Tumor Immunotherapy Using A2A Adenosine Receptor Antagonists




Abstract
:1. Introduction
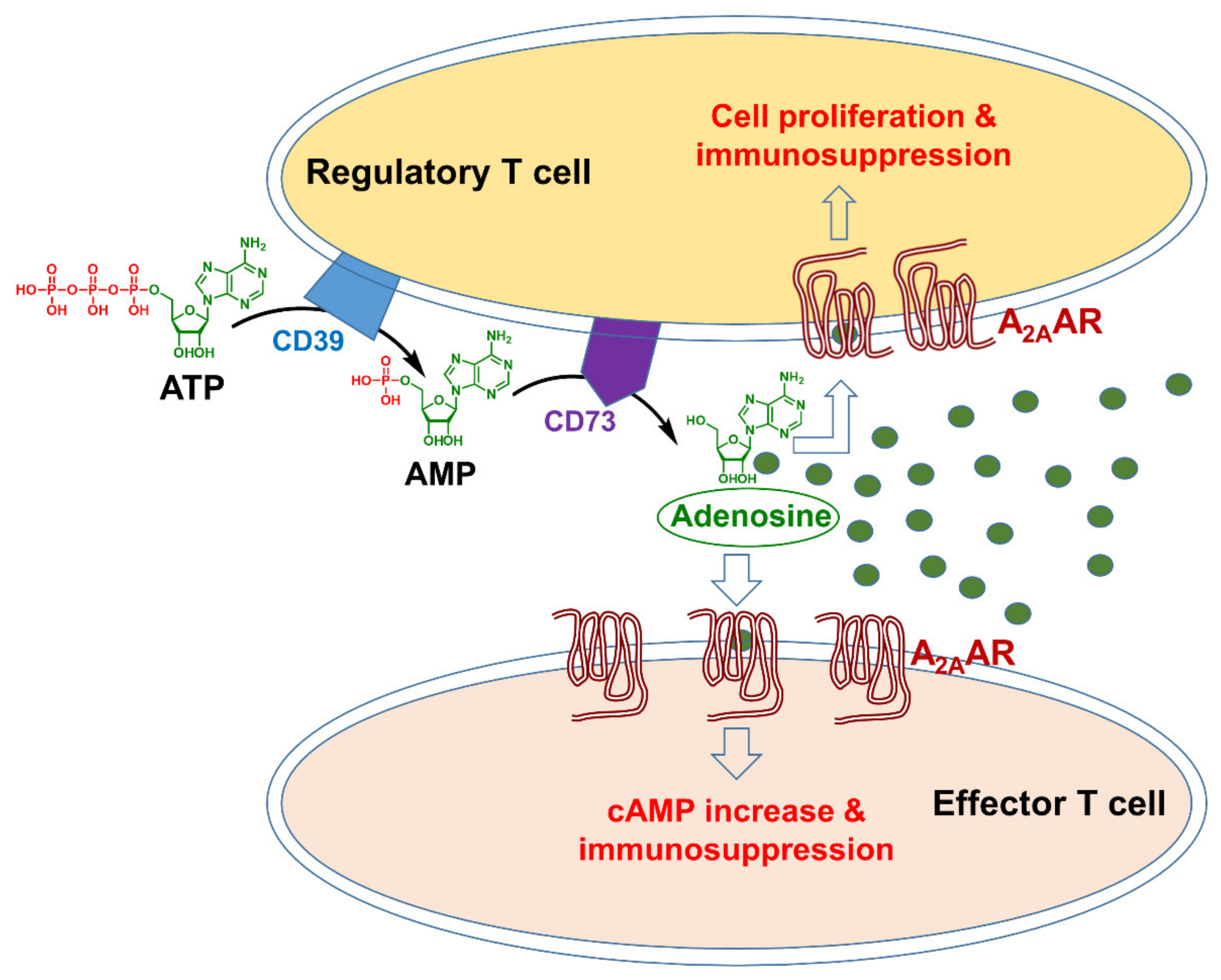
2. Mechanism of Action of A2AAR Antagonists in Immuno-Oncology
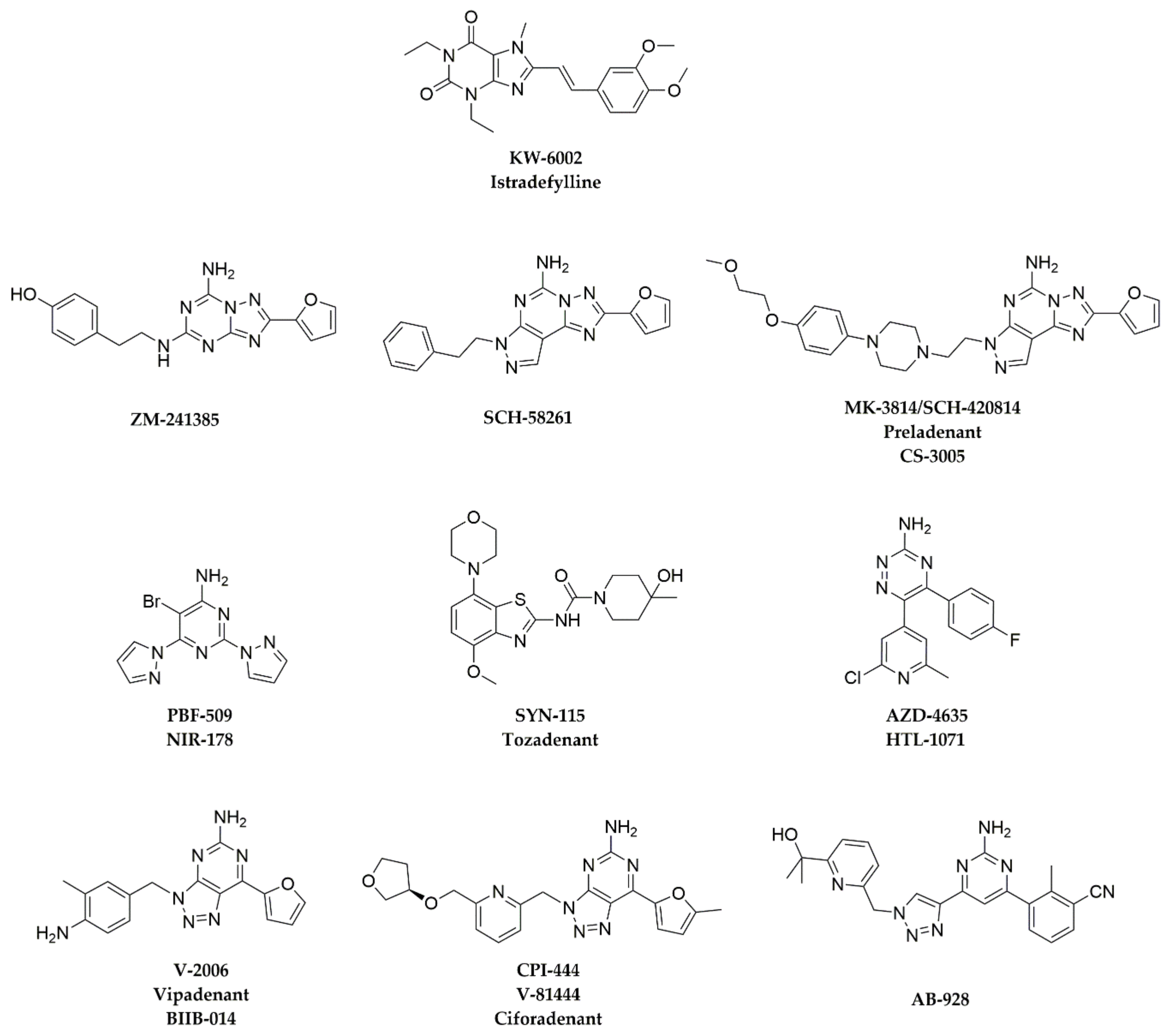
3. A2AAR Antagonists in Preclinical and Clinical Studies
3.1. ZM-241385
3.2. SCH-58261
3.3. MK-3814
3.4. PBF-509
3.5. SYN-115
3.6. AZD-4635
3.7. CPI-444
3.8. AB-928
3.9. EOS-100850
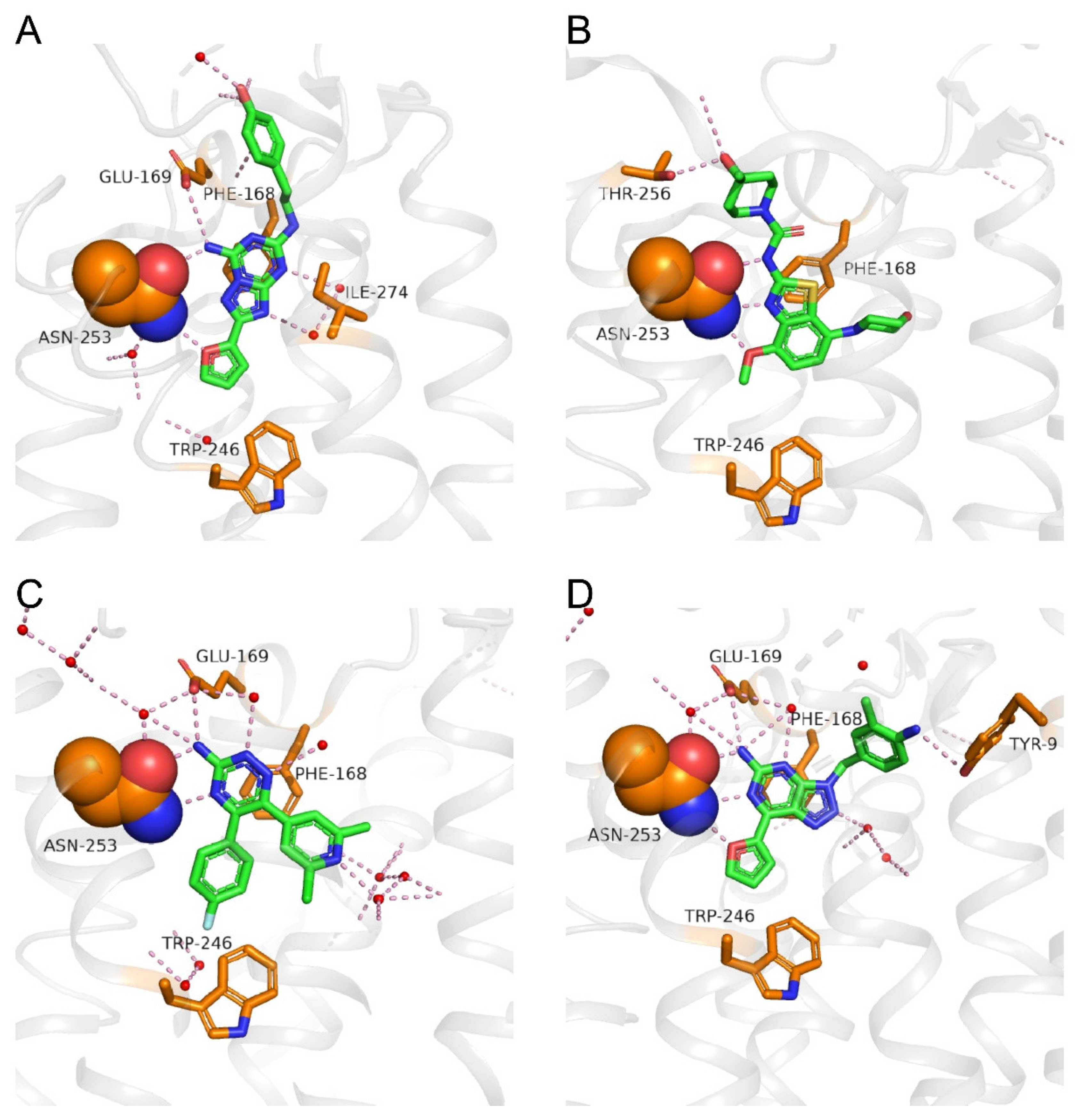
4. Binding Modes of Antagonists in Complexes with A2AAR
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharm. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.C.; Jackson, P.F. Adenosine A2A receptor antagonists and Parkinson’s disease. ACS Chem. Neurosci. 2011, 2, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, M.; Freissmuth, M.; Nanoff, C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell Signal. 2002, 14, 99–108. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Müller, C.E. Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology 2016, 104, 31–49. [Google Scholar] [CrossRef] [Green Version]
- Waarde, A.; Dierckx, R.; Zhou, X.; Khanapur, S.; Tsukada, H.; Ishiwata, K.; Luurtsema, G.; de Vries, E.F.J.; Elsinga, P.H. Potential therapeutic applications of adenosine A2A receptor ligands and opportunities for A2A receptor imaging. Med. Res. Rev. 2018, 38, 5–56. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P.; Curran, M.P. Regadenoson. Am. J. Cardiovasc. Drugs 2011, 10, 65–71. [Google Scholar] [CrossRef]
- Hage, F.G.; Ghimire, G.; Lester, D.; McKay, J.; Bleich, S.; El-Hajj, S.; Iskandrian, A.E. The prognostic value of regadenoson myocardial perfusion imaging. J. Nucl. Cardiol. 2015, 22, 1214–1221. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.Y.; Zhang, X.H.; Zhen, X.C. Development of adenosine A2A receptor antagonists for the treatment of Parkinson’s disease: A recent update and challenge. ACS Chem. Neurosci. 2019, 10, 783–791. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gao, Z.G.; Matricon, P.; Eddy, M.T.; Carlsson, J. Adenosine A2A receptor antagonists: From caffeine to selective non-xanthines. Br. J. Pharmacol. 2020, in press. [Google Scholar] [CrossRef]
