Hepatic Tumor Cell Morphology Plasticity under Physical Constraints in 3D Cultures Driven by YAP–mTOR Axis

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
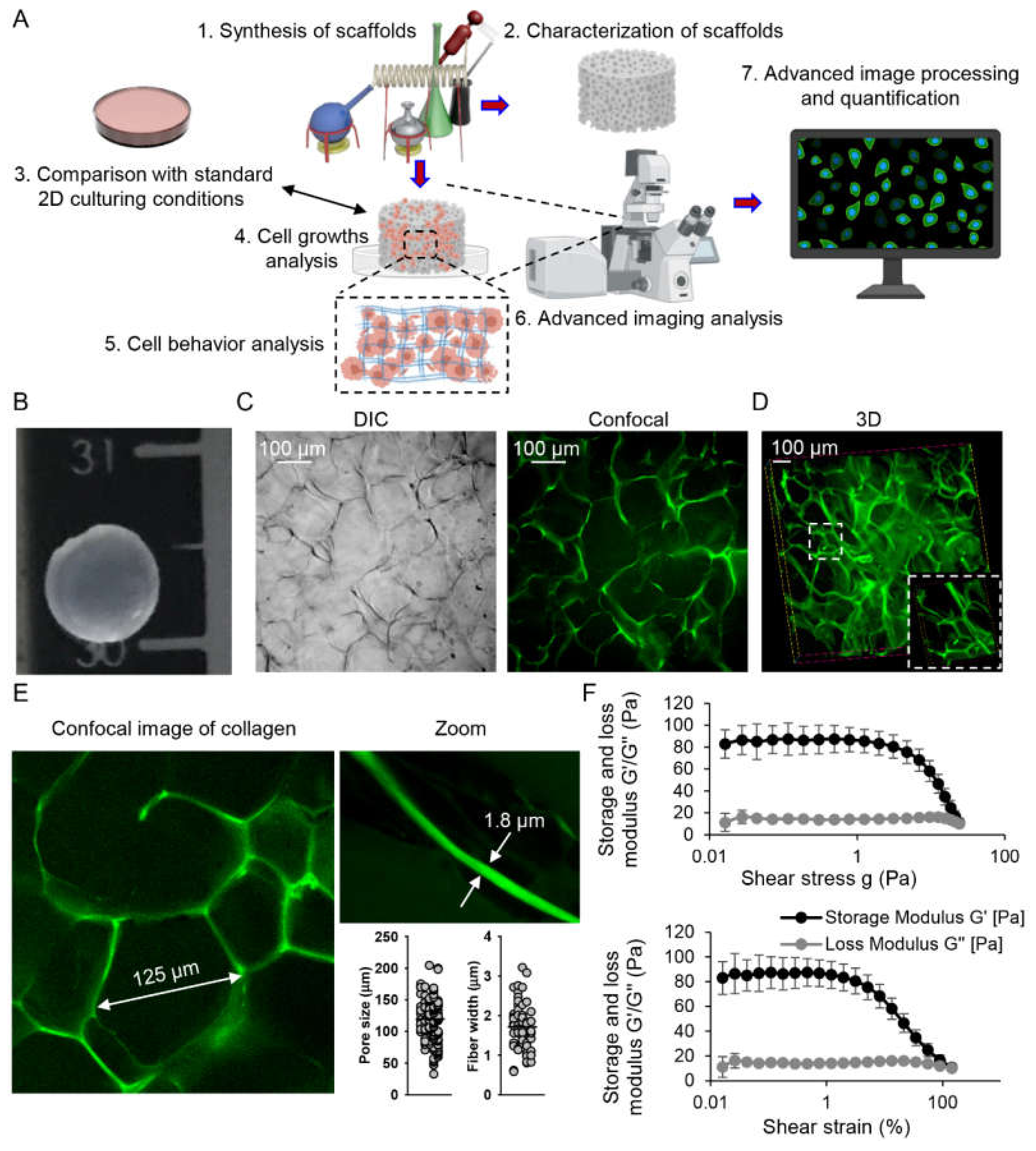
2.1. Characterization of Collagen Scaffolds
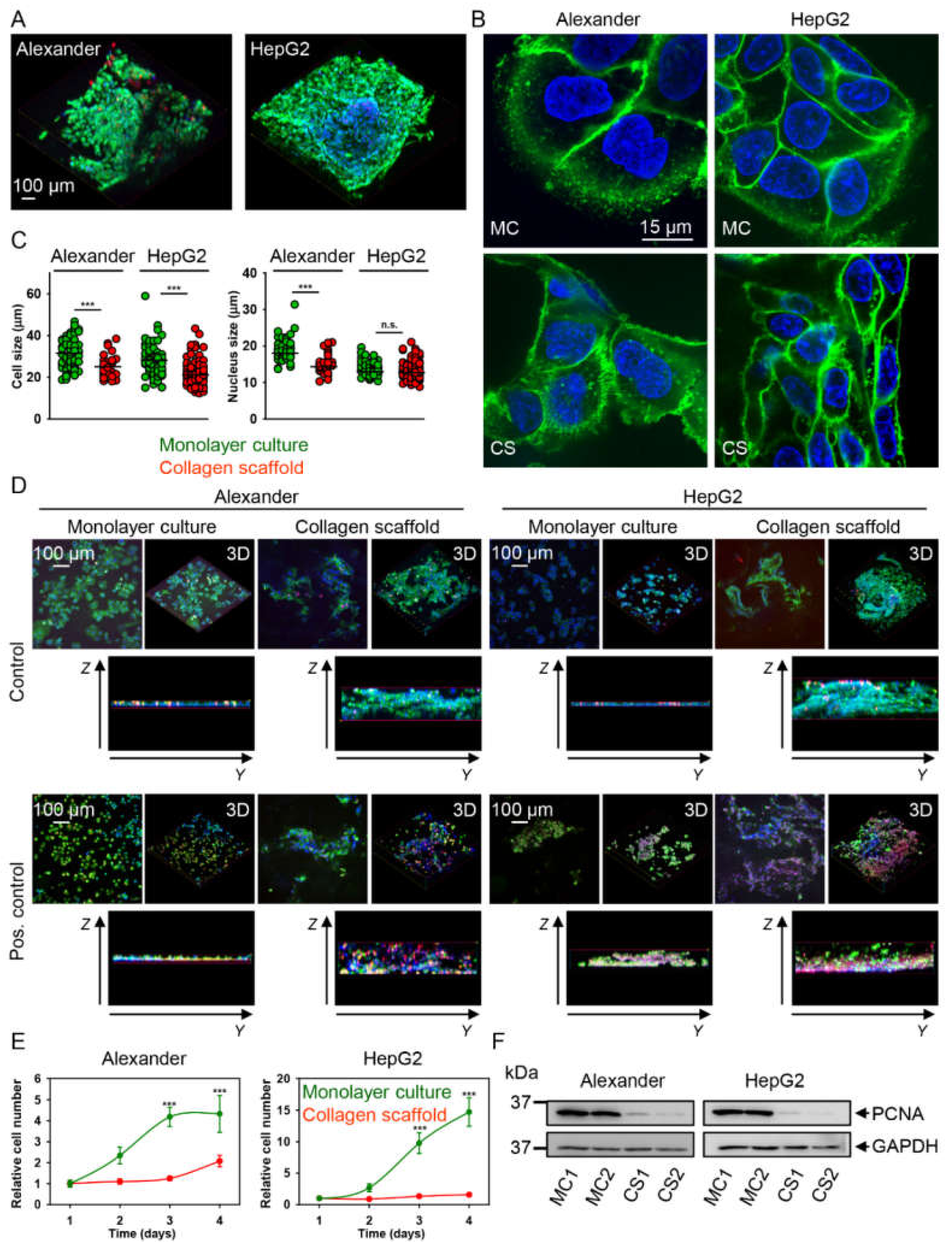
2.2. HepG2 and Alexander Cells Change Their Size and Shape during Growth in Collagen Scaffolds
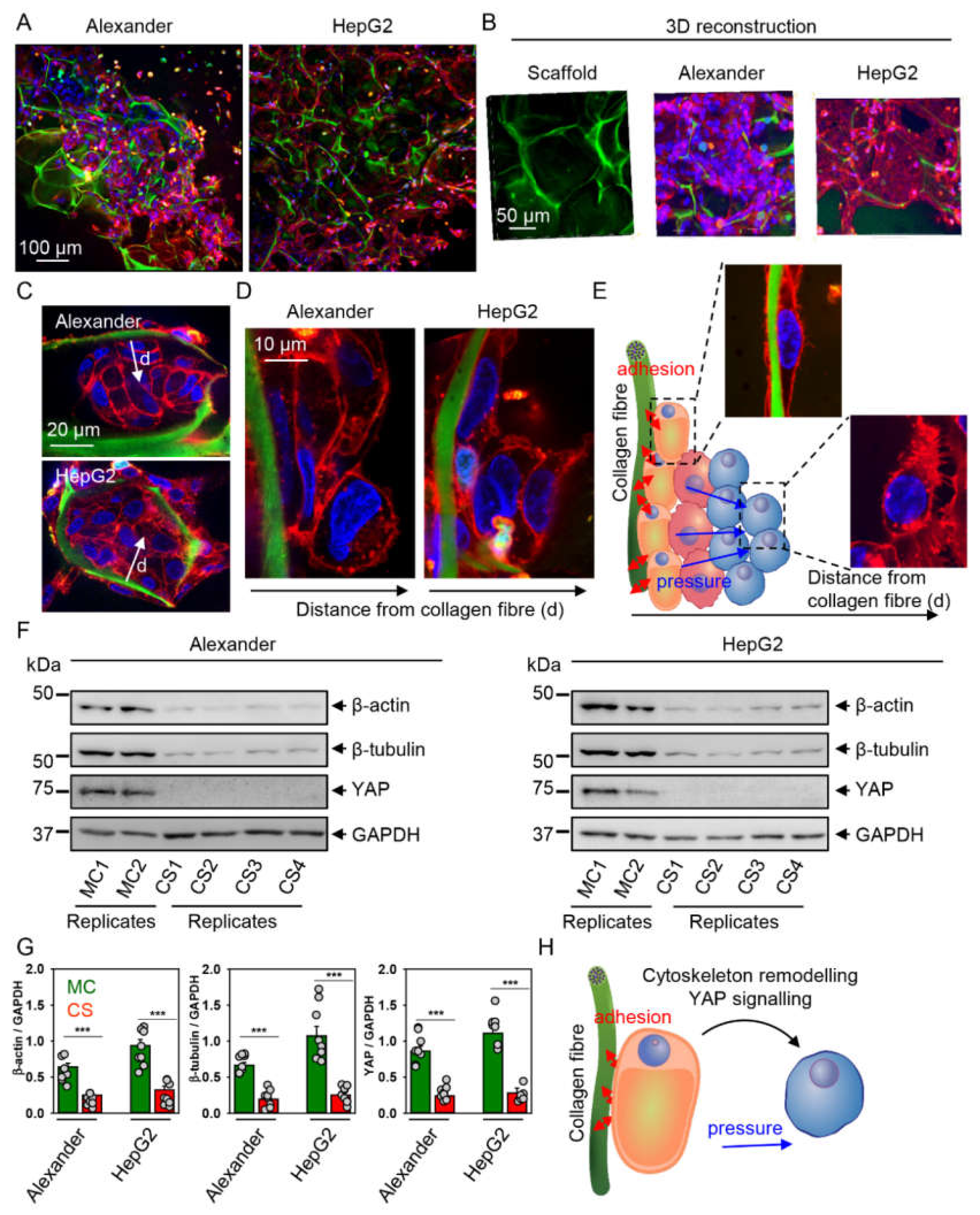
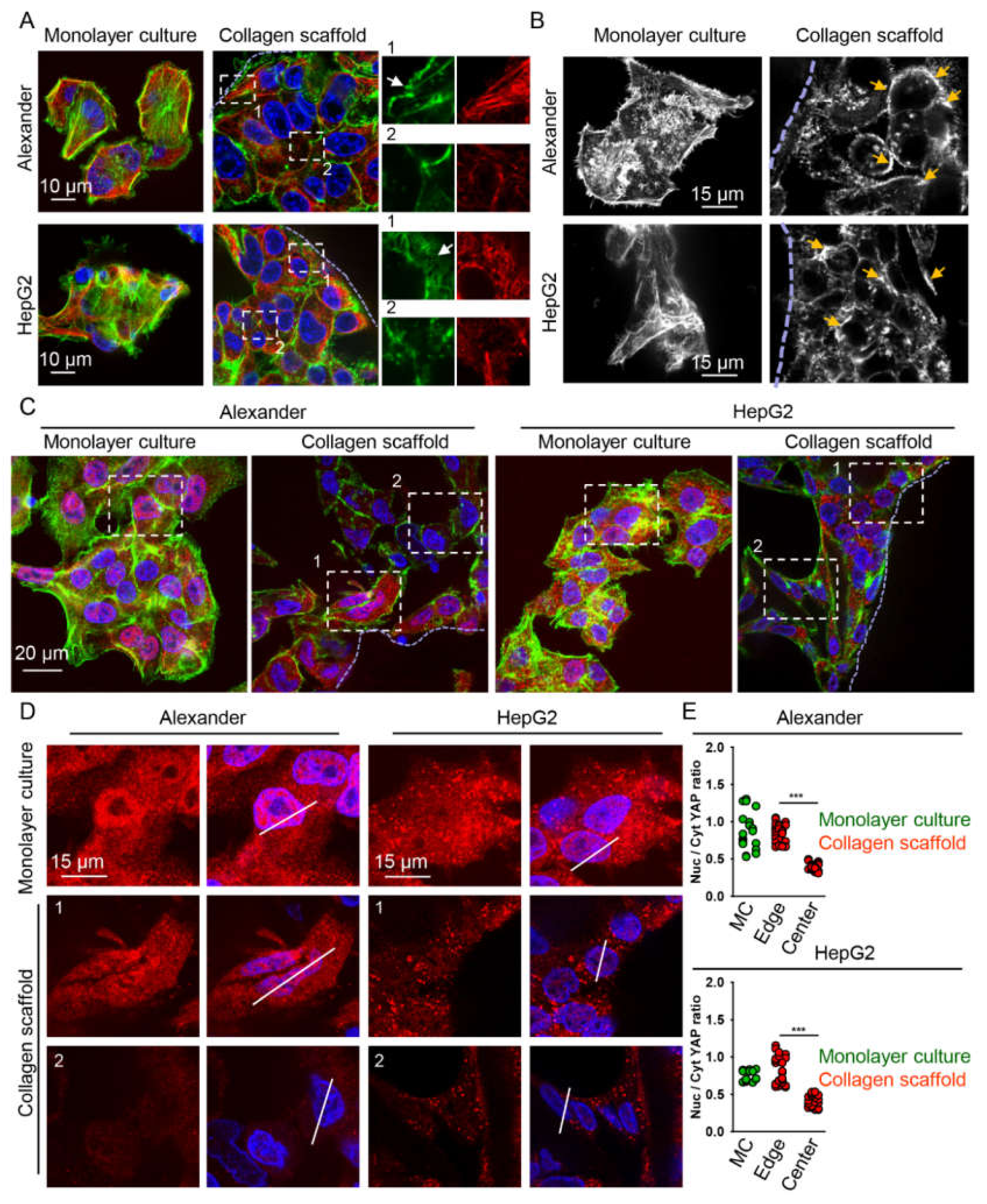
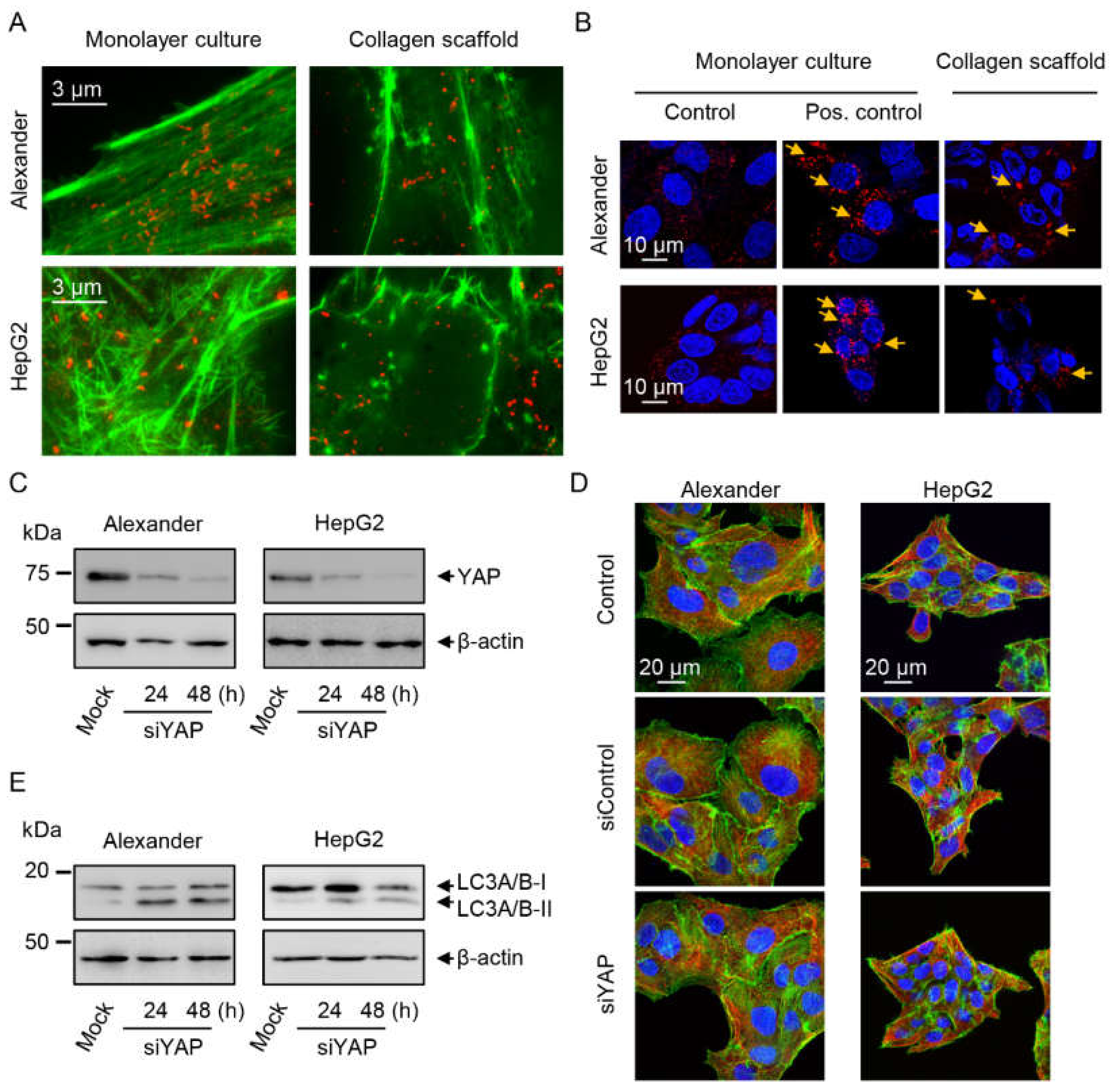
2.3. Cytoskeleton Remodeling and Modulation of YAP Signaling in Cells Grown in Collagen Scaffolds
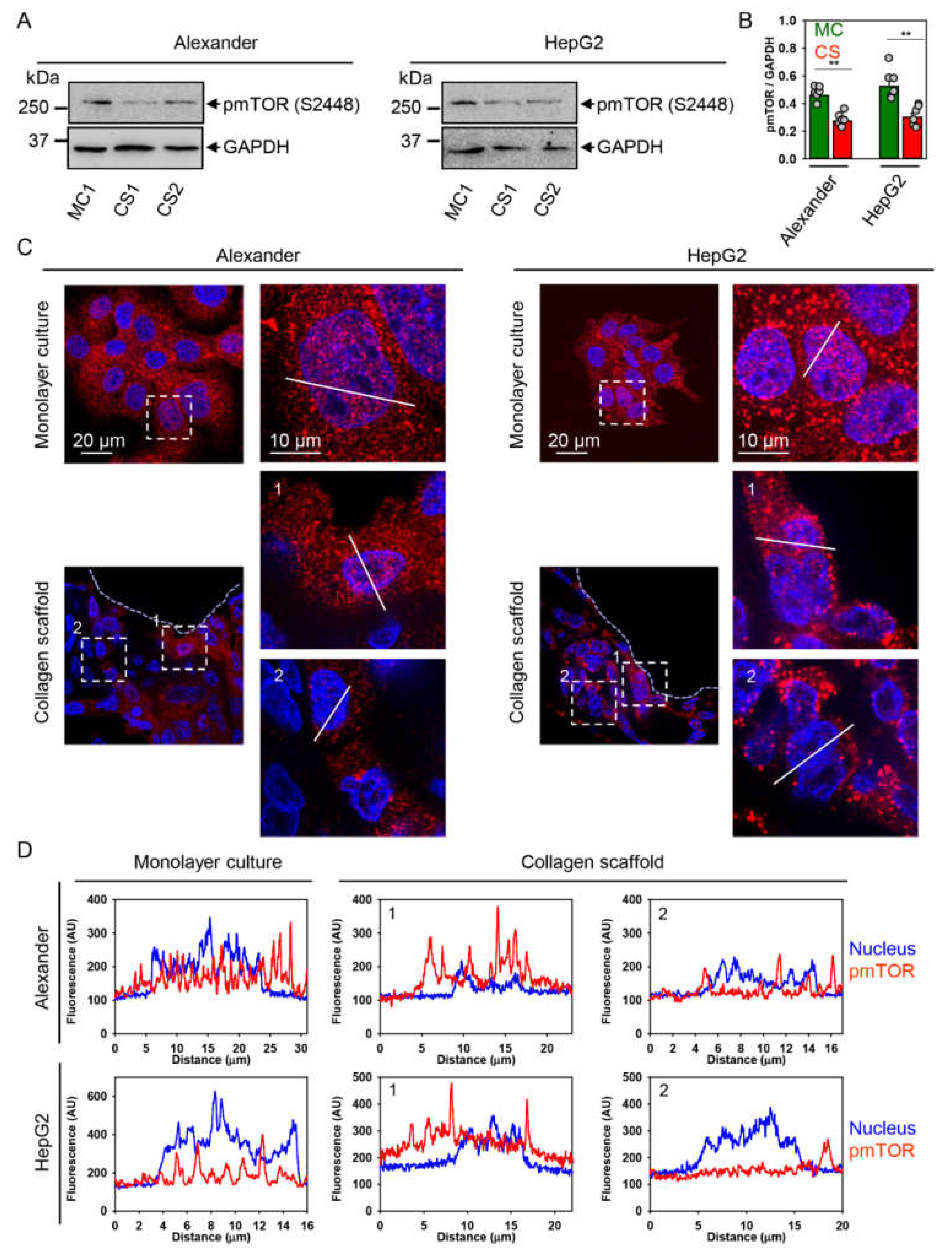
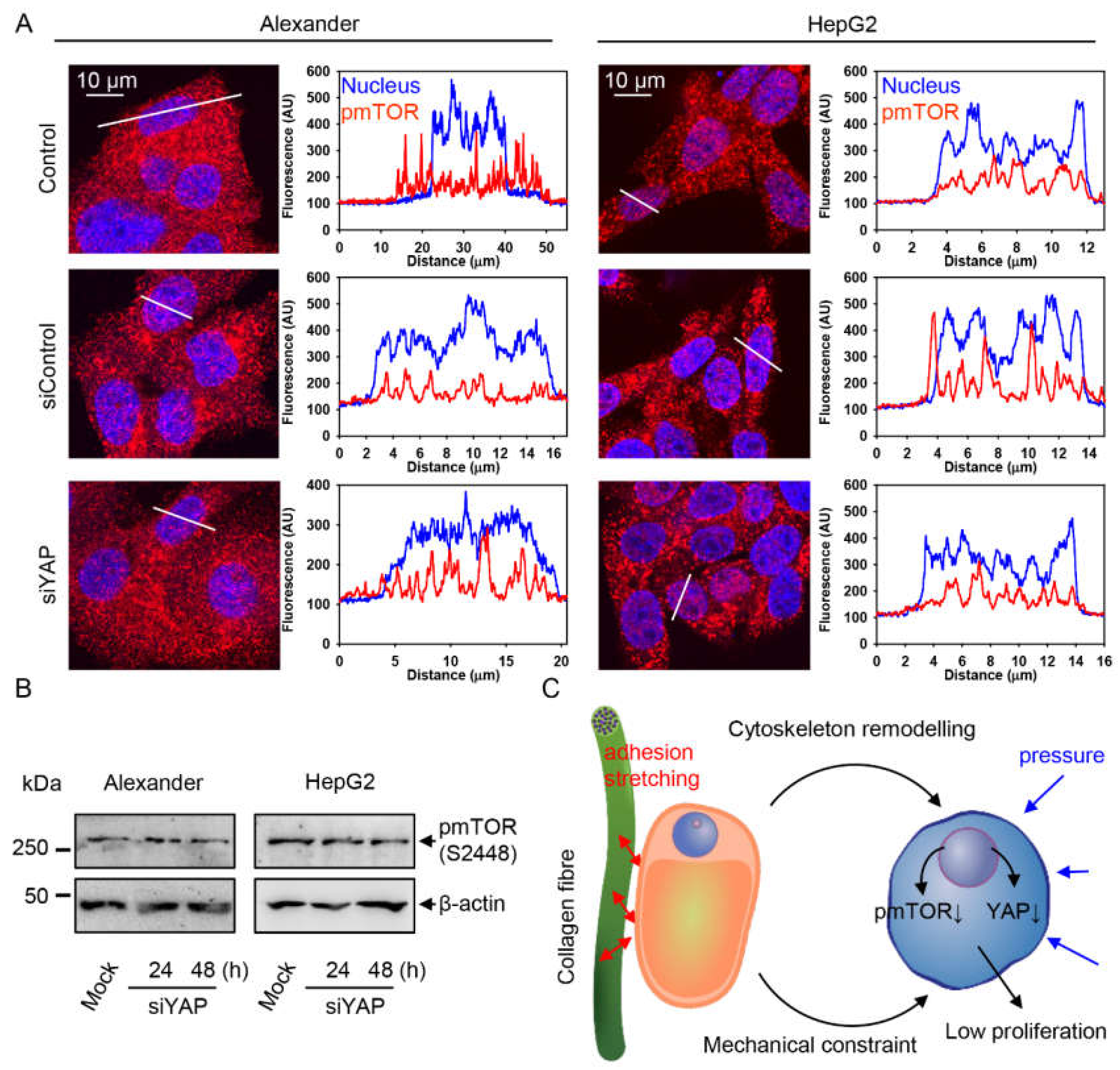
2.4. YAP–mTOR Signaling Interplay under Mechanical Stress in Collagen Scaffolds
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Synthesis and Characterization of Collagen Scaffolds
4.3. Cell Lines and 3D Culturing
4.4. Assessment of Cell Viability
4.5. Cell Proliferation Analysis
4.6. Cell Extracts and Immunoblot Analysis
4.7. Immunofluorescence
4.8. Confocal Microscopy
4.9. Spinning Disk Super-Resolution Microscopy
4.10. Transient Transfection with siRNA
4.11. Quantification of YAP Subcellular Localization
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chaudhuri, O.; Cooper-White, J.; Janmey, P.A.; Mooney, D.J.; Shenoy, V.B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584, 535–546. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, C.S.; Fu, J. Forcing stem cells to behave: A biophysical perspective of the cellular microenvironment. Ann. Rev. Biophys. 2012, 41, 519–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Diaz, M.F.; Price, K.M.; Ozuna, J.A.; Zhang, S.; Sevick-Muraca, E.M.; Hagan, J.P.; Wenzel, P.L. Fluid shear stress activates YAP1 to promote cancer cell motility. Nat. Commun. 2017, 8, 14122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunova, M.; Zablotskii, V.; Dempsey, N.M.; Devillers, T.; Jirsa, M.; Syková, E.; Kubinová, Š.; Lunov, O.; Dejneka, A. Modulation of collective cell behaviour by geometrical constraints. Integr. Biol. 2016, 8, 1099–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartz, M.A.; Lund, A.W. Lymphatic and interstitial flow in the tumour microenvironment: Linking mechanobiology with immunity. Nat. Rev. Cancer 2012, 12, 210–219. [Google Scholar] [CrossRef]
- Masuzaki, R.; Tateishi, R.; Yoshida, H.; Goto, E.; Sato, T.; Ohki, T.; Imamura, J.; Goto, T.; Kanai, F.; Kato, N.; et al. Prospective risk assessment for hepatocellular carcinoma development in patients with chronic hepatitis C by transient elastography. Hepatology 2009, 49, 1954–1961. [Google Scholar] [CrossRef]
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; Van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.J.; Wells, R.G.; Iredale, J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Albhaisi, S.; Sanyal, A.J. Applying Non-Invasive Fibrosis Measurements in NAFLD/NASH: Progress to Date. Pharmaceut. Med. 2019, 33, 451–463. [Google Scholar] [CrossRef]
- Castera, L. Liver stiffness and hepatocellular carcinoma: Liaisons dangereuses? Hepatology 2009, 49, 1793–1794. [Google Scholar] [CrossRef]
