Synthesis, 3D-QSAR, and Molecular Modeling Studies of Triazole Bearing Compounds as a Promising Scaffold for Cyclooxygenase-2 Inhibition

 ,
,  , ,
, ,  and
and
Abstract
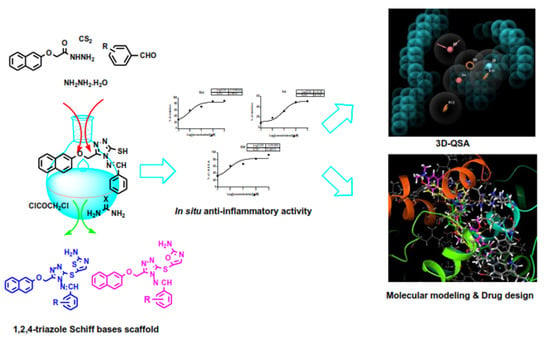
:
1. Introduction
Rational Design of Selective COX-2 Inhibitor
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro Cyclooxygenase Inhibition Assay
2.2.2. In Situ Anti-Inflammatory Activity
2.3. Computational Study
2.3.1. 3D-QSAR
2.3.2. Developing a Pharmacophore Model
2.3.3. 3D-QSAR Contour Map Analysis
2.3.4. Atomic-Based QSAR Validation
2.3.5. Field-Base 3D-QSAR
2.3.6. Field-Base 3D-QSAR Validation
2.4. Molecular Docking Study
2.5. Molecular Electrostatic Potential (MESP) and Molecular Orbital Energy Study
2.6. In Silico ADME Predictive Study
3. Materials and Methods
3.1. Instrument
3.2. Chemicals and Reagents
3.3. Experimental
Chemistry
3.4. Biological Assays
3.4.1. Cyclooxygenase Inhibition Assays
3.4.2. In Situ Anti-Inflammatory Assay
3.5. Molecular Modeling Study
3.5.1. QSAR
3.5.2. Pharmacophore 3D-QSAR Modeling
3.5.3. Model Validation
3.5.4. Molecular Modeling Study
3.6. Computational Study
3.7. Lipinski’s Rule for Drug Likeliness and In Silico ADME Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abuo-Rahma, G.E.-D.A.; Abdel-Aziz, M.; Farag, N.A.; Kaoud, T.S. Novel 1-[4-(Aminosulfonyl) phenyl]-1H-1, 2, 4-triazole derivatives with remarkable selective COX-2 inhibition: Design, synthesis, molecular docking, anti-inflammatory and ulcerogenicity studies. Eur. J. Med. Chem. 2014, 83, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.; Abdelall, E.K.; Fadaly, W.A.; Kamel, G.M. Synthesis, cyclooxygenase inhibition, and anti-inflammatory evaluation of novel diarylheterocycles with a central pyrazole, pyrazoline, or pyridine ring. Med. Chem. Res. 2015, 24, 2632–2644. [Google Scholar] [CrossRef]
- Abdelazeem, A.H.; Abdelatef, S.A.; El-Saadi, M.T.; Omar, H.A.; Khan, S.I.; McCurdy, C.R.; El-Moghazy, S.M. Novel pyrazolopyrimidine derivatives targeting COXs and iNOS enzymes; design, synthesis and biological evaluation as potential anti-inflammatory agents. Eur. J. Pharm. Sci. 2014, 62, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Zebardast, T.; Zarghi, A.; Daraie, B.; Hedayati, M.; Dadrass, O.G. Design and synthesis of 3-alkyl-2-aryl-1, 3-thiazinan-4-one derivatives as selective cyclooxygenase (COX-2) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3162–3165. [Google Scholar] [CrossRef] [PubMed]
- Pairet, M.; Engelhardt, G. Distinct isoforms (COX-1 and COX-2) of cyclooxygenase: Possible physiological and therapeutic implications. Fundam. Clin. Pharmacol. 1996, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Singh, V. Positioning dual inhibitors in the treatment of pain and inflammatory disorders. Inflammopharmacology 2008, 16, 1–15. [Google Scholar] [CrossRef]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Tsatsanis, C.; Androulidaki, A.; Venihaki, M.; Margioris, A.N. Signalling networks regulating cyclooxygenase-2. Int. J. Biochem. Cell Biol. 2006, 38, 1654–1661. [Google Scholar] [CrossRef]
- Salgın-Gökşen, U.; Gökhan-Kelekçi, N.; Göktaş, Ö.; Köysal, Y.; Kılıç, E.; Işık, Ş.; Aktay, G.; Özalp, M. 1-Acylthiosemicarbazides, 1, 2, 4-triazole-5 (4H)-thiones, 1, 3, 4-thiadiazoles and hydrazones containing 5-methyl-2-benzoxazolinones: Synthesis, analgesic-anti-inflammatory and antimicrobial activities. Bioorg. Med. Chem. 2007, 15, 5738–5751. [Google Scholar] [CrossRef]