- Pinna, A. Adenosine A2A receptor antagonists in Parkinson’s disease: Progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 2014, 28, 455–474. [Google Scholar] [CrossRef] [PubMed]
- Dungo, R.; Deeks, E.D. Istradefylline: First global approval. Drugs 2013, 73, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, G.; Zimmerman, T.P.; Hiemstra, K.; Winston, M.; Chu, L. Adenosine inhibition of lymphocyte-mediated cytolysis: Possible role of cyclic adenosine monophosphate. Science 1975, 187, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997, 57, 2602–2605. [Google Scholar] [PubMed]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–919. [Google Scholar] [CrossRef]
- Ohta, A.; Elieser Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, S.M.; Sitkovsky, M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1alpha driven immunosuppression and improve immunotherapies of cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Mediavilla-Varela, M.; Castro, J.; Chiappori, A.; Noyes, D.; Hernandez, D.C.; Allard, B.; Stagg, J.; Antonia, S.J. A novel antagonist of the immune checkpoint protein adenosine A2A receptor restores tumor-infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia 2017, 19, 530–536. [Google Scholar] [CrossRef]
- Willingham, S.B.; Ho, P.Y.; Hotson, A.; Hill, C.; Piccione, E.C.; Hsieh, J.; Liu, L.; Buggy, J.J.; McCaffery, I.; Miller, R.A. A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to anti-PD-L1 and anti-CTLA-4 in preclinical models. Cancer Immunol Res. 2018, 6, 1136–1149. [Google Scholar] [CrossRef] [Green Version]
- Jaakola, V.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.T.; Lane, J.R.; IJzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, B.; Nehme, R.; Warne, T.; Leslie, A.G.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef]