- Choong, K.L.; Wong, Y.H.; Yeong, C.H.; Gnanasuntharam, G.K.; Goh, K.L.; Yoong, B.K.; Pongnapang, N.; Abdullah, B.J.J. Elasticity characterization of liver cancers using shear wave ultrasound elastography: Comparison between hepatocellular carcinoma and liver metastasis. J. Diagn. Med. Sonog. 2017, 33, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Masuzaki, R.; Tateishi, R.; Yoshida, H.; Sato, T.; Ohki, T.; Goto, T.; Yoshida, H.; Sato, S.; Sugioka, Y.; Ikeda, H.; et al. Assessing liver tumor stiffness by transient elastography. Hepatol. Int. 2007, 1, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.P.; Luo, Q.; Deng, B.; Ju, Y.; Song, G.B. Stiffer matrix accelerates migration of hepatocellular carcinoma cells through enhanced aerobic glycolysis via the MAPK-YAP signaling. Cancers 2020, 12, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.L.; Liu, S.; Cui, W.; Shi, Y.; Liu, Q.; Duan, J.J.; Yu, S.C.; Zhang, X.; Cui, Y.H.; Kung, H.F.; et al. Aldehyde dehydrogenase 1A1 circumscribes high invasive glioma cells and predicts poor prognosis. Am. J. Cancer Res. 2015, 5, 1471–1483. [Google Scholar]
- Plodinec, M.; Loparic, M.; Monnier, C.A.; Obermann, E.C.; Zanetti-Dallenbach, R.; Oertle, P.; Hyotyla, J.T.; Aebi, U.; Bentires-Alj, M.; Lim, R.Y.; et al. The nanomechanical signature of breast cancer. Nat. Nanotechnol. 2012, 7, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.J. The role of the extracellular matrix in cancer stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Luo, Q.; Ju, Y.; Song, G. A soft matrix enhances the cancer stem cell phenotype of HCC cells. Int. J. Mol. Sci. 2019, 20, 2831. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Ma, M.; Dong, G.; Yao, R.R.; Li, J.H.; Zheng, Q.D.; Dong, Y.Y.; Ma, H.; Gao, D.M.; Cui, J.F.; et al. Increased matrix stiffness promotes tumor progression of residual hepatocellular carcinoma after insufficient heat treatment. Cancer Sci. 2017, 108, 1778–1786. [Google Scholar] [CrossRef]
- Dong, Y.Y.; Zheng, Q.D.; Wang, Z.M.; Lin, X.H.; You, Y.; Wu, S.F.; Wang, Y.H.; Hu, C.; Xie, X.Y.; Chen, J.; et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J. Hematol. Oncol. 2019, 12, 112. [Google Scholar] [CrossRef] [Green Version]
- Deville, S.S.; Cordes, N. The extracellular, cellular, and nuclear stiffness, a trinity in the cancer resistome-A review. Front. Oncol. 2019, 9, 1376. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.-L. The Hippo–YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.-X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhardt, A.A.; Gayyed, M.F.; Klein, A.P.; Dong, J.; Maitra, A.; Pan, D.; Montgomery, E.A.; Anders, R.A. Expression of Yes-associated protein in common solid tumors. Hum. Pathol. 2008, 39, 1582–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Zhang, Y.; Wu, H.; Barry, E.; Yin, Y.; Lawrence, E.; Dawson, D.; Willis, J.E.; Markowitz, S.D.; Camargo, F.D.; et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 2011, 108, E1312–E1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strippoli, R.; Sandoval, P.; Moreno-Vicente, R.; Rossi, L.; Battistelli, C.; Terri, M.; Pascual-Anton, L.; Loureiro, M.; Matteini, F.; Calvo, E.; et al. Caveolin1 and YAP drive mechanically induced mesothelial to mesenchymal transition and fibrosis. Cell Death Dis. 2020, 11, 647. [Google Scholar] [CrossRef]
- Moroishi, T.; Hansen, C.G.; Guan, K.-L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef]
- Liang, N.; Zhang, C.; Dill, P.; Panasyuk, G.; Pion, D.; Koka, V.; Gallazzini, M.; Olson, E.N.; Lam, H.; Henske, E.P.; et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med. 2014, 211, 2249–2263. [Google Scholar] [CrossRef]