- El-Moghazy, S.M.; Barsoum, F.F.; Abdel-Rahman, H.M.; Marzouk, A.A. Synthesis and anti-inflammatory activity of some pyrazole derivatives. Med. Chem. Res. 2012, 21, 1722–1733. [Google Scholar] [CrossRef]
- Jackson, L.M.; Hawkey, C.J. COX-2 selective nonsteroidal anti-inflammatory drugs. Drugs 2000, 59, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.D.; Fisher, M.C.; Kremer, J.; Chang, H.; Rosenstein, E.D.; Kishimoto, M.; Lee, S.; Yazici, Y.; Kavanaugh, A.; Abramson, S.B. The COX-2 inhibitor market withdrawals and prescribing patterns by rheumatologists in patients with gastrointestinal and cardiovascular risk. Clin. Exp. Rheumatol. 2009, 27, 395–401. [Google Scholar] [PubMed]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. 2011, 10, 655–683. [Google Scholar] [PubMed]
- Amin, N.H.; Mohammed, A.A.; Abdellatif, K.R. Novel 4-methylsulfonylphenyl derivatives as NSAIDS with preferential COX-2 inhibition. Future Med. Chem. 2018, 10, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Akhter, M.W.; Haq, S.E. Synthesis of some new condensed heterocyclic 6-substituted-1, 2, 4-triazolo [3, 4-b]-1, 3, 4-thiadiazole derivatives of 2-naphthoxyacetic acid as potent anti-inflammatory agents with reduced ulcerogenicity. Indian J. Chem. Sect. B-Org. Chem. Incl. Med. Chem. 2017, 56, 1177–1184. [Google Scholar]
- Kharb, R.; Sharma, P.C.; Yar, M.S. Pharmacological significance of triazole scaffold. J. Enzyme Inhib. Med. Chem. 2011, 26, 1–21. [Google Scholar] [CrossRef]
- Thakur, A.; Gupta, P.; Shukla, P.; Verma, A.; Pathak, P. 1, 2, 4-Triazole Scafolds: Recent Advances and Pharmacological Applications. Int. J. Curr. Res. Aca. Rev. 2016, 4, 277. [Google Scholar] [CrossRef]
- Kaplancıklı, Z.A.; Turan-Zitouni, G.; Özdemir, A.; Revial, G. New triazole and triazolothiadiazine derivatives as possible antimicrobial agents. Eur. J. Med. Chem. 2008, 43, 155–159. [Google Scholar] [CrossRef]
- Bayrak, H.; Demirbas, A.; Karaoglu, S.A.; Demirbas, N. Synthesis of some new 1, 2, 4-triazoles, their Mannich and Schiff bases and evaluation of their antimicrobial activities. Eur. J. Med. Chem. 2009, 44, 1057–1066. [Google Scholar] [CrossRef]
- El Shehry, M.; Abu-Hashem, A.; El-Telbani, E. Synthesis of 3-((2, 4-dichlorophenoxy) methyl)-1, 2, 4-triazolo (thiadiazoles and thiadiazines) as anti-inflammatory and molluscicidal agents. Eur. J. Med. Chem. 2010, 45, 1906–1911. [Google Scholar] [CrossRef]
- Khan, I.; Zaib, S.; Ibrar, A.; Rama, N.H.; Simpson, J.; Iqbal, J. Synthesis, crystal structure and biological evaluation of some novel 1, 2, 4-triazolo [3, 4-b]-1, 3, 4-thiadiazoles and 1, 2, 4-triazolo [3, 4-b]-1, 3, 4-thiadiazines. Eur. J. Med. Chem. 2014, 78, 167–177. [Google Scholar] [CrossRef]
- Kumar, B.N.P.; Mohana, K.N.; Mallesha, L. Synthesis and antiproliferative activity of some new fluorinated Schiff bases derived from 1, 2, 4-triazoles. J. Fluor. Chem. 2013, 156, 15–20. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Y.-H.; Liu, Y.; Chen, Y.-R.; Pan, E.-S.; You, W.-W.; Zhao, P.-L. Design, synthesis and biological evaluation of novel 1, 2, 4-triazolo [3, 4-b][1, 3, 4] thiadiazines bearing furan and thiophene nucleus. Eur. J. Med. Chem. 2015, 103, 335–342. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luong, C.; Miller, A.; Barnett, J.; Chow, J.; Ramesha, C.; Browner, M.F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Mol. Biol. 1996, 3, 927–933. [Google Scholar] [CrossRef]
