New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding


Abstract
:
1. Introduction
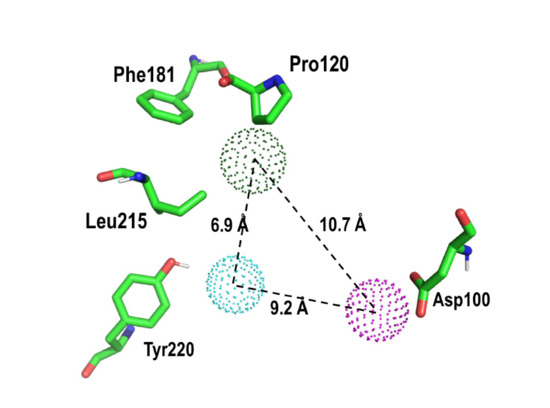
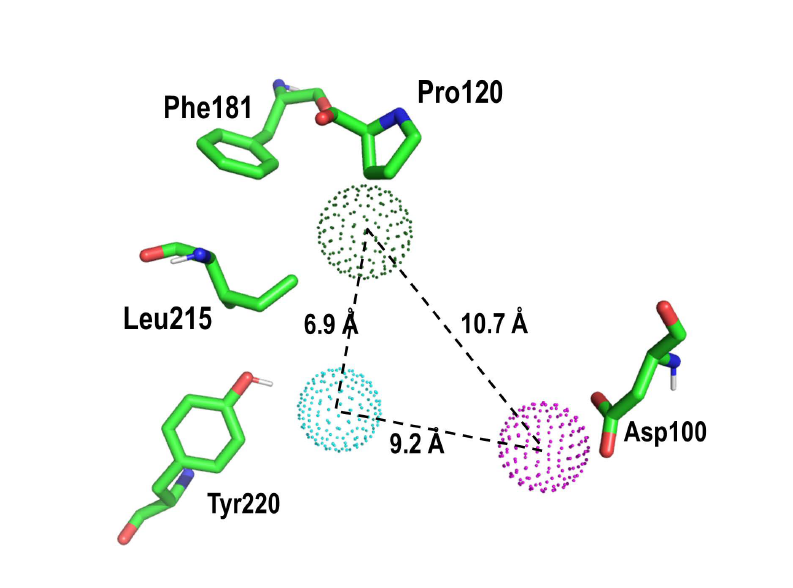
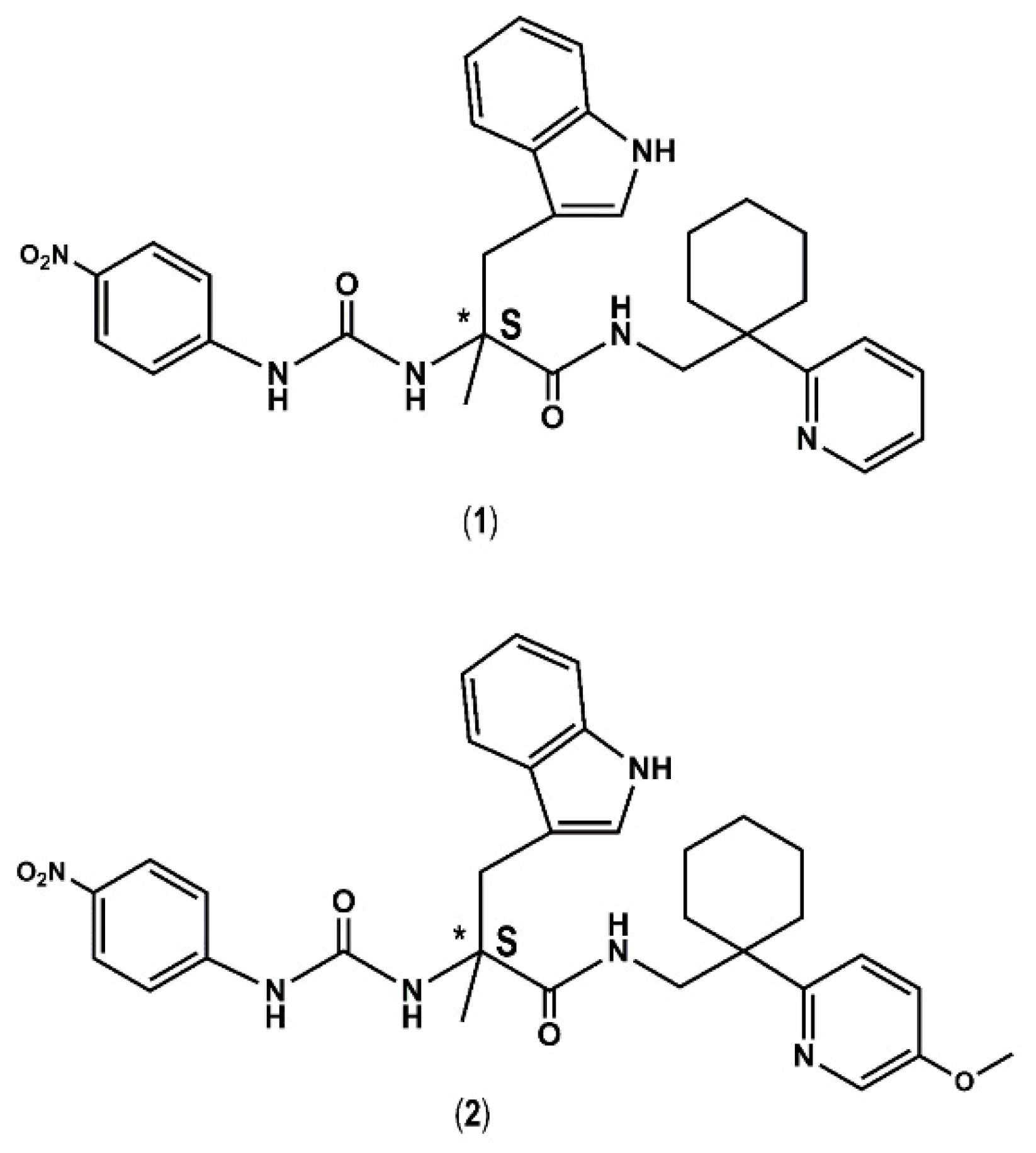
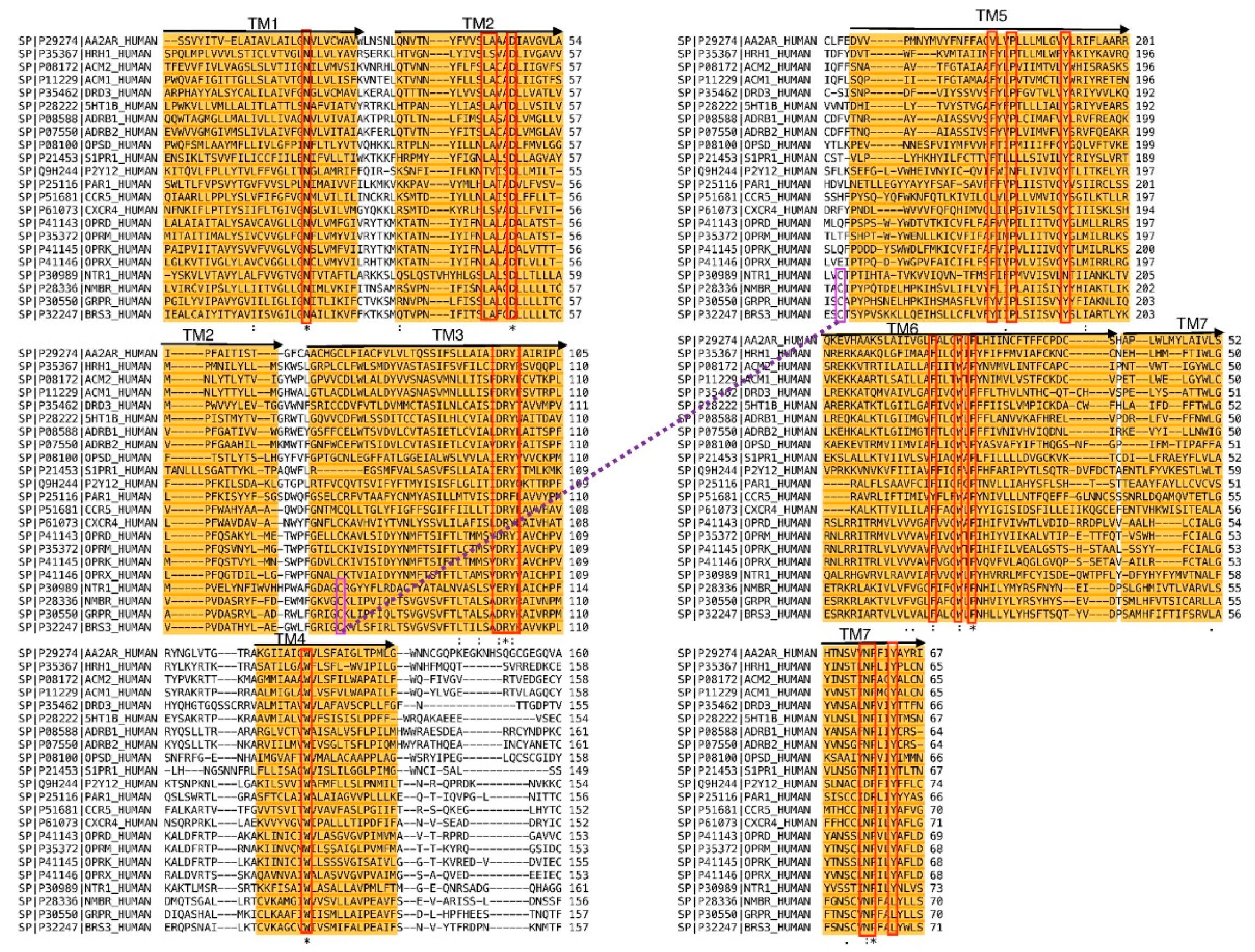
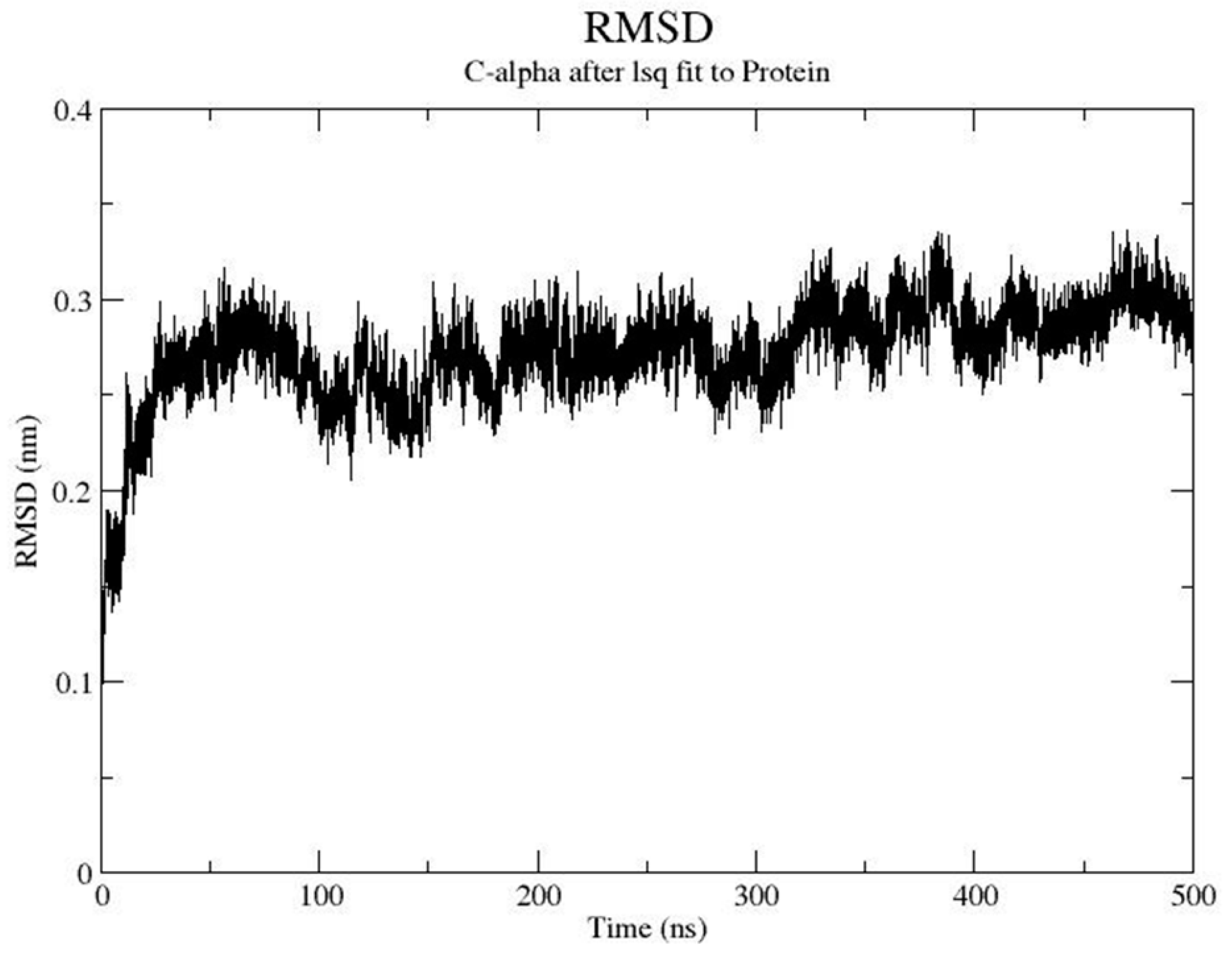
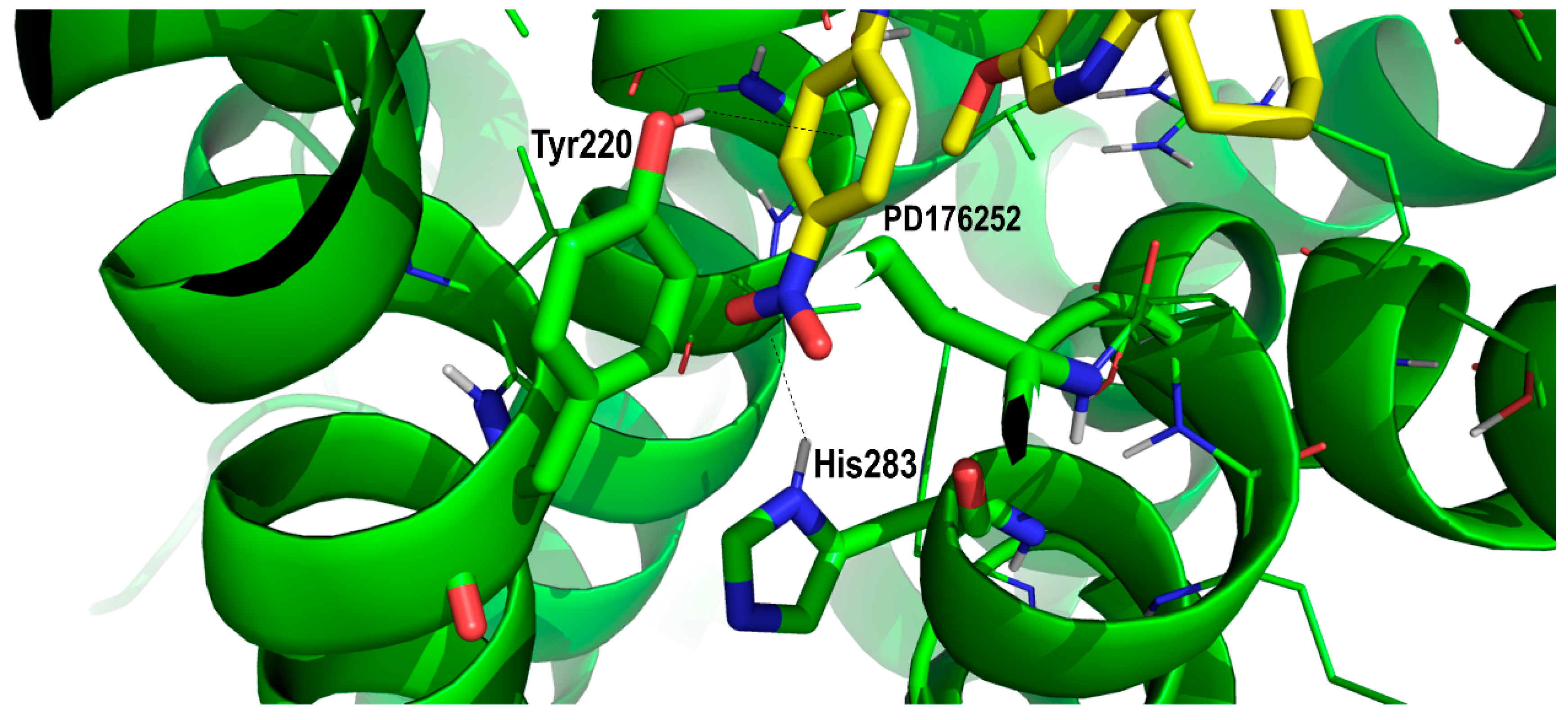
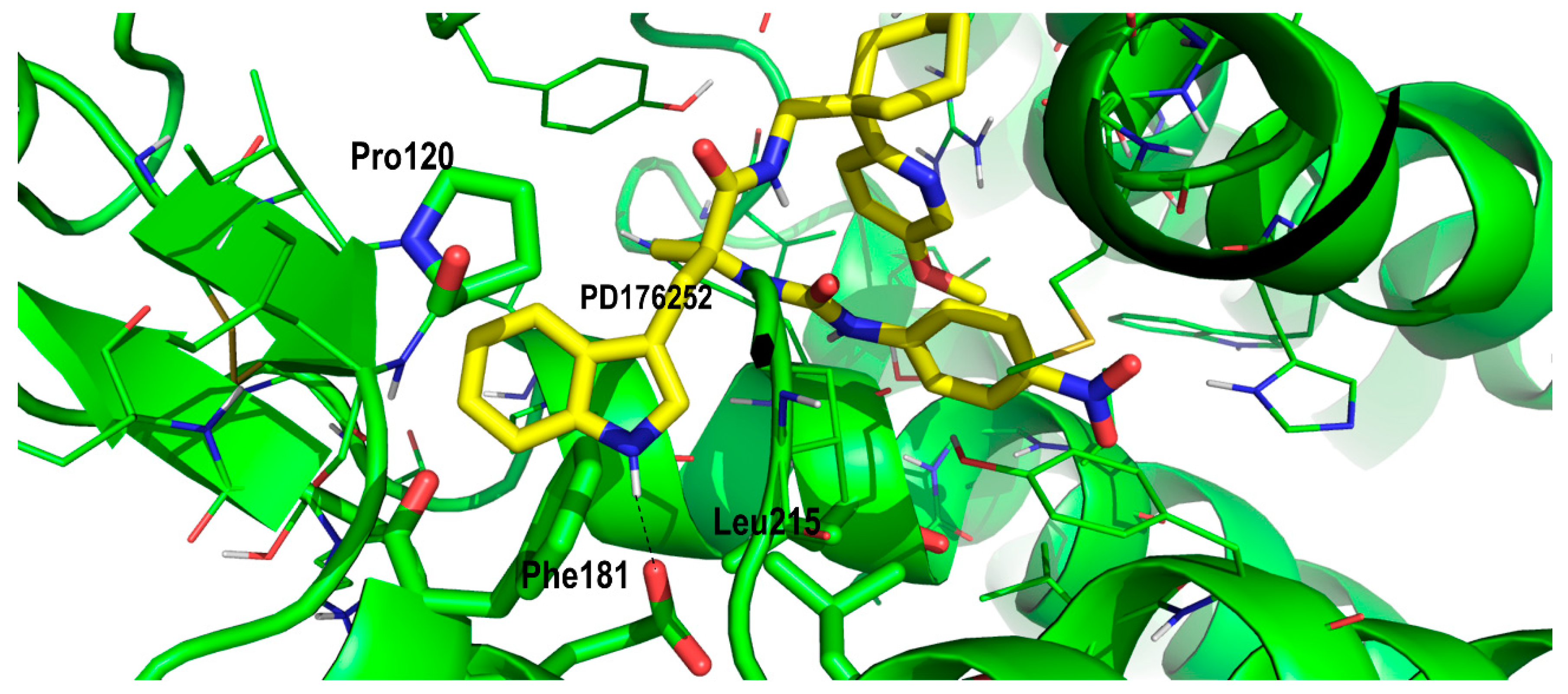
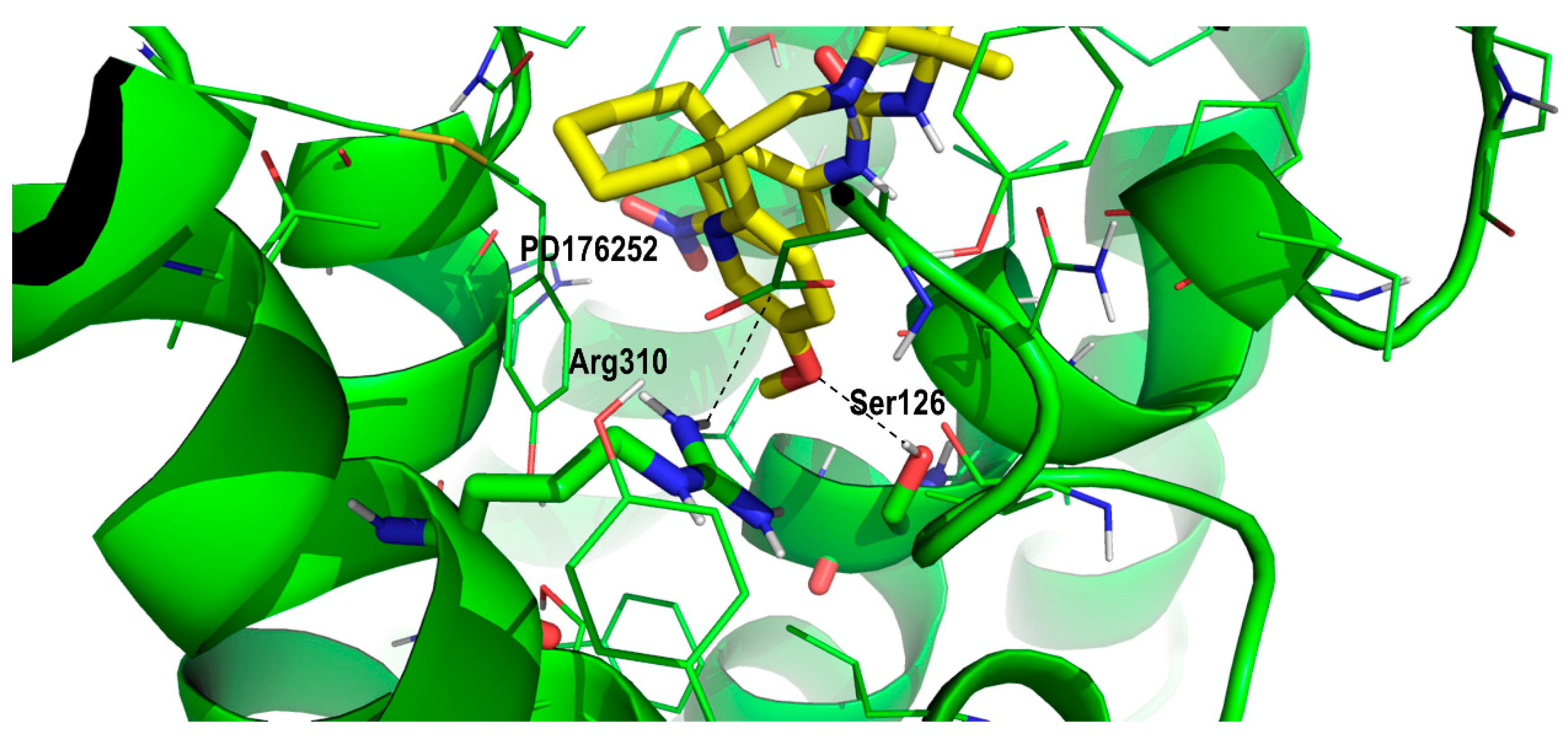
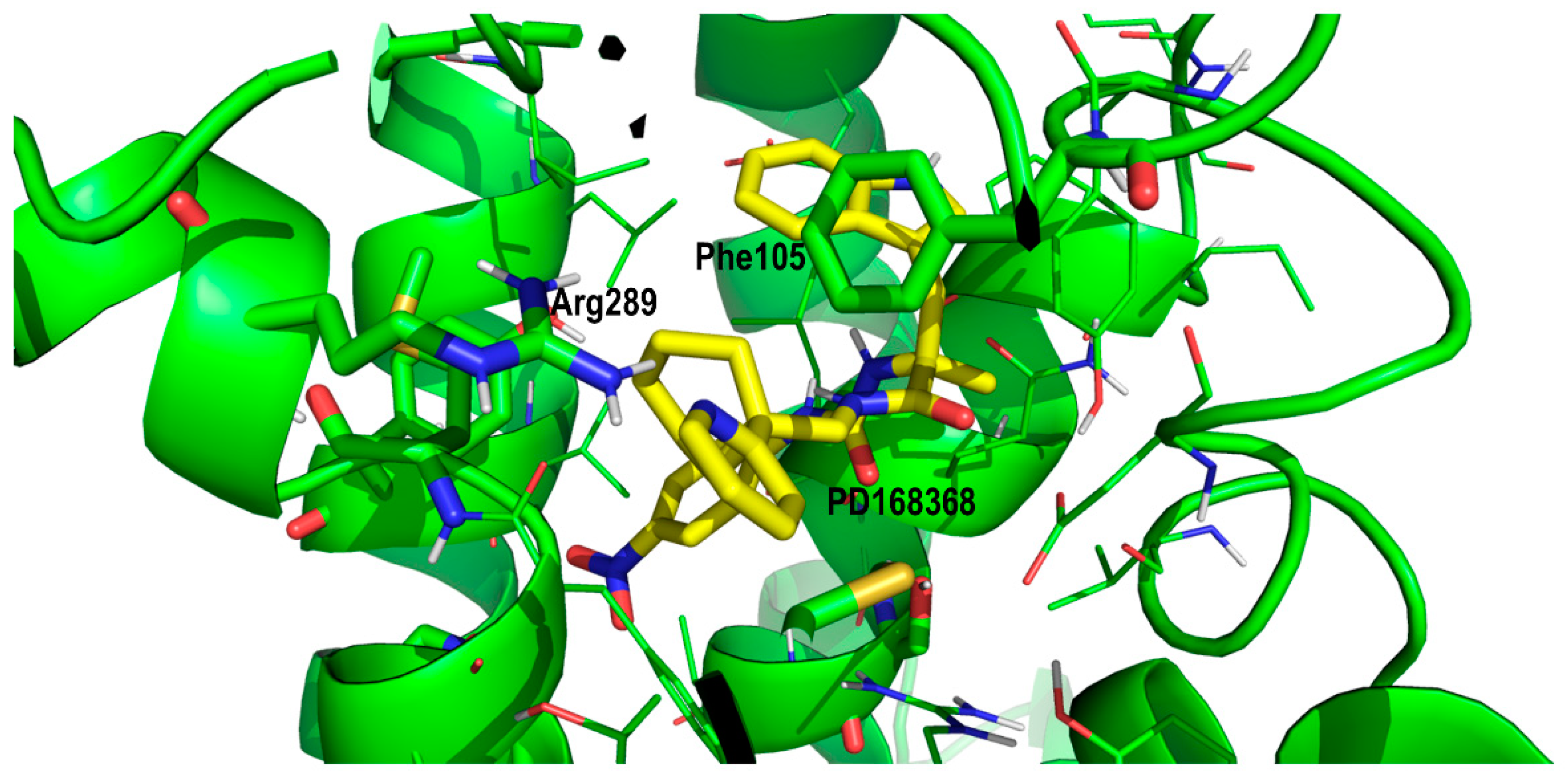
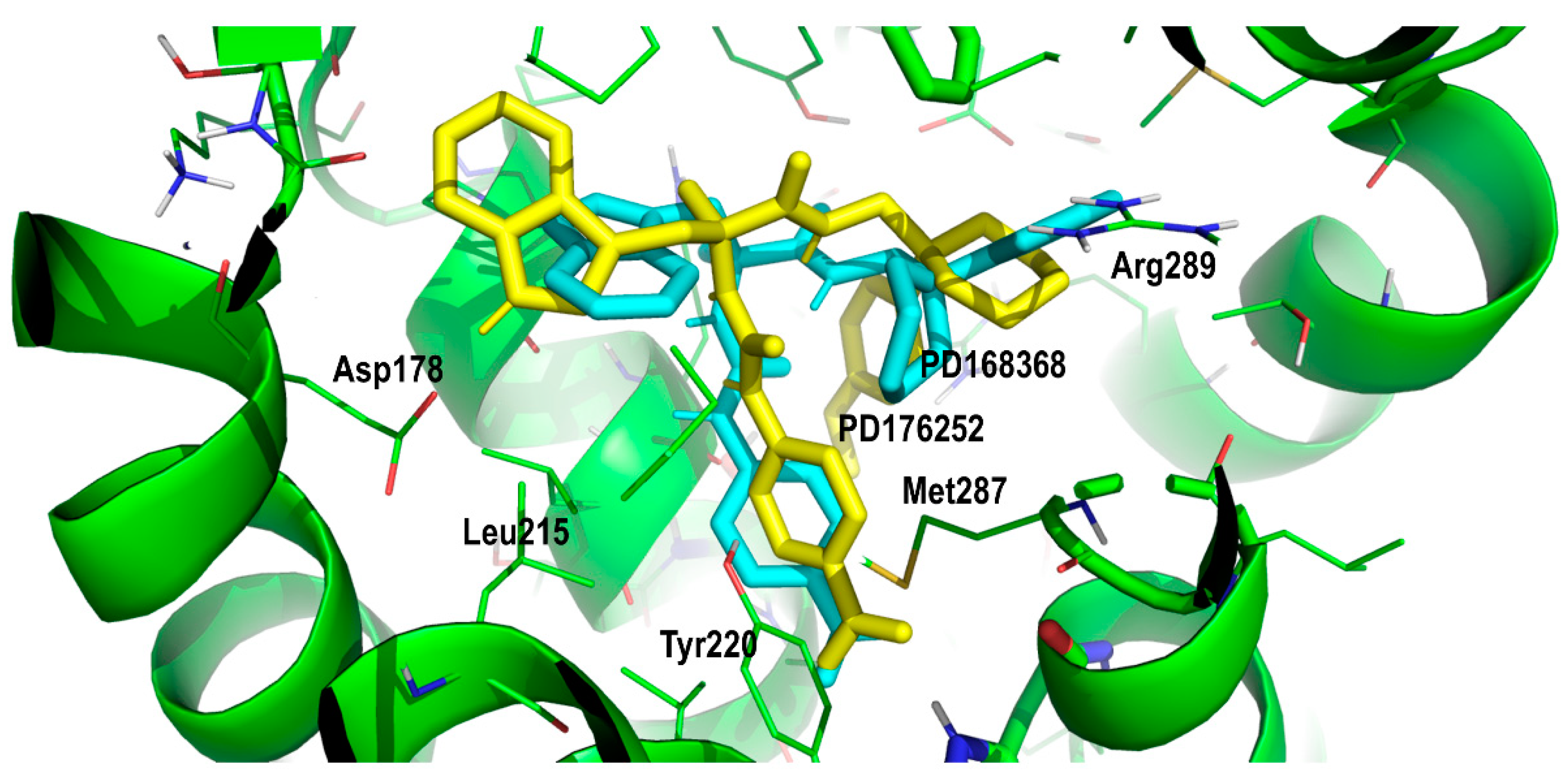
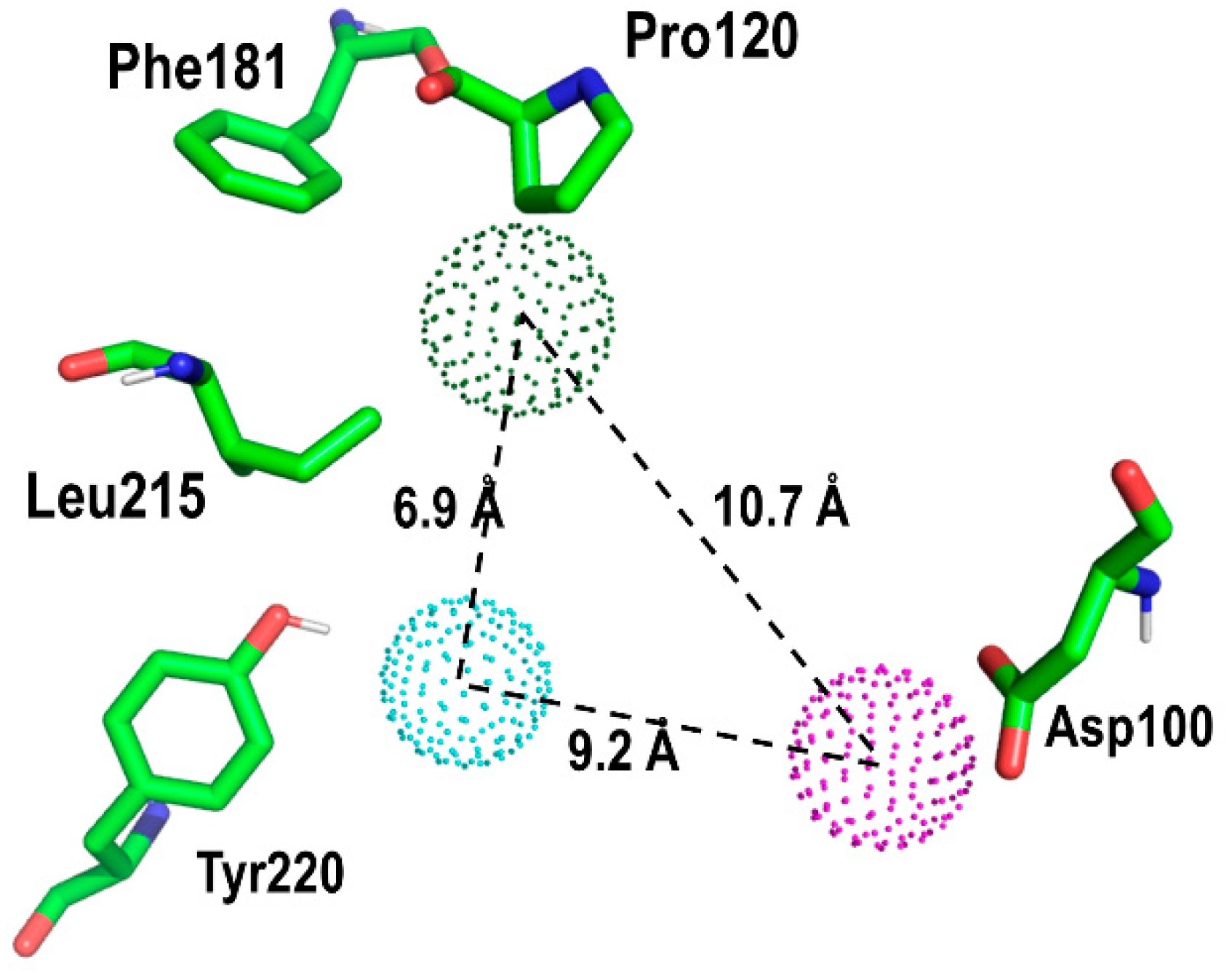
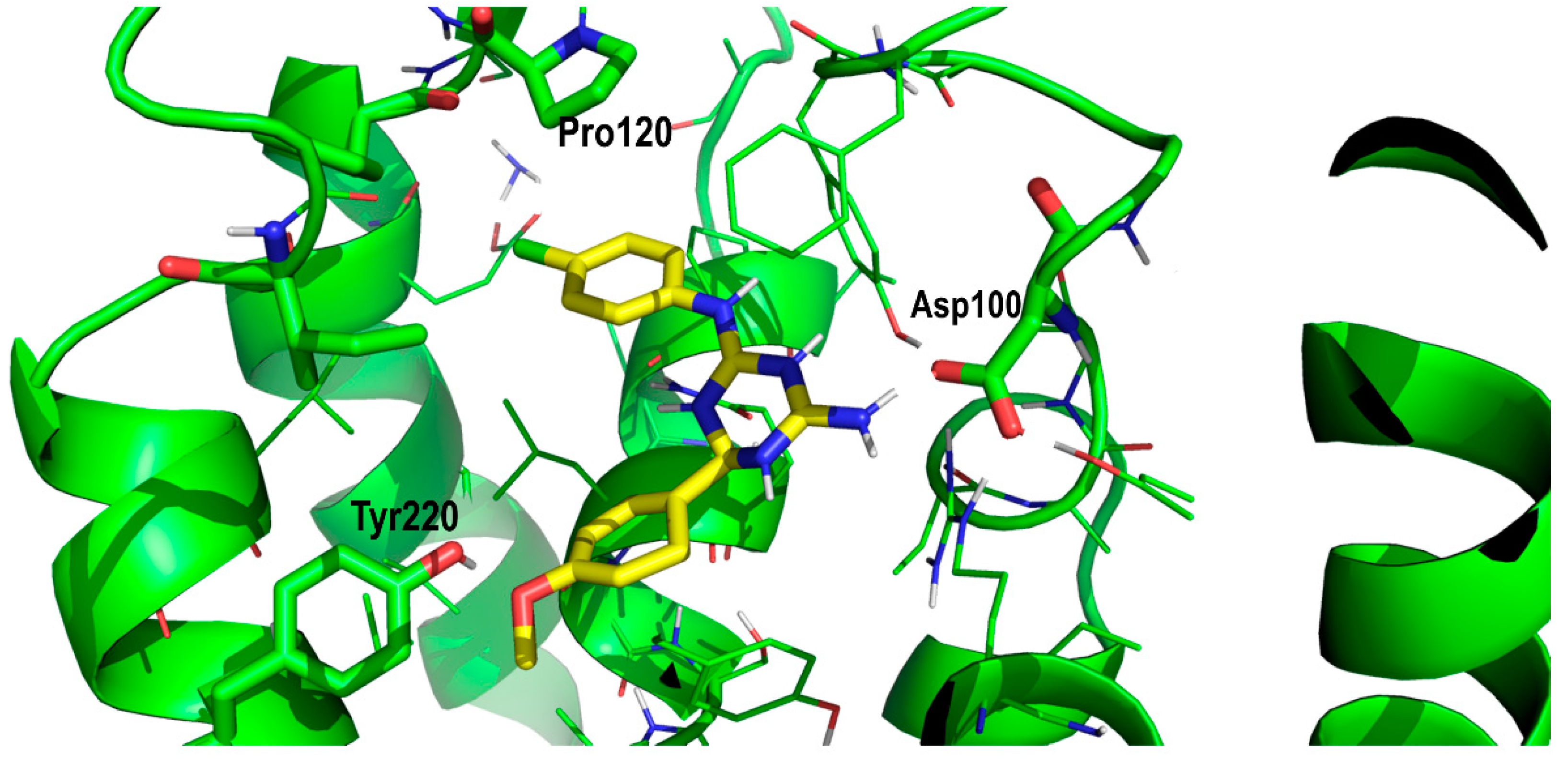
2. Results and Discussion
Proof of Concept
3. Materials and Methods
3.1. Molecular Modeling
3.2. Binding Assays
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Jensen, R.T.; Moody, T.W. Bombesin Peptides (Cancer). In Hand-Book of Biologically Active Peptides; Kastin, A.J., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 506–511. [Google Scholar]
- Ramos-Alvarez, I.; Moreno, P.; Mantey, S.A.; Nakamura, T.; Nuche-Berenguer, B.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Insights into bombesin receptors and ligands: Highlighting recent advances. Peptides 2015, 72, 128–144. [Google Scholar] [CrossRef] [Green Version]
- Weber, H.C. Regulation and signaling of human bombesin receptors and their biological effects. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Flores, D.G.; De Farias, C.B.; Leites, J.; De Oliveira, M.S.; Lima, R.C.; Tamajusuku, A.S.; Leone, L.P.D.; Meurer, L.; Brunetto, A.L.; Schwartsmann, G.; et al. Gastrin releasing peptide receptors regulate proliferation of C6 glioma cells through a phosphatidylinositol 3-kinase-dependent mechanism. Curr. Neurovasc. Res. 2008, 5, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Moreno, P.; Jensen, R.T. Neuropeptides as lung cancer growth factors. Peptides 2015, 72, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Benya, R.V.; Kusui, T.; Pradhan, T.K.; Battey, J.F.; Jensen, R.T. Expression and characterization of cloned human bombesin receptors. Mol. Pharmacol. 1995, 47, 10–20. [Google Scholar] [PubMed]
- Fathi, Z.; Corjay, M.H.; Shapira, H.; Wada, E.; Benya, R.; Jensen, R.; Viallet, J.; Sausville, E.A.; Battey, J.F. BRS-3: A novel bombesin receptor subtype selectively expressed in testis and lungcarcinoma cells. J. Biol. Chem. 1993, 268, 5979–8594. [Google Scholar] [PubMed]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Beny, R.V. International Union of Pharmacology. LXVIII. Mammalian Bombesin Receptors: Nomenclature, Distribution, Pharmacology, Signaling, and Functions in Normal and Disease States. Pharm. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef] [Green Version]
- Moreno, P.; Ramos-Alvarez, I.; Moody, T.W.; Jensen, R.T. Bombesin related peptides/receptors and their promising therapeutic roles in cancer imaging; targeting and treatment. Expert Opin. Ther. Targets 2016, 20, 1055–1073. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Kim, S.R.; Kim, M.K.; Choi, K.S.; Jang, H.O.; Yun, I.; Bae, M.-K. Neuromedin B receptor antagonist suppresses tumor angiogenesis and tumor growth in vitro and in vivo. Cancer Lett. 2011, 312, 117–127. [Google Scholar] [CrossRef]
