Potent Quinoline-Containing Combretastatin A-4 Analogues: Design, Synthesis, Antiproliferative, and Anti-Tubulin Activity

 , ,
, ,  , , ,
, , ,
Abstract
: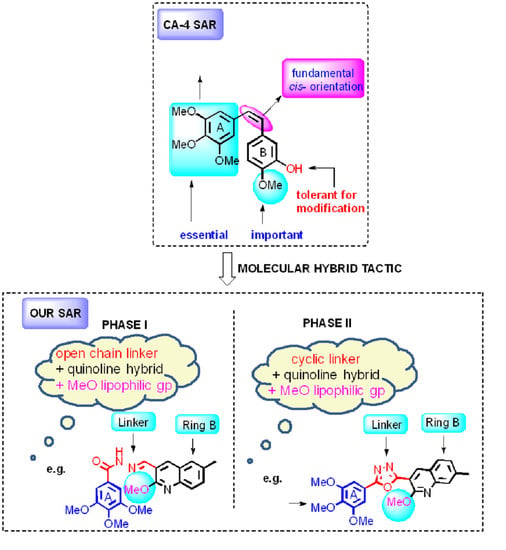
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Results and Discussion
2.2.1. In Vitro Antiproliferative Activities
2.2.2. In Vitro Inhibition of Tubulin Polymerization and Competitive Colchicine-Binding Assays
2.2.3. Cell Cycle Analysis in MCF-7 Cells
2.2.4. Apoptosis Quantification in MCF-7 Cells
2.2.5. Antiproliferative Activity in Non-Cancer Cells
2.2.6. Molecular Modelling for Quinoline Analogues in the Colchicine Binding Site of Tubulin
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. General Procedure for Preparation of Quinoline Analogues 19a–i
N’-[(2-Methoxyquinolin-3-yl)methylene]-3,4,5-trimethoxybenzohydrazide (19a)
N’-[(2-Methoxy-6-methylquinolin-3-yl)methylene]-3,4,5-trimethoxybenzohydrazide (19b)
N’-[(2-Methoxy-7-methylquinolin-3-yl)methylene]3,4,5-trimethoxybenzohydrazide (19c)
N’-[(2-Methoxy-8-methylquinolin-3-yl)methylene]3,4,5-trimethoxybenzohydrazide (19d)
N’-[(2,6-Dimethoxyquinolin-3-yl)methylene]-3,4,5-trimethoxybenzohydrazide (19e)
N’-[(2,7-Dimethoxyquinolin-3-yl)methylene]-3,4,5-trimethoxybenzohydrazide (19f)
N’-[(6-Isopropoxy-2-methoxyquinolin-3-yl)methylene]-3,4,5-trimethoxybenzohydrazide (19g)
N’-[(7-Isopropoxy-2-methoxyquinolin-3-yl)methylene]-3,4,5-trimethoxybenzohydrazide (19h)
N’-[(6-(Benzyloxy)-2-methoxyquinolin-3-yl) methylene]-3,4,5-trimethoxybenzohydrazide (19i)
N’-((7-(Benzyloxy)-2-methoxyquinolin-3-yl)methylene)-3,4,5-trimethoxybenzohydrazide (19j)
3.2.2. General Procedure for Preparation of Quinoline Analogues 20a–i
2-(2-Methoxyquinolin-3-yl)-5-(3,4,5-trimethoxyphenyl)-1,3,4-oxadiazole (20a)
2-(2-Methoxy-6-methylquinolin-3-yl)-5-(3,4,5-trimethoxyphenyl)-1,3,4- oxadiazole (20b)
2-(2-Methoxy-7-methylquinolin-3-yl)-5-(3,4,5-trimethoxyphenyl)-1,3,4- oxadiazole (20c)
2-(2-Methoxy-8-methylquinolin-3-yl)-5-(3,4,5-trimethoxyphenyl)-1,3,4- oxadiazole (20d)
2-(2,6-Dimethoxyquinolin-3-yl)-5-(3,4,5-trimethoxyphenyl)-1,3,4-oxadiazole (20e)
2-(2,7-Dimethoxyquinolin-3-yl)-5-(3,4,5-trimethoxyphenyl)-1,3,4-oxadiazole (20f)
2-(6-Isopropoxy-2-methoxyquinolin-3-yl)-5-(3,4,5-trimethoxyphenyl)-1,3,4-oxadiazole (20g)
2-(7-Isopropoxy-2-methoxyquinolin-3-yl)-5-(3,4,5-trimethoxyphenyl)-1,3,4-oxadiazole (20h)
2-[6-(Benzyloxy)-2-methoxyquinolin-3-yl]-5-(3,4,5-trimethoxyphenyl)-1,3,4-oxadiazole (20i)
2-[7-(Benzyloxy)-2-methoxyquinolin-3-yl]-5-(3,4,5-trimethoxyphenyl)-1,3,4-oxadiazole (20j)