- Hu, J.K.-H.; Du, W.; Shelton, S.J.; Oldham, M.C.; DiPersio, C.M.; Klein, O.D. An FAK-YAP-mTOR signaling axis regulates stem cell-based tissue renewal in mice. Cell Stem Cell 2017, 21, 91–106.e106. [Google Scholar] [CrossRef] [Green Version]
- Bui, T.; Rennhack, J.; Mok, S.; Ling, C.; Perez, M.; Roccamo, J.; Andrechek, E.R.; Moraes, C.; Muller, W.J. Functional redundancy between beta1 and beta3 integrin in activating the IR/Akt/mTORC1 signaling axis to promote ErbB2-driven breast cancer. Cell Rep. 2019, 29, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Srikanth, P.; Castle, A.C.; Nigwekar, S.; Malhotra, R.; Galloway, J.L.; Sykes, D.B.; Rajagopal, J. Modulating cell fate as a therapeutic strategy. Cell Stem Cell 2018, 23, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totaro, A.; Zhuang, Q.; Panciera, T.; Battilana, G.; Azzolin, L.; Brumana, G.; Gandin, A.; Brusatin, G.; Cordenonsi, M.; Piccolo, S. Cell phenotypic plasticity requires autophagic flux driven by YAP/TAZ mechanotransduction. Proc. Natl. Acad. Sci. USA 2019, 116, 17848–17857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrila, J.; Radtke, A.L.; Crabbe, A.; Sarker, S.F.; Herbst-Kralovetz, M.M.; Ott, C.M.; Nickerson, C.A. Organotypic 3D cell culture models: Using the rotating wall vessel to study host-pathogen interactions. Nat. Rev. Microbiol. 2010, 8, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension: How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [Green Version]
- Arriazu, E.; Ruiz de Galarreta, M.; Cubero, F.J.; Varela-Rey, M.; Perez de Obanos, M.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular matrix and liver disease. Antioxid. Redox Signal. 2014, 21, 1078–1097. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Hernandez, A.; Amenta, P.S. The hepatic extracellular matrix. II. Ontogenesis, regeneration and cirrhosis. Virchows Arch. 1993, 423, 77–84. [Google Scholar] [CrossRef]
- Martinez-Hernandez, A.; Amenta, P.S. The hepatic extracellular matrix. I. Components and distribution in normal liver. Virchows Arch. 1993, 423, 1–11. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Barnes, C.P.; Sell, S.A.; Boland, E.D.; Simpson, D.G.; Bowlin, G.L. Nanofiber technology: Designing the next generation of tissue engineering scaffolds. Adv. Drug Deliv. Rev. 2007, 59, 1413–1433. [Google Scholar] [CrossRef] [PubMed]
- Ruoss, M.; Rebholz, S.; Weimer, M.; Grom-Baumgarten, C.; Athanasopulu, K.; Kemkemer, R.; Kass, H.; Ehnert, S.; Nussler, A.K. Development of scaffolds with adjusted stiffness for mimicking disease-related alterations of liver rigidity. J. Funct. Biomater. 2020, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arjmand, A.; Tsipouras, M.G.; Tzallas, A.T.; Forlano, R.; Manousou, P.; Giannakeas, N. Quantification of liver fibrosis—A comparative study. Appl. Sci. Basel 2020, 10, 447. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Shin, J.; Park, H.M.; Kim, Y.G.; Kim, B.G.; Oh, J.W.; Cho, S.W. Liver extracellular matrix providing dual functions of two-dimensional substrate coating and three-dimensional injectable hydrogel platform for liver tissue engineering. Biomacromolecules 2014, 15, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Ruoss, M.; Vosough, M.; Konigsrainer, A.; Nadalin, S.; Wagner, S.; Sajadian, S.; Huber, D.; Heydari, Z.; Ehnert, S.; Hengstler, J.G.; et al. Towards improved hepatocyte cultures: Progress and limitations. Food Chem. Toxicol. 2020, 138, 111188. [Google Scholar] [CrossRef] [PubMed]
