Chemicals, Materials, and Methods
All reagents were purchased from Sigma–Aldrich (St. Louis, MO, USA), Alfa Aesar (Haverhill, MA, USA), or TCI (Tokyo, Japan) in the highest quality commercially available. The solvents were RP grade. The melting points were determined using the Büchi (Flawil, Switzerland) B-540 apparatus. The purity of the products was evaluated unequivocally through MS, 1H NMR, 13C NMR, IR, and [α]20D. The MS (ESI) spectra were recorded with a Waters (Milford, MA, USA) Micromass ZQ spectrometer in a positive mode using a nebulizing nitrogen gas at 400 L/min and a temperature of 250 °C, cone flow 40 mL/min, capillary 3.5 K volts, and cone voltage 60 V; the revelation was performed either in ESI (+) and/or ESI (−), from 100 to 800 units of mass, spectrophotometrically measured using a diode array spectrophotometer. The 1H NMR and 13C NMR spectra were recorded on a Bruker (Billerica, MA, USA) AC 400 or 101 instrument and analyzed using the Top Spin 1.3 (2013) Bruker NMR software package. Chemical shifts were measured using the central peak of the solvent. Purification of the crude material was carried out by column chromatography on silica gel “flash” [230–400 mm, Merck (Darmstadt, Germany)]. The TLC analyses were performed on pre-coated aluminum oxide on aluminum sheets (60 F254, neutral; Merck). The IR spectra were obtained with a Thermo Fisher Scientific (Waltham, MA, USA) Nicolet Avatar 360 spectrophotometer. The optical rotations were measured on the Digital Perkin Elmer (Waltham, MA, USA) 241 polarimeter using a sodium lamp (589 nm) as a light source; concentrations were expressed in g/100 mL, and the length of the cell was 1 dm.
Synthesis of allyl 3,5,6-tri-O-benzyl-α-D-glucofuranoside (12a) and allyl 3,5,6-tri-O-benzyl-β-D-glucofuranoside (
12b). To a solution of
2 (1 g, 2.04 mmol) [obtained by treating
1 (2 g, 9.08 mmol) with 80% NaH (0.9 g, 30 mmol), BnBr (4.66 g, 27.25 mmol) in DMF (40 mL)] at room temperature for 6 h in allyl alcohol (14.6 mL) and under stirring, TsOH (0.062 g, 0.33 mmol) was added. After the reactants were refluxed for 3 h, the mixture was cooled, extracted with CH
2Cl
2, washed with NaHCO
3 sat., dried over Na
2SO
4, filtered and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 9:1) gave
12a and
12b as light yellow oils, which were dried under N
2 atmosphere because of their thermolability (
Rf:
12a 0.4,
12b 0.3; cyclohexane/EtOAc 6:4). Yield:
12a 40% (0.4 g, 0.82 mmol);
12b 45% (0.45 g, 0.92 mmol).
12a: MS (ESI): 490 (M + H
+).