- Susac, L.; Eddy, M.T.; Didenko, T.; Stevens, R.C.; Wüthrich, K. A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl. Acad. Sci. USA 2018, 115, 12733–12738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, M.T.; Lee, M.Y.; Gao, Z.G.; White, K.L.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; et al. Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor. Cell 2018, 172, 68–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasko, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef]
- Moesta, A.K.; Li, X.Y.; Smyth, M.J. Targeting CD39 in cancer. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Colgan, S.P.; Eltzschig, H.K.; Eckle, T.; Thompson, L.F. Physiological roles for ecto-5’-nucleotidase (CD73). Purinergic. Signal. 2006, 2, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Ngiow, S.F.; Madore, J.; Reinhardt, J.; Landsberg, J.; Chitsazan, A.; Rautela, J.; Bald, T.; Barkauskas, D.S.; Ahern, E.; et al. Targeting adenosine in BRAF-mutant melanoma reduces tumor growth and metastasis. Cancer Res. 2017, 77, 4684–4696. [Google Scholar] [CrossRef] [Green Version]
- Kazemi, M.H.; Mohseni, S.R.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell Physiol. 2018, 233, 2032–2057. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Miele, L.; Porta, A.; Pinto, A.; Morello, S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget 2015, 6, 27478–27489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmada, A.; Ahmada, S.; Gloverb, L.; Millera, S.M.; Shannonc, J.M.; Guoa, X.; Franklind, W.A.; Bridgesc, J.P.; Schaacke, J.B.; Colganb, S.P.; et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2α in pulmonary endothelial cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10684–10689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaergaard, J.; Hatfield, S.; Jones, G.; Ohta, A.; Sitkovsky, M. A2A adenosine receptor gene deletion or synthetic A2A antagonist liberate tumor-reactive CD8+ T Cells from tumor-induced immunosuppression. J. Immunol. 