- Lozinsky, V.I. Cryostructuring of polymeric systems. 55. Retrospective view on the more than 40 years of studies performed in the A.N.Nesmeyanov Institute of Organoelement Compounds with respect of the cryostructuring processes in polymeric systems. Gels 2020, 6, 29. [Google Scholar] [CrossRef]
- Lee, J.T.; Chow, K.L. SEM sample preparation for cells on 3D scaffolds by freeze-drying and HMDS. Scanning 2012, 34, 12–25. [Google Scholar] [CrossRef]
- Lunova, M.; Smolkova, B.; Uzhytchak, M.; Janouskova, K.Z.; Jirsa, M.; Egorova, D.; Kulikov, A.; Kubinova, S.; Dejneka, A.; Lunov, O. Light-induced modulation of the mitochondrial respiratory chain activity: Possibilities and limitations. Cell. Mol. Life Sci. 2020, 77, 2815–2838. [Google Scholar] [CrossRef]
- Hayashi, S.; Okada, Y. Ultrafast superresolution fluorescence imaging with spinning disk confocal microscope optics. Mol. Biol. Cell 2015, 26, 1743–1751. [Google Scholar] [CrossRef] [Green Version]
- Nikolova, M.P.; Chavali, M.S. Recent advances in biomaterials for 3D scaffolds: A review. Bioact. Mater. 2019, 4, 271–292. [Google Scholar] [CrossRef]
- Luckert, C.; Schulz, C.; Lehmann, N.; Thomas, M.; Hofmann, U.; Hammad, S.; Hengstler, J.G.; Braeuning, A.; Lampen, A.; Hessel, S. Comparative analysis of 3D culture methods on human HepG2 cells. Arch. Toxicol. 2017, 91, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Abrams, G.A.; Goodman, S.L.; Nealey, P.F.; Franco, M.; Murphy, C.J. Nanoscale topography of the basement membrane underlying the corneal epithelium of the rhesus macaque. Cell Tissue Res. 2000, 299, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.H.; Trapani, L.M.; Sieminski, A.L.; MacKellar, D.; Gong, H.; Kamm, R.D.; Wells, A.; Lauffenburger, D.A.; Matsudaira, P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc. Natl. Acad. Sci. USA 2006, 103, 10889–10894. [Google Scholar] [CrossRef] [Green Version]
- Hayman, M.W.; Smith, K.H.; Cameron, N.R.; Przyborski, S.A. Enhanced neurite outgrowth by human neurons grown on solid three-dimensional scaffolds. Biochem. Biophys. Res. Commun. 2004, 314, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Bavli, D.; Prill, S.; Ezra, E.; Levy, G.; Cohen, M.; Vinken, M.; Vanfleteren, J.; Jaeger, M.; Nahmias, Y. Real-time monitoring of metabolic function in liver-on-chip microdevices tracks the dynamics of mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, E2231–E2240. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.Y.; Wang, J.L.; Jia, J.; Song, N.; Xiang, C.G.; Xu, J.; Hou, Z.Y.; Su, X.H.; Liu, B.; Jiang, T.; et al. Human hepatocytes with drug metabolic function induced from fibroblasts by lineage reprogramming. Cell Stem Cell 2014, 14, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Prieto, A.M.; Skelton, J.K.; Wai, S.N.; Large, E.; Lussignol, M.; Vizcay-Barrena, G.; Hughes, D.; Fleck, R.A.; Thursz, M.; Catanese, M.T.; et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat. Commun. 2018, 9, 682. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Rashbastep, J.; Cederbaum, A.I. Stable expression of human cytochrome-P4502e1 in Hepg2 cells—Characterization of catalytic activities and production of reactive oxygen intermediates. Biochemistry 1993, 32, 6928–6937. [Google Scholar] [CrossRef]