- Vecchio, A.J.; Orlando, B.J.; Nandagiri, R.; Malkowski, M.G. Investigating substrate promiscuity in cyclooxygenase-2: The role of Arg-120 and residues lining the hydrophobic groove. J. Biol. Chem. 2012, 287, 24619–24630. [Google Scholar] [CrossRef] [Green Version]
- Domiati, S.; El-Mallah, A.; Ghoneim, A.; Bekhit, A.; Abd El Razik, H. Evaluation of anti-inflammatory, analgesic activities, and side effects of some pyrazole derivatives. Inflammopharmacology 2016, 24, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.; Lamie, P.F.; Omar, H.A. 3-Methyl-2-phenyl-1-substituted-indole derivatives as indomethacin analogs: Design, synthesis and biological evaluation as potential anti-inflammatory and analgesic agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 318–324. [Google Scholar] [CrossRef]
- Qiao, F.; Yin, Y.; Shen, Y.-N.; Wang, S.-F.; Sha, S.; Wu, X.; Lu, A.-M.; Xu, C.; Zhang, W.-M.; Zhu, H.-L. Synthesis, molecular modeling, and biological evaluation of quinazoline derivatives containing the 1, 3, 4-oxadiazole scaffold as novel inhibitors of VEGFR2. RSC Adv. 2015, 5, 19914–19923. [Google Scholar] [CrossRef]
- Singh, R.; Kashaw, S.; Mishra, V.; Mishra, M.; Rajoriya, V.; Kashaw, V. Design and synthesis of new bioactive 1, 2, 4-Triazoles, potential antitubercular and antimicrobial agents. Indian J. Pharm. Sci. 2018, 80, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Zhaowen, L.; Li, Z.; Chunfen, X.; Yong, Y.; Fanbo, Z.; Kaixun, H. Anticancer activities of some arylcarbamoylalkyltriphenylphosphonium chlorides. Med. Chem. Res. 2007, 16, 380–391. [Google Scholar] [CrossRef]
- Fahim, A.M. Regioselective synthesis of novel fused sulphonamide derivatives utilizing microwave irradiation. Curr. Micro. Chem. 2018, 5, 4–12. [Google Scholar] [CrossRef]
- Roschek, B., Jr.; Fink, R.C.; Li, D.; McMichael, M.; Tower, C.M.; Smith, R.D.; Alberte, R.S. Pro-inflammatory enzymes, cyclooxygenase 1, cyclooxygenase 2, and 5-lipooxygenase, inhibited by stabilized rice bran extracts. J. Med. Food 2009, 12, 615–623. [Google Scholar] [CrossRef]
- Mizushima, Y.; Kobayashi, M. Interaction of anti-inflammatory drugs with serum proteins, especially with some biologically active proteins. J. Pharm. Pharmacol. 1968, 20, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Uddin, M.J.; Banerjee, S.; Duggan, K.; Musee, J.; Kiefer, J.R.; Ghebreselasie, K.; Rouzer, C.A.; Marnett, L.J. Fluorescent indomethacin-dansyl conjugates utilize the membrane-binding domain of cyclooxygenase-2 to block the opening to the active site. J. Biol. Chem. 2019, 294, 8690–8698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrayess, R.; Abdel Aziz, Y.M.; Elgawish, M.S.; Elewa, M.; Elshihawy, H.A.; Said, M.M. Pharmacophore modeling, 3D-QSAR, synthesis, and anti-lung cancer evaluation of novel thieno[2,3-d][1,2,3]triazines targeting EGFR. Arch. Pharm. 2020, 353, e1900108. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Abdelhameed, R.; Elgawish, M.S.; Mira, A.; Ibrahim, A.K.; Ahmed, S.A.; Shimizu, K.; Yamada, K. Anti-choline esterase activity of ceramides from the Red Sea marine sponge Mycale euplectellioides. RSC Adv. 2016, 6, 20422–20430. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Soo, R.A. Dacomitinib in lung cancer: A “lost generation” EGFR tyrosine-kinase inhibitor from a bygone era? Drug Des. Dev. Ther. 2015, 9, 5641–5653. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, Inc. QikProp. 2020. Available online: https://www.schrodinger.com/qikprop (accessed on 4 November 2020).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | IC50 (µM) a | ||