2018, 201, 782–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congreve, M.; Brown, G.A.; Borodovsky, A.; Lamb, M.L. Targeting adenosine A2A receptor antagonism for treatment of cancer. Expert Opin. Drug Discov. 2018, 13, 997–1003. [Google Scholar] [CrossRef]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2A receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Gao, Z.G.; Jacobson, K.A. A2B adenosine receptor and cancer. Int. J. Mol. Sci. 2019, 20, 5139. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Lo, Y.C.; Powell, J.D. A2AR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Cekic, C.; Linden, J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res. 2014, 74, 7239–7249. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, C.; Palomo, I.; Fuentes, E. Role of adenosine A2B receptor overexpression in tumor progression. Life Sci. 2016, 166, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.J.; Tan, J.B.; Becker, A.; Yi, F.F.; Park, T.; Leleti, M.R.; Rosen, B.; Sharif, E.; Debien, L.; Young, S.; et al. Characterization of the potent and selective A2AR antagonist AB928 for the treatment of cancer. Cancer Res. 2017, 77, AM2017–AM4572. [Google Scholar]
- Mittal, D.; Young, A.; Stannard, K.; Yong, M.; Teng, M.W.; Allard, B.; Stagg, J.; Smyth, M.J. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. 2014, 74, 3652–3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, S.; Ichimura, M.; Ikeda, K.; Ishii, A.; Kanda, T.; Koga, K.; Koike, N.; Kurokawa, M.; Kuwana, Y.; Mori, A.; et al. Progress in pursuit of therapeutic A2A antagonists: The adenosine A2A receptor selective antagonist KW-6002: Research and development toward a novel nondopaminergic therapy for Parkinson’s disease. Neurology 2003, 61, S97–S100. [Google Scholar] [CrossRef]
- Poucher, S.M.; Keddie, J.R.; Singh, P.; Stoggall, S.M.; Caulkett, P.W.R.; Jones, G.; Collis, M.G. The in vitro pharmacology of ZM-241385, a potent, non-xanthine, A2A selective adenosine receptor antagonist. Br. J. Pharmacol. 