- Henzel, K.; Thorborg, C.; Hofmann, M.; Zimmer, G.; Leuschner, U. Toxicity of ethanol and acetaldehyde in hepatocytes treated with ursodeoxycholic or tauroursodeoxycholic acid. Biochim. Biophys. Acta 2004, 1644, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Lamas-Paz, A.; Hao, F.J.; Nelson, L.J.; Vazquez, M.T.; Canals, S.; Del Moral, M.G.; Martinez-Naves, E.; Nevzorova, Y.A.; Cubero, F.J. Alcoholic liver disease: Utility of animal models. World J. Gastroenterol. 2018, 24, 5063–5075. [Google Scholar] [CrossRef]
- Taftachi, R.; Ayhan, A.; Ekici, S.; Ergen, A.; Ozen, H. Proliferating-cell nuclear antigen (PCNA) as an independent prognostic marker in patients after prostatectomy: A comparison of PCNA and Ki-67. BJU Int. 2005, 95, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C. PCNA: A silent housekeeper or a potential therapeutic target? Trends Pharmacol. Sci. 2014, 35, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Ihmann, T.; Liu, J.A.; Schwabe, W.; Hausler, P.; Behnke, D.; Bruch, H.P.; Broll, R.; Windhovel, U.; Duchrow, M. High-level mRNA quantification of proliferation marker pKi-67 is correlated with favorable prognosis in colorectal carcinoma. J. Cancer Res. Clin. 2004, 130, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Magana, A.; Blanco, F.J. Human PCNA structure, function, and interactions. Biomolecules 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelman, Z. PCNA: Structure, functions and interactions. Oncogene 1997, 14, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, D.A.; Mullins, D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Vicente, R.; Pavon, D.M.; Martin-Padura, I.; Catala-Montoro, M.; Diez-Sanchez, A.; Quilez-Alvarez, A.; Lopez, J.A.; Sanchez-Alvarez, M.; Vazquez, J.; Strippoli, R.; et al. Caveolin-1 modulates mechanotransduction responses to substrate stiffness through actin-dependent control of YAP. Cell Rep. 2018, 25, 1622–1635. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Gonzalez, M.; Latorre, E.; Arroyo, M.; Trepat, X. Measuring mechanical stress in living tissues. Nat. Rev. Phys. 2020, 2, 300–317. [Google Scholar] [CrossRef]
- Uroz, M.; Wistorf, S.; Serra-Picamal, X.; Conte, V.; Sales-Pardo, M.; Roca-Cusachs, P.; Guimera, R.; Trepat, X. Regulation of cell cycle progression by cell-cell and cell-matrix forces. Nat. Cell Biol. 2018, 20, 646–654. [Google Scholar] [CrossRef]
- Dasgupta, I.; McCollum, D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef] [Green Version]
- Noce, V.; Battistelli, C.; Cozzolino, A.M.; Consalvi, V.; Cicchini, C.; Strippoli, R.; Tripodi, M.; Marchetti, A.; Amicone, L. YAP integrates the regulatory Snail/HNF4 alpha circuitry controlling epithelial/hepatocyte differentiation. Cell Death Dis. 2019, 10, 768. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Cai, Y.; Li, Y.H.; Li, Y.; Hu, N.; Ma, S.; Hu, S.Y.; Zhu, P.J.; Wang, W.H.; Zhou, H. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 2018, 14, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, J. Regulation of Hippo signaling by actin remodeling. BMB Rep. 2018, 51, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.T.; Chen, J.X.; Lim, Y.B.; Finch-Edmondson, M.L.; Seshachalam, V.P.; Qin, L.; Jiang, T.; Low, B.C.; Singh, H.; Lim, C.T.; et al. YAP regulates actin dynamics through ARHGAP29 and promotes metastasis. Cell Rep. 2017, 19, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Frith, J.E.; Kusuma, G.D.; Carthew, J.; Li, F.; Cloonan, N.; Gomez, G.A.; Cooper-White, J.J. Mechanically-sensitive miRNAs bias human mesenchymal stem cell fate via mTOR signalling. Nat. Commun. 2018, 9, 257. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.S.; Liu, Y.W. Mechanical stretch induces mTOR recruitment and activation at the phosphatidic acid-enriched macropinosome in muscle cell. Front. Cell Dev. Biol. 2019, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [Green Version]