1H NMR (CDCl
3)
δ: 3.61 (dd, 1H,
J6b–5 = 5.8 Hz,
J6b–6a = 10.6 Hz, H
6b), 3.78 (dd, 1H,
J6a–5 = 2.0 Hz,
J6a–6b = 10.6 Hz, H
6a), 3.95 (ddd, 1H,
J5–6a = 2.0 Hz,
J5–6b = 5.8 Hz,
J5–4 = 8.4 Hz, H
5), 4.00 (dd, 1H,
J3–2 = 1.9 Hz,
J3–4 = 4.4 Hz, H
3), 4.01 (dddd, 1H,
J7b–9a =
J7b–9b = 1.5 Hz,
J7b–8 = 6.2 Hz,
J7b–7a = 12.8 Hz, H
7b OCH
HCHCH
2), 4.22 (dddd, 1H,
J7a–9a =
J7a–9b = 1.5 Hz,
J7a–8 = 5.2 Hz,
J7a–7b = 12.8 Hz, H
7a OC
HHCHCH
2), 4.26 (dd, 1H,
J2–3 = 1.9 Hz,
J2–1 = 4.5 Hz, H
2), 4.27 (dd, 1H,
J4–3 = 4.4 Hz,
J4–5 = 8.4 Hz, H
4), 4.45 (d, 1H,
J = 11.5 Hz, OC
H2C
6H
5), 4.47 (d, 1H,
J = 11.5 Hz, OC
H2C
6H
5), 4.49 (d, 1H,
J = 12.2 Hz, OC
H2C
6H
5), 4.53 (d, 1H,
J = 12.2 Hz, OC
H2C
6H
5), 4.63 (d, 1H,
J = 11.5 Hz, OC
H2C
6H
5), 4.72 (d, 1H,
J = 11.5 Hz, OC
H2C
6H
5), 5.11 (d, 1H,
J1–2 = 4.5 Hz, H
1), 5.12 (dddd, 1H,
J9b–9a =
J9b–7a =
J9b–7b = 1.5 Hz,
J9b–8 = 10.2 Hz, H
9b OCH
2CHCH
H), 5.19 (dddd, 1H,
J9a–9b =
J9a–7a =
J9a–7b = 1.5 Hz,
J9a–8 = 17.2 Hz, H
9a OCH
2CHC
HH), 5.83 (dddd, 1H,
J8–7a = 5.2 Hz,
J8–7b = 6.2 Hz,
J8–b = 10.2 Hz,
J8–9a = 17.2 Hz, H
8 OCH
2C
HCH
2), 7.24–7.37 (m, 15H, Ar
H) ppm.
13C NMR (CDCl
3)
δ: 69.2, 71.2, 71.7, 72.6, 73.4, 76.1, 77.9, 83.9, 100.4, 117.7, 127.5, 127.5, 127.6, 128.1, 128.2, 128.3, 133.7, 137.9, 138.6, 138.9 ppm. IR (Nujol) ν = 3550, 1454, 1372, 1206, 1057 cm
−1. [α]
20D = –56.8 (c 0.3, CHCl
3).
12b: MS (ESI): 490 (M + H
+).
1H NMR (CDCl
3)
δ: 3.63 (dd, 1H,
J6b–5 = 5.3 Hz,
J6b–6a = 10.7 Hz, H
6b), 3.80 (dd, 1H,
J6a–5 = 2.0 Hz,
J6a–6b = 10.7 Hz, H
6a), 3.90 (dddd, 1H,
J7b–9a =
J7b–9b = 1.5 Hz,
J7b–8 = 6.0 Hz,
J7b–7a = 13.0 Hz, H
7b OCH
HCHCH
2), 4.00 (ddd, 1H,
J5–6a = 2.0 Hz,
J5–6b = 5.3 Hz,
J5–4 = 8.8 Hz, H
5), 4.00 (dd, 1H,
J3–2 = 1.0 Hz,
J3–4 = 5.0 Hz, H
3), 4.14 (dddd, 1H,
J7a–9a =
J7a–9b = 1.5 Hz,
J7a–8 = 5.0 Hz,
J7a–7b = 13.0 Hz, H
7a OC
HHCHCH
2), 4.21 (dd, 1H,
J2–1 ≅
J2–3 = 1.0 Hz, H
2), 4.33 (dd, 1H,
J4–3 = 5.0 Hz,
J4–5 = 8.8 Hz, H
4), 4.43 (d, 1H,
J = 11.5 Hz, OC
H2C
6H
5), 4.47 (d, 1H,
J = 11.5 Hz, OC
H2C
6H
5), 4.52 (s, 2H, OC
H2C
6H
5), 4.57 (d, 1H,
J = 12.2 Hz, OC
H2C
6H
5), 4.67 (d, 1H,
J = 11.5 Hz, OC
H2C
6H
5), 4.87 (d, 1H,
J1–2 = 1.0 Hz, H
1), 5.10 (dddd, 1H,
J9b–9a =
J9b–7a =
J9b–7b = 1.5 Hz,
J9b–8 = 10.5 Hz, H
9b OCH
2CHCH
H), 5.22 (dddd, 1H,
J9a–9b =
J9a–7a =
J9a–7b = 1.5 Hz,
J9a–8 = 17.0 Hz, H
9a OCH
2CHC
HH), 5.83 (dddd, 1H,
J8–7a = 5.0 Hz,
J8–7b = 6.0 Hz,
J8–9b = 10.5 Hz,
J8–9a = 17.0 Hz, H
8 OCH
2C
HCH
2), 7.24–7.37 (m, 15H, Ar
H) ppm.