3.3. Biochemical Evaluation of Activity
3.3.1. Cell Culture
3.3.2. Cell Viability Assay
3.3.3. Tubulin Polymerization Assay
3.3.4. Colchicine Site Competitive Binding Assay
3.3.5. Cell Cycle Analysis
3.3.6. Annexin V/PI Apoptotic Assay
3.4. Computational Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Desai, A.; Mitchison, T.J. Microtubule Polymerization Dynamics. Annu. Rev. Cell Dev. Biol. 1997, 13, 83–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Erratum: Structure of the αβ Tubulin Dimer by Electron Crystallography. Nature 1998, 393, 191. [Google Scholar] [CrossRef] [Green Version]
- Downing, K.H.; Nogales, E. Tubulin Structure: Insights into Microtubule Properties and Functions. Curr. Opin. Struct. Biol. 1998, 8, 785–791. [Google Scholar] [CrossRef]
- Kline-Smith, S.L.; Walczak, C.E. Mitotic Spindle Assembly and Chromosome Segregation. Mol. Cell 2004, 15, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Vindya, N.; Sharma, N.; Yadav, M.; Ethiraj, K. Tubulins-the target for anticancer therapy. Curr. Top. Med. Chem. 2015, 15, 73–82. [Google Scholar] [CrossRef]
- Kamath, P.R.; Sunil, D.; Ajees, A.A. Synthesis of Indole–Quinoline–Oxadiazoles: Their Anticancer Potential and Computational Tubulin Binding Studies. Res. Chem. Intermed. 2016, 42, 5899–5914. [Google Scholar] [CrossRef]
- Khelifi, I.; Naret, T.; Renko, D.; Hamze, A.; Bernadat, G.; Bignon, J.; Lenoir, C.; Dubois, J.; Brion, J.-D.; Provot, O.; et al. Design, Synthesis and Anticancer Properties of Iso Combretaquinolines as Potent Tubulin Assembly Inhibitors. Eur. J. Med. Chem. 2017, 127, 1025–1034. [Google Scholar] [CrossRef]
- Zweifel, M.; Jayson, G.C.; Reed, N.S.; Osborne, R.; Hassan, B.; Ledermann, J.; Shreeves, G.; Poupard, L.; Lu, S.-P.; Balkissoon, J.; et al. Phase II Trial of Combretastatin A4 Phosphate, Carboplatin, and Paclitaxel in Patients With Platinum-Resistant Ovarian Cancer. Ann. Oncol. 2011, 22, 2036–2041. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Brockway, O.; Dandavati, A.; Tzou, S.; Sjoholm, R.; Nickols, A.; Babu, B.; Chavda, S.; Satam, V.; Hartley, R.M.; et al. Design and Synthesis of Novel Enhanced Water Soluble Hydroxyethyl Analogs of Combretastatin A-4. Bioorg. Med. Chem. Lett. 2011, 21, 2087–2091. [Google Scholar] [CrossRef]
- Cooney, M.M.; Ortiz, J.; Bukowski, R.M.; Remick, S.C. Novel Vascular Targeting/Disrupting Agents: Combretastatin A4 Phosphate and Related Compounds. Curr. Oncol. Rep. 2005, 7, 90–95. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT00060242?term=NCT00060242&rank=1 (accessed on 16 October 2020).