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing mTOR complexes in regulation of Glioblastoma Multiforme and its stem cells. Adv. Biol. Regul. 2013, 53, 202–210. [Google Scholar] [CrossRef]
- Jiang, Y. mTOR goes to the nucleus. Cell Cycle 2010, 9, 868. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Clister, T.L.; Lowry, P.R.; Seldin, M.M.; Wong, G.W.; Zhang, J. Dynamic visualization of mTORC1 activity in living cells. Cell Rep. 2015, 10, 1767–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobashi, Y.; Suzuki, S.; Sato, E.; Hamada, Y.; Yanagawa, T.; Ooi, A. EGFR-dependent and independent activation of Akt/mTOR cascade in bone and soft tissue tumors. Mod. Pathol. 2009, 22, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Tsang, C.K.; Watkins, M.; Bertram, P.G.; Zheng, X.F. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature 2006, 442, 1058–1061. [Google Scholar] [CrossRef]
- Uzhytchak, M.; Smolkova, B.; Lunova, M.; Jirsa, M.; Frtus, A.; Kubinova, S.; Dejneka, A.; Lunov, O. Iron oxide nanoparticle-induced autophagic flux Is regulated by interplay between p53-mTOR axis and Bcl-2 signaling in hepatic cells. Cells 2020, 9, 1015. [Google Scholar] [CrossRef] [Green Version]
- Audet-Walsh, E.; Dufour, C.R.; Yee, T.; Zouanat, F.Z.; Yan, M.; Kalloghlian, G.; Vernier, M.; Caron, M.; Bourque, G.; Scarlata, E.; et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev. 2017, 31, 1228–1242. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.Y.; Zhen, Y.Y.; Yuen, C.M.; Fan, R.; Chen, Y.T.; Sheu, J.J.; Chen, Y.L.; Wang, C.J.; Sun, C.K.; Yip, H.K. The mTOR-FAK mechanotransduction signaling axis for focal adhesion maturation and cell proliferation. Am. J. Transl. Res. 2017, 9, 1603–1617. [Google Scholar]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Kang, J.; Fu, C.Y. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Doyle, A.D.; Yamada, K.M. Mechanosensing via cell-matrix adhesions in 3D microenvironments. Exp. Cell Res. 2016, 343, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Yu, C.; Wang, P.; Xu, W.; Wan, X.; Lai, C.S.E.; Liu, J.; Koroleva-Maharajh, A.; Chen, S. Rapid 3D bioprinting of decellularized extracellular matrix with regionally varied mechanical properties and biomimetic microarchitecture. Biomaterials 2018, 185, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Mooney, D.J. Extracellular matrix stiffness causes systematic variations in proliferation and chemosensitivity in myeloid leukemias. Proc. Natl. Acad. Sci. USA 2016, 113, 12126–12131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achterberg, V.F.; Buscemi, L.; Diekmann, H.; Smith-Clerc, J.; Schwengler, H.; Meister, J.J.; Wenck, H.; Gallinat, S.; Hinz, B. The nano-scale mechanical properties of the extracellular matrix regulate dermal fibroblast function. J. Investig. Dermatol. 2014, 134, 1862–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavo, M.; Fato, M.; Penuela, L.; Beltrame, F.; Raiteri, R.; Scaglione, S. Microenvironment complexity and matrix stiffness regulate breast cancer cell activity in a 3D in vitro model. Sci. Rep. 2016, 6, 35367. [Google Scholar] [CrossRef] [Green Version]