13C NMR (CDCl
3)
δ: 68.9, 70.8, 72.0, 72.5, 73.4, 76.7, 77.0, 77.3, 77.3, 78.6, 79.9, 83.0, 107.8, 117.0, 127.6, 127.7, 128.2, 128.3, 128.3, 134.2, 134.3, 138.0, 138.6, 138.9 ppm. IR (Nujol) ν = 3423, 1462, 1398, 1061 cm
−1. [α]
20D = +19.5 (c 0.2, CHCl
3). The physico-chemical data are in agreement with those reported in the literature [
28].
Synthesis of 1,2,3,4-tetra-O-acetyl-α
-D-xylopyranose (
16a). To a solution of
15 (1 g, 6.66 mmol) in pyridine (5 g, 4.5 mL, 62 mmol), Ac
2O was added (5.5 g, 4.5 mL, 52.74 mmol). The mixture was stirred at 0 °C for 6 h, extracted with Et
2O and washed with H
2O and CuSO
4 saturated solution. The combined organic layers were dried with Na
2SO
4, filtered and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 9:1) gave
16a as a yellow oil which was dried under N
2 atmosphere because of its thermolability. Yield: 75% (1.59 g, 5 mmol). MS (ESI): 336 (M + NH
4+).
1H NMR (CDCl
3)
δ: 2.04–2.19 (m, 12H, C
H3CO), 3.72 (dd, 1H,
J5b–4 =
J5b–5a = 11.0 Hz, H
5b), 3.95 (dd, 1H,
J5a–4 = 6.0 Hz,
J5a–5b = 11.0 Hz, H
5a), 5.04 (dd, 1H,
J2–1 = 3.5 Hz,
J2–3 = 10.0 Hz, H
2), 5.05 (ddd, 1H,
J4–5a = 6.0 Hz,
J4–3 = 10.0 Hz,
J4–5b = 11.0 Hz, H
4), 5.48 (dd, 1H,
J3–2 =
J3–4 = 10.0 Hz, H
3), 6.27 (d, 1H,
J1–2 = 3.5 Hz, H
1) ppm.
13C NMR (CDCl
3)
δ: 20.4, 20.6, 20.7, 20.8, 60.6, 68.7, 69.3, 89.2, 168.9, 169.6, 169.7, 170.1 ppm. IR (film) ν = 1766, 1232, 1128 cm
−1. [α]
20D = +88.0 (c 0.3, CHCl
3).
1H NMR,
13C NMR, and IR data are in agreement with those reported in the literature [
21].
Synthesis of 2,3,4-tri-O-acetyl-D-xylopyranose (
17). To a solution of
16a (1 g, 3.14 mmol) in dry DMF (9 mL), AcONH
4 (0.485 g, 6.29 mmol) was added and the mixture was stirred at room temperature for 22 h, extracted with EtOAc, and washed with H
2O and saturated aq. NH
4Cl solution. The combined organic layers were dried with Na
2SO
4 and concentrated. Purification of the oily residue by column chromatography (cyclohexane/EtOAc 7:3) gave
17 as a white solid. Yield 70% (0.607 g, 2.2 mmol). Mp: 142–144 °C (EtOAc/petroleum ether). MS (ESI): 299 (M + Na
+). [α]
20D = +69.0 (c 0.2, CHCl
3).
1H NMR,
13C NMR, and IR data are in agreement with those reported in the literature [
29].
Synthesis of 2,3,4-tri-O-acetyl-D-xylopyranosyl-α-trichloroacetimidate (
14a). To a solution of
17 (0.407 g, 1.48 mmol) and CCl
3CN (1.25 g, 0.86 mL, 8.83 mmol) in dry CH
2Cl
2 (14 mL) at 0 °C and under N
2 atmosphere, DBU was added (0.09 g, 0.88 mL, 0.59 mmol) and the mixture was stirred at 0 °C for 1 h and then concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 9:1) gave
14a as a yellow oil (
Rf: 0.5, cyclohexane/EtOAc 7:3). Yield: 60% (0.37 g, 0.86 mmol). MS (ESI): 437 (M + NH
4+).