- Ohsumi, K.; Hatanaka, T.; Nakagawa, R.; Fukuda, Y.; Morinaga, Y.; Suga, Y.; Nihei, Y.; Ohishi, K.; Akiyama, Y.; Tsuji, T. Synthesis and Antitumor Activities of Amino Acid Prodrugs of Amino-Combretastatins. Anti-Cancer Drug Des. 1999, 14, 539–548. [Google Scholar]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, A.S.; Genazzani, A.A. Medicinal Chemistry of Combretastatin A4: Present and Future Directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, R.; Prota, A.E.; Bargsten, K.; Cavalli, A.; Steinmetz, M.O. Structural Basis of Cis-and Trans-Combretastatin Binding to Tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Tarade, D.; Ma, D.; Pignanelli, C.; Mansour, F.; Simard, D.; Berg, S.V.D.; Gauld, J.; McNulty, J.; Pandey, S. Structurally Simplified Biphenyl Combretastatin a4 Derivatives Retain In Vitro Anti-Cancer Activity Dependent on Mitotic Arrest. PLoS ONE 2017, 12, e0171806. [Google Scholar] [CrossRef]
- Das, B.C.; Tang, X.-Y.; Rogler, P.; Evans, T. Design and Synthesis of 3,5-Disubstituted Boron-Containing 1,2,4-Oxadiazoles as Potential Combretastatin A-4 (CA-4) Analogs. Tetrahedron Lett. 2012, 53, 3947–3950. [Google Scholar] [CrossRef] [Green Version]
- Kaffy, J.; Monneret, C.; Mailliet, P.; Commerçon, A.; Pontikis, R. 1,3-Dipolar Cycloaddition Route to Novel Isoxazole-Type Derivatives Related to Combretastatin A-4. Tetrahedron Lett. 2004, 45, 3359–3362. [Google Scholar] [CrossRef]
- Mahal, K.; Biersack, B.; Schruefer, S.; Resch, M.; Ficner, R.; Schobert, R.; Mueller, T. Combretastatin A-4 Derived 5-(1-Methyl-4-Phenyl-Imidazol-5-Yl)Indoles With Superior Cytotoxic and Anti-Vascular Effects on Chemoresistant Cancer Cells and Tumors. Eur. J. Med. Chem. 2016, 118, 9–20. [Google Scholar] [CrossRef]
- Cury, N.M.; Mühlethaler, T.; Laranjeira, A.B.A.; Canevarolo, R.R.; Zenatti, P.P.; Lucena-Agell, D.; Barasoain, I.; Song, C.; Sun, D.; Dovat, S.; et al. Structural Basis of Colchicine-Site targeting Acylhydrazones active against Multidrug-Resistant Acute Lymphoblastic Leukemia. iScience 2019, 21, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Medarde, M.; Maya, A.B.; Pérez-Melero, C. Review ArticleNaphthalene Combretastatin Analogues: Synthesis, Cytotoxicity and Antitubulin Activity. J. Enzym. Inhib. Med. Chem. 2004, 19, 521–540. [Google Scholar] [CrossRef]
- Salum, L.B.; Mascarello, A.; Canevarolo, R.R.; Altei, W.F.; Laranjeira, A.B.; Neuenfeldt, P.D.; Stumpf, T.R.; Chiaradia-Delatorre, L.D.; Vollmer, L.L.; Daghestani, H.N.; et al. N-(1′-naphthyl)-3,4,5- Trimethoxybenzohydrazide as Microtubule Destabilizer: Synthesis, Cytotoxicity, Inhibition of Cell Migration and in Vivo Activity Against Acute Lymphoblastic Leukemia. Eur. J. Med. Chem. 2015, 96, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Afzal, O.; Kumar, S.; Haider, R.; Ali, R.; Kumar, R.; Jaggi, M.; Bawa, S. A Review on Anticancer Potential of Bioactive Heterocycle Quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef] [PubMed]
- Penthala, N.R.; Janganati, V.; Bommagani, S.; Crooks, P.A. Synthesis and Evaluation of a Series of Quinolinyl Trans-Cyanostilbene Analogs as Anticancer Agents. MedChemComm 2014, 5, 886–890. [Google Scholar] [CrossRef]