- Barkan, D.; Green, J.E.; Chambers, A.F. Extracellular matrix: A gatekeeper in the transition from dormancy to metastatic growth. Eur. J. Cancer 2010, 46, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Zou, D.; Dong, L.; Li, C.; Yin, Z.; Rao, S.; Zhou, Q. The m(6)A eraser FTO facilitates proliferation and migration of human cervical cancer cells. Cancer Cell Int. 2019, 19, 321. [Google Scholar] [CrossRef] [Green Version]
- Ramaiahgari, S.C.; Den Braver, M.W.; Herpers, B.; Terpstra, V.; Commandeur, J.N.; Van de Water, B.; Price, L.S. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch. Toxicol. 2014, 88, 1083–1095. [Google Scholar] [CrossRef]
- Yang, N.; Chen, T.; Wang, L.; Liu, R.; Niu, Y.; Sun, L.; Yao, B.; Wang, Y.; Yang, W.; Liu, Q.; et al. CXCR4 mediates matrix stiffness-induced downregulation of UBTD1 driving hepatocellular carcinoma progression via YAP signaling pathway. Theranostics 2020, 10, 5790–5801. [Google Scholar] [CrossRef]
- Hui, L.; Zhang, J.; Ding, X.; Guo, X.; Jiang, X. Matrix stiffness regulates the proliferation, stemness and chemoresistance of laryngeal squamous cancer cells. Int. J. Oncol. 2017, 50, 1439–1447. [Google Scholar] [CrossRef]
- Gao, J.; He, L.; Zhou, L.; Jing, Y.; Wang, F.; Shi, Y.; Cai, M.; Sun, J.; Xu, H.; Jiang, J.; et al. Mechanical force regulation of YAP by F-actin and GPCR revealed by super-resolution imaging. Nanoscale 2020, 12, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.G.; Dedoussis, G.V.Z.; Spanakos, G.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. An improved fluorescence assay for the determination of lymphocyte-mediated cytotoxicity using flow-cytometry. J. Immunol. Methods 1994, 177, 101–111. [Google Scholar] [CrossRef]
- Lynnyk, A.; Lunova, M.; Jirsa, M.; Egorova, D.; Kulikov, A.; Kubinova, S.; Lunov, O.; Dejneka, A. Manipulating the mitochondria activity in human hepatic cell line Huh7 by low-power laser irradiation. Biomed. Opt. Express 2018, 9, 1283–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunov, O.; Zablotskii, V.; Churpita, O.; Lunova, M.; Jirsa, M.; Dejneka, A.; Kubinova, S. Chemically different non-thermal plasmas target distinct cell death pathways. Sci. Rep. 2017, 7, 600. [Google Scholar] [CrossRef] [Green Version]
- Lunov, O.; Syrovets, T.; Rocker, C.; Tron, K.; Nienhaus, G.U.; Rasche, V.; Mailander, V.; Landfester, K.; Simmet, T. Lysosomal degradation of the carboxydextran shell of coated superparamagnetic iron oxide nanoparticles and the fate of professional phagocytes. Biomaterials 2010, 31, 9015–9022. [Google Scholar] [CrossRef]
- Smolkova, B.; Lunova, M.; Lynnyk, A.; Uzhytchak, M.; Churpita, O.; Jirsa, M.; Kubinova, S.; Lunov, O.; Dejneka, A. Non-thermal plasma, as a new physicochemical source, to induce redox imbalance and subsequent cell death in liver cancer cell lines. Cell. Physiol. Biochem. 2019, 52, 119–140. [Google Scholar]
- Levada, K.; Pshenichnikov, S.; Omelyanchik, A.; Rodionova, V.; Nikitin, A.; Savchenko, A.; Schetinin, I.; Zhukov, D.; Abakumov, M.; Majouga, A.; et al. Progressive lysosomal membrane permeabilization induced by iron oxide nanoparticles drives hepatic cell autophagy and apoptosis. Nano Converg. 2020, 7, 17. [Google Scholar] [CrossRef]