1H NMR (CDCl
3)
δ: 2.03 (s, 3H, C
H3CO), 2.06 (s, 3H, C
H3CO), 2.07 (s, 3H, C
H3CO), 3.82 (dd, 1H,
J5a–4 =
J5a–5b = 11.0 Hz, H
5a), 4.00 (dd, 1H,
J5b–4 = 5.9 Hz,
J5b–5a = 11.0 Hz, H
5b), 5.08 (dd, 1H,
J2–1 = 3.6 Hz,
J2–3 = 10.0 Hz, H
2), 5.11 (ddd, 1H,
J4–5b = 5.9 Hz,
J4–5a = 7.5 Hz,
J4–3 = 11.0 Hz, H
4), 5.58 (dd, 1H,
J3–2 =
J3–4 = 10.0 Hz, H
3), 6.50 (d, 1H,
J1–2 = 3.6 Hz, H
1), 8.67 [s, 1H, C(N
H)CCl
3] ppm.
13C NMR (CDCl
3)
δ: 20.5, 20.6, 20.7, 60.8, 68.6, 69.3, 69.9, 76.7, 77.0, 77.3, 93.1, 160.9, 169.8 ppm. IR (film): ν = 3481, 2500, 1757, 1487, 1218, 1046 cm
−1. [α]
20D = +45.2 (c 0.2, CHCl
3). The physico-chemical data are in agreement with those reported in the literature [
22].
Synthesis of α-allyl 3,5,6-tri-O-benzyl-(2,3,4-tri-O-acetyl-2-O-β-D-xylopyranosyl)-D-glucofuranoside (19a). A mixture of 12a (0.35 g, 0.71 mmol) and 14a (0.358 g, 0.86 mmol) in dry CH2Cl2 (8.75 mL) under N2 atmosphere and in the presence of activated 4Å molecular sieves (0.8 g) was stirred for 30 min at room temperature, cooled at −20 °C and TMSOTf (6 mg, 5 μL, 0.03 mmol) was then added. The mixture was stirred again at room temperature for 6 h, quenched with Et3N, filtered on Celite®, and concentrated. Purification of the residue by column chromatography (petroleum ether/Et2O 6:4) gave 19a as a colorless oil (Rf 0.4, petroleum ether/Et2O 1:1). Yield: 45% (0.24 g, 0.32 mmol). MS (ESI): 766 (M + NH4+). 1H NMR (CDCl3) δ: 1.99 (s, 3H, CH3CO), 2.03 (s, 3H, CH3CO), 2.08 (s, 3H, CH3CO), 3.37 (dd, 1H, J5’b–4’ = 7.0 Hz, J5’b–5’a = 12.2 Hz, H5’b), 3.71 (dd, 1H, J6b–5 = 6.1 Hz, J6b–6a = 10.5 Hz, H6b) 3.84 (dd, 1H, J6a–5 = 2.4 Hz, J6a–6b = 10.5 Hz, H6a), 3.97–4.05 (m, 3H, H2, H5, H7b), 4.18–4.23 (m, 1H, H7a), 4.22 (dd, 1H, J5’a–4’ = 4.5 Hz, J5’a–5’b = 12.2 Hz, H5’a), 4.28 (dd, 1H, J3–2 = 5.0 Hz, J3–4 = 6.5 Hz, H3), 4.33 (dd, 1H, J4–3 ≅ J4–5 = 6.5 Hz, H4), 4.51–4.58 (m, 5H, OCH2C6H5), 4.63 (d, 1H, J1’–2’ = 5.9 Hz, H1’) 4.80 (d, 1H, J = 11.6 Hz, OCH2C6H5), 4.93 (ddd, 1H, J4’–5’a = 4.5 Hz, J4’–3’ ≅ J4’–5’b = 7.0 Hz, H4’), 4.99 (dd, 1H, J2’–1’ = 5.9 Hz, J2’–3’ = 8.0 Hz, H2’), 5.02 (d, 1H, J1–2 = 4.3 Hz, H1), 5.12 (dd, 1H, J3’–4’ = 7.0 Hz, J3’–2’ = 8.0 Hz, H3’), 5.16 (dddd, 1H, J9b–9a = J9b–7a = J9b–7b = 1.6 Hz, J9b–8 = 10.5 Hz, H9b OCH2CHCHH), 5.30 (dddd, 