- Zhou, Y.; Yan, W.; Cao, D.; Shao, M.; Li, D.; Wang, F.; Yang, Z.; Chen, Y.; He, L.; Wang, T.; et al. Design, Synthesis and Biological Evaluation of 4-Anilinoquinoline Derivatives as Novel Potent Tubulin Depolymerization Agents. Eur. J. Med. Chem. 2017, 138, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.A.; Ducki, S.; Hirst, N.; McGown, A.T. Tubulin and Microtubules as Targets for Anticancer Drugs. Prog. Cell Cycle Res. 2003, 5, 309–326. [Google Scholar]
- Ansari, M.; Shokrzadeh, M.; Karima, S.; Rajaei, S.; Fallah, M.; Ghassemi-Barghi, N.; Ghasemian, M.; Emami, S. New Thiazole-2(3H)-Thiones Containing 4-(3,4,5-Trimethoxyphenyl) Moiety as Anticancer Agents. Eur. J. Med. Chem. 2020, 185, 111784. [Google Scholar] [CrossRef]
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More Than Just Vascular Targeting Agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef]
- Agut, R.; Falomir, E.; Murga, J.; Martín-Beltrán, C.; Gil-Edo, R.; Pla, A.; Carda, M.; Marco, J.A. Synthesis of Combretastatin A-4 and 3′-Aminocombretastatin A-4 derivatives with Aminoacid Containing Pendants and Study of their Interaction with Tubulin and as Downregulators of the VEGF, hTERT and c-Myc Gene Expression. Molecules 2020, 25, 660. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Pérez, M.-J.; Priego, E.-M.; Bueno, O.; Martins, M.S.; Canela, M.-D.; Liekens, S.; Liekens, S. Blocking Blood Flow to Solid Tumors by Destabilizing Tubulin: An Approach to Targeting Tumor Growth. J. Med. Chem. 2016, 59, 8685–8711. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, S.; Hadizadeh, F.; Eisvand, F.; Mosaffa, F.; Ghodsi, R. Synthesis, Structure-Activity Relationship and Molecular Docking Studies of Novel Quinoline-Chalcone Hybrids as Potential Anticancer Agents and Tubulin Inhibitors. J. Mol. Struct. 2020, 1202, 127310. [Google Scholar] [CrossRef]
- Chaudhary, V.; Venghateri, J.B.; Dhaked, H.P.S.; Bhoyar, A.S.; Guchhait, S.K.; Panda, D. Novel Combretastatin-2-aminoimidazole Analogues as Potent Tubulin Assembly Inhibitors: Exploration of Unique Pharmacophoric Impact of Bridging Skeleton and Aryl Moiety. J. Med. Chem. 2016, 59, 3439–3451. [Google Scholar] [CrossRef] [PubMed]
- Shobeiri, N.; Rashedi, M.; Mosaffa, F.; Zarghi, A.; Ghandadi, M.; Ghasemi, A.; Ghodsi, R. Synthesis and Biological Evaluation of Quinoline Analogues of Flavones as Potential Anticancer Agents and Tubulin Polymerization Inhibitors. Eur. J. Med. Chem. 2016, 114, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, F.; Shuai, W.; Sun, H.; Yao, H.; Ma, C.; Xu, S.; Yao, H.; Zhu, Z.; Yang, D.-H.; et al. Discovery of Novel Quinoline–Chalcone Derivatives as Potent Antitumor Agents with Microtubule Polymerization Inhibitory Activity. J. Med. Chem. 2019, 62, 993–1013. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, M.A.; El-Emam, A.A.; El-Gohary, N.S. 1,4,5,6,7,8-Hexahydroquinolines and 5,6,7,8-Tetrahydronaphthalenes: A New Class of Antitumor Agents Targeting the Colchicine Binding Site of Tubulin. Bioorg. Chem. 2020, 99, 103831. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Liang, Y.; Zhou, P.; Cheng, J.; Ding, K.; Wang, Y. Design, Synthesis, Antitumor Activities and Biological Studies of Novel Diaryl Substituted Fused Heterocycles as Dual Ligands Targeting Tubulin and Katanin. Eur. J. Med. Chem. 2019, 178, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight Into Tubulin Regulation From a Complex With Colchicine and a Stathmin-Like Domain. Nat. Cell Biol. 