- Gonzalez, N.; Moreno, P.; Jensen, R.T. Bombesin receptor subtype 3 as a potential target for obesity and diabetes. Expert Opin. Ther. Targets 2015, 19, 1153–1170. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Nissao, E.; Jackson, I.L.; Leong, W.; Dancy, L.; Cuttitta, F.; Vujaskovic, Z.; Sunday, M.E. Radiation-Induced Lung Injury Is Mitigated by Blockade of Gastrin-Releasing Peptide. Am. J. Pathol. 2013, 182, 1248–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehling, S.; Fukuyama, T.; Ko, M.C.; Olivry, T.; Bäumer, W. Neuromedin B Induces Acute Itch in Mice via the Activation of Peripheral Sensory Neurons. Acta Derm. Venereol. 2019, 99, 587–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristau, M.; Devin, C.; Oiry, C.; Chaloin, O.; Amblard, M.; Bernad, N.; Heitz, A.; Fehrentz, J.A.; Martinez, J. Synthesis and Biological Evaluation of Bombesin Constrained Analogues. J. Med. Chem. 2000, 43, 2356–2361. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, N.; Mantey, S.A.; Pradhan, T.K.; Sancho, V.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Characterization of putative GRP- and NMB-receptor antagonist’s interaction with human receptors. Peptides 2009, 30, 1473–1486. [Google Scholar] [CrossRef] [Green Version]
- Perez, J.J.; Corcho, F.; Llorens, O. Molecular modeling in the design of peptidomimetics and peptide surrogates. Curr. Med. Chem. 2002, 9, 2209–2229. [Google Scholar] [CrossRef]