|---|---|---|---|
| COX-1 | COX-2 | COX-2 S.I. b | |
| 9a | 6.93 | 0.22 | 31.5 |
| 9b | 7.67 | 0.19 | 40.4 |
| 9c | 9.43 | 0.13 | 72.5 |
| 9d | 7.97 | 0.15 | 53.1 |
| 10a | 9.72 | 0.13 | 74.8 |
| 10b | 9.89 | 0.14 | 70.6 |
| 10c | 10.12 | 0.12 | 84.3 |
| 10d | 10.35 | 0.11 | 94.1 |
| 11a | 12.37 | 0.08 | 154.6 |
| 11b | 10.73 | 0.09 | 119.2 |
| 11c | 12.63 | 0.05 | 252.6 |
| 11d | 11.53 | 0.06 | 192.2 |
| 12a | 9.83 | 0.1 | 98.3 |
| 12b | 12.13 | 0.06 | 202.2 |
| 12c | 12.47 | 0.04 | 311.8 |
| 12d | 13 | 0.04 | 325.0 |
| Celecoxib | 14.7 | 0.05 | 294.0 |
| PLS | SD | R2 | R2 CV | Stability | F | P | RMSE | Q2 | Pearson-r |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.1018 | 0.7786 | 0.2547 | 0.714 | 45.7 | 1.34 × 10−5 | 0.27 | −0.19 | 0.5598 |
| 2 | 0.0555 | 0.9393 | 0.1111 | 0.202 | 92.8 | 5.01 × 10−8 | 0.24 | 0.0566 | 0.639 |
| 3 | 0.0464 | 0.9611 | 0.1983 | 0.242 | 90.6 | 4.86 × 10−8 | 0.25 | −0.0618 | 0.6058 |
| In | Ligand Name | QSAR Set | Activity | Predicted Activity | Activity | Prediction Error |
|---|---|---|---|---|---|---|
| 1 | 9a | training | 0.657 | 0.753 | Inactive | 0.096 |
| 2 | 9b | training | 0.721 | 0.741 | Inactive | 0.02 |
| 3 | 9c | training | 0.886 | 0.809 | Inactive | −0.076 |
| 4 | 9d | training | 0.824 | 0.787 | Inactive | −0.036 |
| 5 | 10a | test | 0.886 | 1.011 | Inactive | 0.125 |
| 6 | 10b | training | 0.853 | 0.884 | Inactive | 0.031 |
| 7 | 10c | training | 0.92 | 0.92 | Inactive | 0.002 |
| 8 | 10d | training | 0.958 | 0.97 | Inactive | 0.012 |
| 9 | 11a | training | 1.097 | 1.097 | Active | 0.001 |
| 10 | 11b | training | 1.046 | 1.046 | Active | 0.001 |
| 11 | 11c | test | 1.31 | 1.101 | Active | −0.208 |
| 12 | 11d | training | 1.221 | 1.216 | Active | −0.004 |
| 13 | 12a | training | 1 | 0.989 | Active | −0.01 |
| 14 | 12b | test | 1.221 | 1.019 | Active | −0.201 |
| 15 | 12c | training | 1.397 | 1.396 | Active | 0 |
| 16 | 12d | test | 1.397 | 0.973 | Active | −0.423 |
| 17 | Dansyl-Indomethacin (1) | test | 0.77 | 0.700 | −0.069 | |
| 18 | Dansyl-Indomethacin (2) | test | 0.12 | 0.127 | 0.007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elrayess, R.; Elgawish, M.S.; Elewa, M.; Nafie, M.S.; Elhady, S.S.; Yassen, A.S.A. Synthesis, 3D-QSAR, and Molecular Modeling Studies of Triazole Bearing Compounds as a Promising Scaffold for Cyclooxygenase-2 Inhibition. Pharmaceuticals 2020, 13, 370. https://doi.org/10.3390/ph13110370
Elrayess R, Elgawish MS, Elewa M, Nafie MS, Elhady SS, Yassen ASA. Synthesis, 3D-QSAR, and Molecular Modeling Studies of Triazole Bearing Compounds as a Promising Scaffold for Cyclooxygenase-2 Inhibition. Pharmaceuticals. 2020; 13(11):370. https://doi.org/10.3390/ph13110370
Chicago/Turabian StyleElrayess, Ranza, Mohamed Saleh Elgawish, Marwa Elewa, Mohamed S. Nafie, Sameh S. Elhady, and Asmaa S. A. Yassen. 2020. "Synthesis, 3D-QSAR, and Molecular Modeling Studies of Triazole Bearing Compounds as a Promising Scaffold for Cyclooxygenase-2 Inhibition" Pharmaceuticals 13, no. 11: 370. https://doi.org/10.3390/ph13110370
APA StyleElrayess, R., Elgawish, M. S., Elewa, M., Nafie, M. S., Elhady, S. S., & Yassen, A. S. A. (2020). Synthesis, 3D-QSAR, and Molecular Modeling Studies of Triazole Bearing Compounds as a Promising Scaffold for Cyclooxygenase-2 Inhibition. Pharmaceuticals, 13(11), 370. https://doi.org/10.3390/ph13110370