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.S.; Bokhtia, R.M.; Al-Mahmoudy, A.M.; Taher, E.S.; Alawadh, M.A.; Elagawany, M.; Abdel-Aal, E.H.; Panda, S.; Gouda, A.M.; Asfour, H.Z.; et al. Design, Synthesis and Biological Evaluation of Novel 5-((Substituted Quinolin-3-Yl/1-Naphthyl) Methylene)-3-Substituted Imidazolidin-2,4-Dione as HIV-1 Fusion Inhibitors. Bioorganic Chem. 2020, 99, 103782. [Google Scholar] [CrossRef]
- Karkara, B.B.; Mishra, S.S.; Singh, B.N.; Panda, G. Synthesis of 2-Methoxy-3-(Thiophen-2-Ylmethyl)Quinoline Containing Amino Carbinols as Antitubercular Agents. Bioorg. Chem. 2020, 99, 103775. [Google Scholar] [CrossRef]
- Insuasty, D.; Abonia, R.; Insuasty, B.; Quiroga, J.; Laali, K.K.; Nogueras, M.; Cobo, J. Microwave-Assisted Synthesis of Diversely Substituted Quinoline-Based Dihydropyridopyrimidine and Dihydropyrazolopyridine Hybrids. ACS Comb. Sci. 2017, 19, 555–563. [Google Scholar] [CrossRef]
- Ibrahim, T.S.; Seliem, I.A.; Ibrahim, T.S.; Al-Mahmoudy, A.M.M.; Abdel-Samii, Z.K.M.; Alhakamy, N.A.; Asfour, H.Z.; Elagawany, M. An Efficient Greener Approach for N-acylation of Amines in Water Using Benzotriazole Chemistry. Molecules 2020, 25, 2501. [Google Scholar] [CrossRef]
- Agha, K.A.; Abo-Dya, N.E.; Ibrahim, T.S.; Abdel-Aal, E.H.; Abdel-Samii, Z.K. N-Acylbenzotriazole: Convenient Approach for Protecting Group-Free Monoacylation of Symmetric Diamines. Mon. Chem. 2020, 151, 589–598. [Google Scholar] [CrossRef]
- Agha, K.A.; Abo-Dya, N.E.; Ibrahim, T.S.; Abdel-Aal, E.H. Efficient Synthesis of N-Acylbenzotriazoles Using Tosyl Chloride: En Route to Suberoylanilide Hydroxamic Acid (SAHA). ARKIVOC 2016, 3, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, H.; Killian, B.J.; Jabeen, F.; Pillai, G.G.; Berman, H.M.; Mathelier, M.; Sibble, A.J.; Yeung, J.; Zhou, W.; et al. Synthesis, Characterization and Energetic Properties of 1,3,4-Oxadiazoles. Eur. J. Org. Chem. 2015, 2015, 5183–5188. [Google Scholar] [CrossRef]
- Wang, X.; Yu, H.; Xu, P.; Zheng, R. A Facile Synthesis of Acylhydrazines from Acylbenzotriazoles. J. Chem. Res. 2005, 2005, 595–597. [Google Scholar] [CrossRef]
- Wienecke, A.; Bacher, G. Indibulin, a Novel Microtubule Inhibitor, Discriminates between Mature Neuronal and Nonneuronal Tubulin. Cancer Res. 2009, 69, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, M.; Anwar, S.; Elgamal, F.; Ahmed, E.R.; Aly, O.M. Potent Combretastatin A-4 Analogs Containing 1,2,4-Triazole: Synthesis, Antiproliferative, Anti-Tubulin Activity, and Docking Study. Eur. J. Med. Chem. 2019, 183, 111697. [Google Scholar] [CrossRef]
- Tahir, S.K.; Kovar, P.; Rosenberg, S.H.; Ng, S.-C. Rapid Colchicine Competition-Binding Scintillation Proximity Assay Using Biotin-Labeled Tubulin. Biotechniques 2000, 29, 156–160. [Google Scholar] [CrossRef]
- Moussa, S.A.; Osman, E.E.A.; Eid, N.M.; Abou-Seri, S.M.; El Moghazy, S.M. Design and Synthesis of Novel 5-(4-Chlorophenyl)Furan Derivatives With Inhibitory Activity on Tubulin Polymerization. Future Med. Chem. 2018, 10, 1907–1924. [Google Scholar] [CrossRef]
- Molecular Operating Environment; Version 2019.0102; Chemical Computing Group: Montreal, ON, Canada, 2019.