- Jelinek, M.; Kocourek, T.; Jurek, K.; Jelinek, M.; Smolkova, B.; Uzhytchak, M.; Lunov, O. Preliminary study of Ge-DLC nanocomposite biomaterials prepared by laser codeposition. Nanomaterials 2019, 9, 451. [Google Scholar] [CrossRef] [Green Version]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.-L.; et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 2017, 171, 1397–1410. [Google Scholar] [CrossRef]
- Dell, R.B.; Holleran, S.; Ramakrishnan, R. Sample size determination. ILAR J. 2002, 43, 207–213. [Google Scholar] [CrossRef]
- Jonkman, J.; Brown, C.M.; Wright, G.D.; Anderson, K.I.; North, A.J. Tutorial: Guidance for quantitative confocal microscopy. Nat. Protoc. 2020, 15, 1585–1611. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kitaoka, M. A beginner’s guide to rigor and reproducibility in fluorescence imaging experiments. Mol. Biol. Cell 2018, 29, 1519–1525. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Description | Diffusion Times (ms) and Fractions 1 | |
|---|---|---|
| Scaffold Pores | Collagen Matrix | |
| Alexa 647 in collagen scaffold (sample 1) | 0.35 ± 0.06 (26 ± 9)% | 0.63 ± 0.19 (45 ± 6)% |
| Alexa 647 in collagen scaffold (sample 2) | 0.32 ± 0.06 (17 ± 3)% | 0.60 ± 0.08 (38 ± 4)% |
| Alexa 647 in collagen scaffold (sample 3) | 0.22 ± 0.02 (21 ± 2)% | 0.43 ± 0.15 (45 ± 5)% |
| Alexa 647 in collagen scaffold (sample 4) | 0.86 ± 0.37 (14 ± 2)% | 0.91 ± 0.33 (42 ± 2)% |
| Dextran 3000-Alexa 488 (sample 3) | 1.32 ± 0.78 (27 ± 17)% | 3.39 ± 1.27 (47 ± 11)% |
| Dextran 3000-Alexa 488 (sample 3) | 1.49 ± 0.70 (23 ± 8)% | 44.65 ± 17.80 (8 ± 1)% |
| Dextran 10,000-Alexa 488 (sample 4) | 0.57 ± 0.51 (28 ± 3)% | 2.02 ± 0.40 (25 ± 4)% |
| Dextran 10,000-Alexa 488 (sample 4) | 0.81 ± 0.44 (16 ± 4)% | 3.35 ± 0.45 (37 ± 6)% |
| DOPC-Bodipy in POPC liposomes (sample 1) | 9.46 ± 4.96 | 90.74 ± 56.49 |
| DOPC-Bodipy in POPC liposomes (sample 2) | 1.86 ± 0.12 | 125.46 ± 99.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frtús, A.; Smolková, B.; Uzhytchak, M.; Lunova, M.; Jirsa, M.; Hof, M.; Jurkiewicz, P.; Lozinsky, V.I.; Wolfová, L.; Petrenko, Y.; et al. Hepatic Tumor Cell Morphology Plasticity under Physical Constraints in 3D Cultures Driven by YAP–mTOR Axis. Pharmaceuticals 2020, 13, 430. https://doi.org/10.3390/ph13120430
Frtús A, Smolková B, Uzhytchak M, Lunova M, Jirsa M, Hof M, Jurkiewicz P, Lozinsky VI, Wolfová L, Petrenko Y, et al. Hepatic Tumor Cell Morphology Plasticity under Physical Constraints in 3D Cultures Driven by YAP–mTOR Axis. Pharmaceuticals. 2020; 13(12):430. https://doi.org/10.3390/ph13120430
Chicago/Turabian StyleFrtús, Adam, Barbora Smolková, Mariia Uzhytchak, Mariia Lunova, Milan Jirsa, Martin Hof, Piotr Jurkiewicz, Vladimir I. Lozinsky, Lucie Wolfová, Yuriy Petrenko, and et al. 2020. "Hepatic Tumor Cell Morphology Plasticity under Physical Constraints in 3D Cultures Driven by YAP–mTOR Axis" Pharmaceuticals 13, no. 12: 430. https://doi.org/10.3390/ph13120430
APA StyleFrtús, A., Smolková, B., Uzhytchak, M., Lunova, M., Jirsa, M., Hof, M., Jurkiewicz, P., Lozinsky, V. I., Wolfová, L., Petrenko, Y., Kubinová, Š., Dejneka, A., & Lunov, O. (2020). Hepatic Tumor Cell Morphology Plasticity under Physical Constraints in 3D Cultures Driven by YAP–mTOR Axis. Pharmaceuticals, 13(12), 430. https://doi.org/10.3390/ph13120430