1H, J9a–9b = J9a–7a = J9a–7b = 1.6 Hz, J9a–8 = 17.2 Hz, H9a OCH2CHCHH), 5.89 (dddd, 1H, J8–7a = 5.1 Hz, J8–7b = 6.1 Hz, J8–9b = 10.5 Hz, J8–9a = 17.2 Hz, H8 OCH2CHCH2), 7.26–7.33 (m, 15 H, ArH) ppm. 13C NMR (CDCl3) δ: 20.6, 20.7, 20.8, 61.7, 68.6, 68.8, 70.1, 70.6, 72.5, 72.6, 73.3, 76.0, 76.7, 76.8, 81.0, 84.9, 99.5, 100.7, 117.0, 122.4, 127.5, 127.6, 128.2, 128.4, 134.2, 137.8, 138.6, 138.8, 169.1, 169.9, 170.0 ppm. IR (CHCl3): ν = 3064, 1756, 1454, 1372, 1221, 1057 cm−1. [α]20D = +17.6 (c = 0.2, MeOH).
Synthesis of β-allyl 3,5,6-tri-O-benzyl-(2,3,4-tri-O-acetyl-2-O-β-D-xylopyranosyl)-D-glucofuranoside (19b). A mixture of 12b (0.35 g, 0.71 mmol) and 14a (0.358 g, 0.86 mmol) in dry CH2Cl2 (8.75 mL) under N2 atmosphere and in the presence of activated 4Å molecular sieves (0.8 g) was stirred for 30 min at room temperature, cooled at −20 °C and TMSOTf (6 mg, 5 μL, 0.03 mmol) was then added. The mixture was stirred again at room temperature for 3.5 h, quenched with Et3N, filtered on Celite®, and concentrated. Purification of the residue by column chromatography (petroleum ether/Et2O 6:4) gave 19b as a colorless oil (Rf 0.4, petroleum ether/Et2O 1:1). Yield: 60% (0.321 g, 0.43 mmol). MS (ESI): 766 (M + NH4+). 1H NMR (CDCl3) δ: 1.97 (s, 3H, CH3CO), 2.02 (s, 3H, CH3CO), 2.06 (s, 3H, CH3CO), 3.23 (dd, 1H, J5’b–4’ = 9.2 Hz, J5’b–5’a = 12.0 Hz, H5’b), 3.68 (dd, 1H, J6b–5 = 5.2 Hz, J6b–6a = 10.5 Hz, H6b), 3.88 (dd, 1H, J6a–5 = 2.0 Hz, J6a–6b = 10.5 Hz, H6a), 3.94 (dd, 1H, J3–2 = 1.2 Hz, J3–4 = 5.0 Hz, H3), 3.97 (dddd, 1H, J7b–9a = J7b–9b = 1.5 Hz, J7b–8 = 6.0 Hz, J7b–7a = 13.0 Hz, H7b, OCHHCHCH2), 4.06 (dd, 1H, J5’a–4’ = 5.2 Hz, J5’a–5’b = 12.0 Hz, H5’a), 4.06 (ddd, 1H, J5–6a = 2.0 Hz, J5–6b = 5.2, J5–4 = 9.5 Hz, H5), 4.14 (dd, 1H, J2–1 = Hz, J2–3 = 1.2 Hz, H2), 4.19 (dd, 1H, J4–3 = 5.0 Hz, J4–5 = 9.5 Hz, H4), 4.19 (dddd, 1H, J7a–9a = J7a–9b = 1.5 Hz, J7a–8 = 5.0, J7a–7b = 13.0 Hz, H7a OCHHCHCH2), 4.24 (d, 1H, J1’–2’ = 7.2 Hz, H1’), 4.52 (d, 1H, J = 11.2 Hz, OCH2C6H5), 4.53 (d, 1H, J = 12.0 Hz, OCH2C6H5), 4.59 (s, 2H, OCH2C6H5), 4.60 (d, 1H, J = 12.0 Hz, OCH2C6H5), 4.79 (d, 1H, J = 11.2 Hz, OCH2C6H5), 4.83 (dd, 1H, J2’–1’ = 7.2 Hz, J2’–3’ = 8.5 Hz, H2’), 4.91 (ddd, 1H, J4’–5’a = 5.2 Hz, J4’–3’ = 8.5 Hz, J4’–5’b = 9.2 Hz, H4’), 5.01 (d, 1H, J1–2 = 1.2 Hz, H1), 5.08 (dd, 1H, J3’–2’ = J3’–4’ = 8.5 Hz, H3’), 5.18 (dddd, 1H, J9b–9a = J9b–9a = J9b–7b = 1.5 Hz, J9b–8 = 10.5 Hz, H9b OCH2CHCHH), 5.28 (dddd, 1H, J9a–9b = J9a–7a = J9a–7b = 1.5 Hz, J9a–8 = 17.0 Hz, H9a OCH2CHCHH), 5.89 (dddd, 1H, J8–7a = 5.0 Hz, J8–7b = 6.0 Hz, J8–9b = 10.5 Hz, J8–9a = 17.0 Hz, H8 OCH2CHCH2), 7.25–7.35 (m, 15 H, ArH) ppm. 13C NMR (MeOD) δ: 19.1, 19.1, 19.3, 61.9, 68.5, 68.8, 69.8, 70.9, 71.8, 71.8, 71.8, 73.0, 76.4, 79.9, 80.4, 84.6, 100.1, 106.6, 115.7, 117.1, 127.2, 127.4, 127.4, 127.5, 127.8, 127.9, 127.9, 128.1, 134.2, 137.9, 138.4, 138.7, 169.6, 170.0, 170.2 ppm. IR (CHCl3): ν = 3064, 1756, 1454, 1372, 1221, 1057 cm−1. [α]20D = −12.8 (c 0.2, MeOH).
Synthesis of β-allyl 3,5,6-tri-O-benzyl-2-O-β-D-xylopyranosyl-D-glucofuranoside (22b). To a solution of 19b (0.21 g, 0.29 mmol) in dry MeOH (62 mL) MeONa was added (0.038 g, 0.70 mmol) and the mixture was stirred at room temperature for 3 h, then concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 1:9) gave 22b as a pale-yellow oil (Rf: 0.3, EtOAc). Yield: 90% (0.161 g, 0.26 mmol). MS (ESI): 605 (M + H+). 1H NMR (CDCl3) δ: 2.51 (br s, 1H, OH), 2.77 (br s, 1H, OH), 2.94 (br s, 1H, OH), 3.19 (dd, 1H, J5’b-4’ = 9.2, J5’b-5’a = 12.0 Hz, H5’b), 3.29 (dd, 1H, J2’-1’ = 6.5 Hz, J2’-3’ = 8.0 Hz, H2’), 3.43 (dd, 1H, J3’-2’ = J3’-4’ = 8.0 Hz, H3’), 3.67 (ddd, J4’-5’a = 4.8 Hz, J4’-3’ = 8.0 Hz, J4’-5’b = 9.2 Hz, H4’), 3.70 (dd, 1H, J6b-5 = 4.5 Hz, J6b–6a = 10.8 Hz, H6b), 3.88 (dd, 1H, J6a–5 = 2.0 Hz, J6a–6b = 10.8 Hz, H6a), 3.95 (dd, 1H, J5’a–4’ = 4.8 Hz, J5’a–5’b = 12.0 Hz, H5’a), 3.98 (dddd, 1H, J7b–9a = J7b–9b = 1.5 Hz, J7b–8 = 6.0 Hz, J7b–7a = 13.0 Hz, H7b OCHHCHCH2), 4.04 (dd, 1H, J3–2 = 1.0 Hz, J3–4 = 4.8 Hz, H3), 4.06 (ddd, 1H, J5–6a = 2.0 Hz, J5–6b = 4.5 Hz, J5–4 = 9.2 Hz, H5), 4.08 (d, 1H, J1’–2’ = 6.5 Hz, H1’), 4.19 (dddd, 1H, J7a–9a = J7a–9b = 1.5 Hz, J7a–8 = 5.0 Hz, J7a–7b = 13.0 Hz, H7a OCHHCHCH2), 4.23 (dd, 1H, J2–1 = J2–3 = 1.0 Hz, H2), 4.35 (dd, 1H, J4–3 = 4.8 Hz, J4–5 = 9.2 Hz, H4), 4.51 (d, 1H, J = 11.2 Hz, OCH2C6H5), 4.56 (d, 1H, J = 12.5 Hz, OCH2C6H5), 4.58 (d, 1H, J = 12.5 Hz, OCH2C6H5), 4.61 (d, 1H, J = 12.5 Hz, OCH2C6H5), 4.62 (d, 1H, J = 12.5 Hz, OCH2C6H5), 4.75 (d, 1H, J = 11.2 Hz, OCH2C6H5), 5.06 (d, 1H, J1-2 = 1.0 Hz, H1), 5.17 (dddd, 1H, J9b–9a = J9b–7a = J9b–7b = 1.6 Hz, J9b–8 = 10.5 Hz, H9b OCH2CHCHH), 5.28 (dddd, 1H, J9a–9b = J9a–7a = J9a–7b = 1.6 Hz, J9a–8 = 17.0 Hz, H9a OCH2CHCHH), 5.90 (dddd, 1H, J8–7a = 5.0 Hz, J8–7b = 6.0 Hz, J8–9b = 10.5 Hz, J8–9a = 17.0 Hz, H8 OCH2CHCH2), 7.28–7.37 (m, 15H, ArH) ppm. 13C NMR (CDCl3) δ: 64.7, 68.9, 69.4, 70.3, 72.4, 72.4, 72.4, 72.6, 73.4, 75.1, 79.9, 80.5, 84.6, 101.9, 106.6, 117.0, 127.5, 127.6, 127.7, 127.7, 127.9, 128.0, 128.3, 128.3, 128.4, 134.1, 137.7, 138.7, 138.7 ppm. IR (CHCl3): ν = 3415, 3019, 1785, 1450, 1368, 1045 cm−1. [α]20D = –19.6 (c = 0.2, MeOH).
Synthesis of 3,5,6-tri-O-benzyl-2-O-β-D-xylopyranosyl-α-D-glucofuranoside and 3,5,6-tri-O-benzyl-2-O-β-D-xylopyranosyl-β-D-glucofuranoside (23). To a solution of 22b (0.07 g, 0.12 mmol) in MeCOONa (0.034 g, 0.42 mmol) and MeCOOH (0.22 mL), PdCl2 (0.04 g, 0.22 mmol), and H2O (11 μL) were added. The mixture was stirred at room temperature for 12 h, filtered on Celite®, and washed with EtOAc. The filtrate was washed with a saturated NaHCO3 solution and extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. Purification of the residue by column chromatography (EtOAc/cyclohexane 8:2) gave 23 (α/β 2:3) as a colorless oil (Rf: 23a 0.2, 23b 0.3; EtOAc/MeOH 99:1). Yield: 42% (0.027 g, 0.05 mmol). MS (ESI): 600 (M + NH4+). 1H NMR (MeOD) δ: 3.11–3.21 (m, 3H), 3.23–3.30 (m, 3H), 3.44–3.54 (m, 3H), 3.66–3.73 (m, 2H), 3.82–3.91 (m, 3H), 3.93–3.98 (m, 2H), 4.08–4.11 (m, 1H), 4.11–4.14 (4H), 4.19 (dd, 1H, J1 = 2.6 Hz, J2 = 4.0 Hz), 4.25 (dd, 1H, J1 = 2.5 Hz, J2 = 4.6 Hz), 4.28 (d, 1H, J = 4.0 Hz), 4.30 (dd, 1H, J1 = J2 = 2.1 Hz), 4.38 (dd, 1H, J1 = 4.6 Hz, J2 = 7.9 Hz), 4.50–4.77 (m, 10H, OCH2C6H5), 5.25 (d, 1H, J = 1.0 Hz, H1 β-anomer), 5.39 (d, 1H, J = 4.0 Hz, H1 α-anomer), 7.26–7.38 (m, 30H, ArH) ppm. 13C NMR (MeOD) δ: 65.6, 65.6, 69.7, 69.7, 70.2, 70.2, 71.8, 71.9, 71.9, 71.9, 72.9, 73.0, 73.0, 73.3, 75.9, 76.4, 76.5, 76.9, 79.8, 80.7, 80.8, 81.6, 85.0, 85.0, 96.5, 102.3, 102.8, 103.0, 127.1, 127.1, 127.2, 127.3, 127.3, 127.4, 127.4, 127.4, 127.5, 127.7, 127.8, 127.8, 127.9, 127.9, 128.0, 128.0, 128.1, 128.1, 137.9, 138.0, 138.4, 138.4, 138.8, 138.8 ppm. IR (Nujol): ν = 3399, 1458, 1376, 1041 cm−1. [α]20D = −32.6 (c = 0.3, MeOH).