- XQuartz 2.7.11; X.org Foundation: Portland, OR, USA, 2016.
- MMFF94 Force Field (mmff94). Available online: https://open-babel.readthedocs.io/en/latest/Forcefields/mmff94.html (accessed on 28 April 2020).
- Halgren, T.A. Merck Molecular Force Field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound N°. | R | Antiproliferative Activities IC50 µM a | Inhibition of Tubulin Polymerization b IC50 µM | |||
|---|---|---|---|---|---|---|
| HL-60 | MCF-7 | HCT-116 | HeLa | |||
| 19a | H | 0.013 ± 0.009 | 0.030 ± 0.001 | 0.308 ± 0.012 | 0.093 ± 0.006 | 4.69 |
| 19b | 6-CH3 | 0.045 ± 0.002 | 0.072 ± 0.004 | 0.371 ± 0.002 | 0.028 ± 0.001 | 1.48 |
| 19c | 7-CH3 | 4.210 ± 0.0062 | 0.703 ± 0.002 | 3.830 ± 0.026 | 1.151 ± 0.254 | 35.64 |
| 19d | 8-CH3 | 1.810 ± 0.004 | 1.097 ± 0.049 | 0.450 ± 0.022 | 0.107 ± 0.006 | 2.26 |
| 19e | 6-OCH3 | 0.088 ± 0.002 | 0.031 ± 0.007 | 0.812 ± 0.020 | 0.124 ± 0.019 | 3.647 |
| 19f | 7-OCH3 | 0.150 ± 0.062 | 0.044 ± 0.002 | 0.366 ± 0.030 | 0.167 ± 0.009 | 8.96 |
| 19g | 6-OCH(CH3)2 | 0.137 ± 0.007 | 0.078 ± 0.003 | 0.233 ± 0.017 | 0.014 ± 0.004 | 3.04 |
| 19h | 7-OCH(CH3)2 | 0.040 ± 0.003 | 0.026 ± 0.002 | 0.022 ± 0.001 | 0.038 ± 0.003 | 1.32 |
| 19i | 6-OCH2Ph | 0.983 ± 0.042 | 0.530 ± 0.001 | 0.138 ± 0.004 | 0.152 ± 0.004 | 4.08 |
| 19j | 7-OCH2Ph | 0.585 ± 0.051 | 0.204 ± 0.001 | 0.067 ± 0.012 | 0.082 ± 0.001 | 3.34 |
| 20a | H | 1.551 ± 0.032 | 3.480 ± 0.001 | 4.030 ± 0.013 | 1.960 ± 0.003 | 17.55 |
| 20b | 6-CH3 | 0.336 ± 0.011 | 0.185 ± 0.007 | 0.378 ± 0.002 | 0.616 ± 0.002 | 8.83 |
| 20c | 7-CH3 | 0.012 ± 0.009 | 0.094 ± 0.007 | 0.024 ± 0.009 | 0.210 ± 0.002 | 2.41 |
| 20d | 8-CH3 | 0.147 ± 0.018 | 0.076 ± 0.006 | 0.087 ± 0.009 | 0.117 ± 0.014 | 6.62 |
| 20e | 6-OCH3 | 0.166 ± 0.075 | 0.094 ± 0.008 | 0.179 ± 0.001 | 0.144 ± 0.010 | 6.45 |
| 20f | 7-OCH3 | 0.055 ± 0.001 | 0.44 ± 0.005 | 0.199 ± 0.007 | 0.095 ± 0.001 | 3.25 |
| 20g | 6-OCH(CH3)2 | 0.270 ± 0.015 | 0.044 ± 0.003 | 0.086 ± 0.003 | 0.071 ± 0.003 | 2.31 |