- Perez, J.J. Designing Peptidomimetics. Curr. Top. Med. Chem. 2018, 18, 566–590. [Google Scholar] [CrossRef]
- Ashwood, V.; Brownhill, V.; Higginbottom, M.; Horwell, D.C.; Hughes, J.; Lewthwaite, R.A.; McKnight, A.T.; Pinnock, R.D.; Pritchard, M.C.; Suman-Chauhan, N.; et al. PD 176252-The First High Affinity Non-peptide Gastrin-Releasing Peptide (BB2) Receptor Antagonist. Bioorg. Med. Chem. Lett. 1998, 8, 2589–2594. [Google Scholar] [CrossRef]
- Carrieri, A.; Lacivita, E.; Belviso, B.D.; Caliandro, R.; Mastrorilli, P.; Gallo, V.; Niso, M.; Leopoldo, M. Structural determinants in the binding of BB2 receptor ligands: In silico, x-ray and NMR studies in PD176252 analogues. Curr. Top. Med. Chem. 2017, 17, 1599–1610. [Google Scholar] [CrossRef]
- Moody, T.W.; Mantey, S.A.; Moreno, P.; Nakamura, T.; Lacivita, E.; Leopoldo, M.; Jensen, R.T. ML-18 is a non-peptide bombesin receptor subtype-3 antagonist which inhibits lung cancer growth. Peptides 2015, 64, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Moody, T.W.; Tashakkori, N.; Mantey, S.A.; Moreno, P.; Ramos-Alvarez, I.; Leopoldo, M.; Jensen, R.T. AM-37 and ST-36 are Small Molecule Bombesin Receptor Antagonists. Front. Endocrinol. 2017, 8, 176. [Google Scholar] [CrossRef] [Green Version]