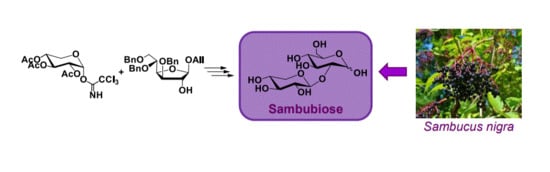
Synthesis of 2-O-β-D-xylopyranosyl-D-α-glucopyranose and 2-O-β-D-xylopyranosyl-D-β-glucopyranose (
8, sambubiose). To a solution of
23 (0.09 g, 0.15 mmol) in EtOH (9 mL), Pd/C 10% (0.018 g) was added and the mixture was hydrogenated under stirring at room temperature at 1 atm for 6 h, then filtered on Celite
®, washed with EtOH, and kept overnight. The crystals obtained were then filtered and triturated to obtain
8 (sambubiose) as a white solid (
Rf: 0.2, CH
3CN/H
2O 9:1). Yield 51% (0.024 g, 0.08 mmol). Mp: 132–134 °C (petroleum ether). MS (ESI): 313 (M + 1), 311 (M-1).
1H NMR (DMSO
-d
6)
δ: 3.02–3.05 (m, 2H, H
5, H
2’), 3.08–3.17 (m, 3H, H
2, H
3’, H
5’a), 3.23–3.28 (m, 1H, H
4’), 3.41–3.47 (m, 1H, H
6a), 3.54–3.68 (m, 4H, H
3, H
4, H
6b, H
5’b), 4.25 (d, 1H,
J1’–2’ =
7.2 Hz, H
1’), 4.39 (dd, 1H,
JOH6–6a ≅
JOH6–6b = 5.5 Hz, OH
6), 4.77 (d, 1H,
J = 3.2 Hz, OH), 4.93 (d, 1H,
J = 5.6 Hz, OH), 4.98 (d, 1H,
J = 5.2 Hz, OH
4’), 5.00 (d, 1 H,
J = 4.8 Hz, OH), 5.02–5.05 (m, 2H, H
1, OH) 6.34 (d, 1H,
J = 4.5 Hz, OH
1) ppm.
13C NMR (DMSO-d
6)
δ: 61.5 (C6), 66.1 (C5’), 69.9 (C4’), 70.5 (C5), 72.1 (C3), 72.4 (C4), 74.2 (C2’), 76.7 (C3’), 82.7 (C2), 92.0 (C1), 106.4 (C1’). α-anomer
13C NMR (D
2O)
δ: 60.4 (C6), 65.0 (C5’), 69.2 (C4’), 69.4 (C5), 71.0 (C3), 71.7 (C4), 73.1 (C2’), 75.5 (C3’), 80.7 (C2), 91.7 (C1), 104.7 (C1’). β-anomer
13C NMR (D
2O)
δ: 60.6 (C6), 65.1 (C5’), 69.1 (C4’), 69.4 (C5), 73.4 (C2’), 75.5, 75.69, 75.64, 81.8 (C2), 94.6 (C1), 103.6 (C1’). IR (Nujol): ν = 3400, 2840, 1070 cm
−1. [α]
20D = −5.0 (c = 0.03, MeOH). [
10]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