| 20h | 7-OCH(CH3)2 | 0.109 ± 0.006 | 0.042 ± 0.006 | 0.146 ± 0.009 | 0.193 ± 0.002 | 5.80 |
| 20i | 6-OCH2Ph | 0.043 ± 0.002 | 0.039 ± 0.003 | 0.082 ± 0.001 | 0.138 ± 0.003 | 4.60 |
| 20j | 7-OCH2Ph | 0.029 ± 0.007 | 0.028 ± 0.003 | 0.129 ± 0.009 | 0.069 ± 0.003 | 2.29 |
| CA-4 | - | 0.076 ± 0.004 | 0.019 ± 0.004 | 0.026 ± 0.001 | 0.064 ± 0.004 | 2.17 |
| Compound | Colchicine Binding a (% ± SD) | |
|---|---|---|
| 1 µM Drug | 5 µM Drug | |
| 19b | 79 ± 1.6 | 83± 0.54 |
| 19h | 80 ± 0.4 | 86 ± 0.13 |
| 20c | 69 ± 0.8 | 73 ± 0.27 |
| 20j | 75 ± 1.5 | 80 ± 0.26 |
| CA-4 | 86 ± 0.9 | 97 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, T.S.; Hawwas, M.M.; Malebari, A.M.; Taher, E.S.; Omar, A.M.; O'Boyle, N.M.; McLoughlin, E.; Abdel-Samii, Z.K.; Elshaier, Y.A.M.M. Potent Quinoline-Containing Combretastatin A-4 Analogues: Design, Synthesis, Antiproliferative, and Anti-Tubulin Activity. Pharmaceuticals 2020, 13, 393. https://doi.org/10.3390/ph13110393
Ibrahim TS, Hawwas MM, Malebari AM, Taher ES, Omar AM, O'Boyle NM, McLoughlin E, Abdel-Samii ZK, Elshaier YAMM. Potent Quinoline-Containing Combretastatin A-4 Analogues: Design, Synthesis, Antiproliferative, and Anti-Tubulin Activity. Pharmaceuticals. 2020; 13(11):393. https://doi.org/10.3390/ph13110393
Chicago/Turabian StyleIbrahim, Tarek S., Mohamed M. Hawwas, Azizah M. Malebari, Ehab S. Taher, Abdelsattar M. Omar, Niamh M. O'Boyle, Eavan McLoughlin, Zakaria K. Abdel-Samii, and Yaseen A. M. M. Elshaier. 2020. "Potent Quinoline-Containing Combretastatin A-4 Analogues: Design, Synthesis, Antiproliferative, and Anti-Tubulin Activity" Pharmaceuticals 13, no. 11: 393. https://doi.org/10.3390/ph13110393
APA StyleIbrahim, T. S., Hawwas, M. M., Malebari, A. M., Taher, E. S., Omar, A. M., O'Boyle, N. M., McLoughlin, E., Abdel-Samii, Z. K., & Elshaier, Y. A. M. M. (2020). Potent Quinoline-Containing Combretastatin A-4 Analogues: Design, Synthesis, Antiproliferative, and Anti-Tubulin Activity. Pharmaceuticals, 13(11), 393. https://doi.org/10.3390/ph13110393