- Tokita, K.; Hocart, S.J.; Katsuno, T.; Mantey, S.A.; Coy, D.H.; Jensen, R.T. Tyrosine 220 in the 5th Transmembrane Domain of the Neuromedin B Receptor Is Critical for the High Selectivity of the Peptoid Antagonist PD168368. J. Biol. Chem. 2001, 276, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Jutila, M.A.; Quinn, M.T. Gastrin-Releasing Peptide/Neuromedin B Receptor Antagonists PD176252, PD168368, and Related Analogs Are Potent Agonists of Human Formyl-Peptide Receptors. Mol. Pharmacol. 2011, 79, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Shuttleworth, S.J.; Connors, R.V.; Chai, A.; Coward, P. Discovery and optimization of a novel Neuromedin B receptor antagonist. Bioorg. Med. Chem. Lett. 2009, 19, 4264–4267. [Google Scholar] [CrossRef]
- Martinez, A.; Zudaire, E.; Julian, M.; Moody, T.W.; Cuttitta, F. Gastrin-releasing peptide (GRP) induces angiogenesis and the specific GRP blocker 77427 inhibits tumor growth in vitro and in vivo. Oncogene 2005, 24, 4106–4113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, P.; Mantey, S.A.; Nuche-Berenguer, B.; Reitman, M.L.; Gonzalez, N.; Coy, D.H.; Jensen, R.T. Comparative pharmacology of bombesin receptor subtype-3 nonpeptide agonist MK-5046, a universal peptide agonist; and peptide antagonist Bantag-1 for human bombesin receptors. J. Pharmacol. Exp. Ther. 2013, 347, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Lupala, C.S.; Rasaifar, B.; Gomez-Gutierrez, P.; Perez, J.J. Using Molecular Dynamics for the refinement of atomistic models of GPCRs by homology modeling. J. Biomol. Struct. Dyn. 2018, 36, 2436–2448. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A New Approach for Rapid Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Venkatakrishnana, A.J.; Maa, A.K.; Fonseca, R.; Latorraca, N.R.; Kelly, B.; Betz, R.M.; Asawa, C.; Kobilka, B.K.; Dror, R.O. Diverse GPCRs exhibit conserved water networks for stabilization and activation. Proc. Natl. Acad. Sci. USA 2019, 116, 3288–3293. [Google Scholar] [CrossRef] [Green Version]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar]
- Molecular Operating Environment (MOE), Version 2019.01; Chemical Computing Group UCL: Montreal, QC, Canada, 2020.
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.J. Managing molecular diversity. Chem. Soc. Rev. 2005, 34, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Jenkins, J.L.; Scheiber, J.; Chetan, S.; Sukuru, K.; Glick, M.; Davies, J.W. How Similar Are Similarity Searching Methods? A Principal Component Analysis of Molecular Descriptor Space. J. Chem. Inf. Model. 2009, 49, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Lipkus, A.H. A proof of the triangular inequality for the Tanimoto distance. J. Math. Chem. 1999, 26, 263–265. [Google Scholar] [CrossRef]
- Jarvis, R.A.; Patrick, E.A. Clustering Using a Similarity Measure Based on Shared Near Neighbors. IEEE Trans. Comp. 1973, 22, 1025–1034. [Google Scholar] [CrossRef]
- Ohki-Hamazaki, H.; Iwabuchi, M.; Maekawa, F. Development and function of bombesin-like peptides and their receptors. Int. J. Dev. Biol. 2005, 49, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.; Singh, D.K.; Meena, S.; Datta, D.; Siddiqi, M.I.; Banerjee, D. Pharmacophore-Based Screening and Identification of Novel Human Ligase I Inhibitors with Potential Anticancer Activity. J. Chem. Inf. Model. 2014, 54, 781–792. [Google Scholar] [CrossRef]
