, W.L.; Hellman, L.M.; Boggess, B.; et al. Cell-Wall Remodeling by the Zinc-Protease AmpDh3 from Pseudomonas aeruginosa. J. Am. Chem. Soc. 2013, 135, 12604–12607. [Google Scholar] [CrossRef] [Green Version]
- Vocadlo, D.J.; Davies, G.J.; Laine, R.; Withers, S.G. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 2001, 412, 835–838. [Google Scholar] [CrossRef]
- Maeda, R.; Matsumoto, M.; Kondo, K. Enzymatic hydrolysis reaction of water-soluble chitin derivatives with egg white lysozyme. J. Ferment. Bioeng. 1997, 84, 478–479. [Google Scholar] [CrossRef]
- Amano, K.I.; Ito, E. Action of lysozyme on partially deacetylated chitin. Eur. J. Biochem. 1978, 85, 97–104. [Google Scholar] [CrossRef]
- Raymond, J.B.; Price, N.P.; Pavelka, M.S. A method for the enzymatic synthesis and HPLC purification of the peptidoglycan precursor UDP-N-acetylmuramic acid. Fems Microbiol. Lett. 2003, 229, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.Y.; Huang, S.H.; Chang, Y.C.; Cheng, W.C.; Cheng, T.J.R.; Wong, C.H. Enzymatic Synthesis of Lipid II and Analogues. Angew. Chem. Int. Ed. 2014, 53, 8060–8065. [Google Scholar] [CrossRef] [PubMed]
- Unsleber, S.; Borisova, M.; Mayer, C. Enzymatic synthesis and semi-preparative isolation of N-acetylmuramic acid 6-phosphate. Carbohydr. Res. 2017, 445, 98–103. [Google Scholar] [CrossRef] [PubMed]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Queda, F.; Covas, G.; Filipe, S.R.; Marques, M.M.B. Assembly of Peptidoglycan Fragments—A Synthetic Challenge. Pharmaceuticals 2020, 13, 392. https://doi.org/10.3390/ph13110392
Queda F, Covas G, Filipe SR, Marques MMB. Assembly of Peptidoglycan Fragments—A Synthetic Challenge. Pharmaceuticals. 2020; 13(11):392. https://doi.org/10.3390/ph13110392
Chicago/Turabian StyleQueda, Fausto, Gonçalo Covas, Sérgio R. Filipe, and M. Manuel B. Marques. 2020. "Assembly of Peptidoglycan Fragments—A Synthetic Challenge" Pharmaceuticals 13, no. 11: 392. https://doi.org/10.3390/ph13110392
APA StyleQueda, F., Covas, G., Filipe, S. R., & Marques, M. M. B. (2020). Assembly of Peptidoglycan Fragments—A Synthetic Challenge. Pharmaceuticals, 13(11), 392. https://doi.org/10.3390/ph13110392