- Gomez-Gutierrez, P.; Campos, P.M.; Perez, J.J. Identification of a Novel Inhibitory Allosteric Site of MAP Kinases. PLoS ONE 2016, 11, e0167379. [Google Scholar] [CrossRef]
- Noinaj, N.; White, J.F.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; et al. The crystal structure of the neurotensin receptor NTS1 in complex with neurotensin (8–13). Nature 2012, 490, 508–513. [Google Scholar]
- Costanzi, S.; Skorski, M.; Deplano, A.; Habermehl, B.; Mendoza, M.; Wang, K.; Biederman, M.; Dawson, J.; Gao, J. Homology modeling of a Class A GPCR in the inactive conformation: A quantitative analysis of the correlation between model/template sequence identity and model accuracy. J. Mol. Graph. Model. 2016, 70, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Nayeem, A.; Sitkoff, D.; Krystek, S., Jr. A comparative study of available software for high-accuracy homology modeling: From sequence alignments to structural models. Protein Sci. 2006, 15, 808–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavasotto, C.N.; Palomba, D. Expanding the horizons of G protein-coupled receptor structure-based ligand discovery and optimization using homology models. Chem. Commun. 2015, 51, 13576–13594. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordomi, A.; Edholm, O.; Perez, J.J. Effect of different treatments of long-range interactions and sampling conditions in molecular dynamic simulations of rhodopsin embedded in a dipalmitoyl phosphatidylcholine bilayer. J. Comput. Chem. 2007, 28, 1017–1030. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast flexible and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Kaminski, G.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound# | Chemical Structure | BB1R(Neuromendin B Receptor) Radioligand Displacement (%) | BB2R(Gastrin-Releasing Peptide Receptor) Radioligand Displacement (%) |
|---|---|---|---|
| 1 |  | 19.7 | 0.0 |
| 2 |  | 24.3 | 0.0 |
| 3 |  | 28.1 | 0.0 |
| 4 |  | 30.5 | 0.0 |
| 5 |  | 38.0 | 10.5 |
| 6 |  | 16.1 | 0.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasaeifar, B.; Gomez-Gutierrez, P.; Perez, J.J. New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding. Pharmaceuticals 2020, 13, 197. https://doi.org/10.3390/ph13080197
Rasaeifar B, Gomez-Gutierrez P, Perez JJ. New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding. Pharmaceuticals. 2020; 13(8):197. https://doi.org/10.3390/ph13080197
Chicago/Turabian StyleRasaeifar, Bahareh, Patricia Gomez-Gutierrez, and Juan J. Perez. 2020. "New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding" Pharmaceuticals 13, no. 8: 197. https://doi.org/10.3390/ph13080197
APA StyleRasaeifar, B., Gomez-Gutierrez, P., & Perez, J. J. (2020). New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding. Pharmaceuticals, 13(8), 197. https://doi.org/10.3390/ph13080197