1995, 115, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- De Zwart, M.; Vollinga, R.C.; Beukers, M.W.; Sleegers, D.F.; von Frijtag Drabbe Künze, J.K.; de Groote, M.; IJzerman, A.P. Potent antagonists for the human adenosine A2B receptor. Derivatives of the triazolotriazine adenosine receptor antagonist ZM-241385 with high affinity. Drug Develop. Res. 1999, 48, 95–103. [Google Scholar] [CrossRef]
- Ongini, E.; Dionisotti, S.; Irenius, S.G.E.; Fredholm, B.B. Comparison of CGS-5943, ZM-241385 and SCH-58261 as antagonists at human adenosine receptors. Naunyn Schmiedeberg’s Arch. Pharmacol. 1999, 359, 7–10. [Google Scholar] [CrossRef]
- Neustadt, B.R.; Hao, J.; Lindo, N.; Greenlee, W.J.; Stamford, A.W.; Tulshian, D.; Ongini, E.; Hunter, J.; Monopoli, A.; Bertorelli, R.; et al. Potent, selective, and orally active adenosine A2A receptor antagonists: Arylpiperazine derivatives of pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines. Bioorg. Med. Chem. Lett. 2007, 17, 1376–1380. [Google Scholar] [CrossRef]
- Flohr, A.; Moreau, J.; Poli, S.M.; Riemer, C.; Steward, L. 4-Hydroxy-4-methyl-piperidine-1-carboxylic acid (4-methoxy-7-morpholin-4-yl-benzothiazol-2-yl)-amide. US Patent 20,050,261,289, 24 November 2005. [Google Scholar]
- Borodovsky, A.; Wang, Y.; Ye, M.; Shaw, J.C.; Sachsenmeier, K.F.; Deng, N.; DelSignore, K.J.; Fretland, A.J.; Clarke, J.D.; Goodwin, R.J.; et al. Abstract 5580: Preclinical pharmacodynamics and antitumor activity of AZD-4635, a novel adenosine 2A receptor inhibitor that reverses adenosine mediated T cell suppression. AACR Annu. Meet. 2017. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Bamford, S.J.; Botting, R.; Comer, M.; Denny, S.; Gaur, S.; Griffin, M.; Jordan, A.M.; Knight, A.R.; Lerpiniere, J.; et al. Antagonists of the human A2A adenosine receptor. 4. Design, synthesis, and preclinical evaluation of 7-aryltriazolo[4,5-d]pyrimidines. J. Med. Chem. 2009, 52, 33–47. [Google Scholar] [CrossRef]
- Uustare, A.; Vonk, A.; Terasmaa, A.; Fuxe, K.; Rinken, A. Kinetic and functional properties of [3H] ZM-241385, a high affinity antagonist for adenosine A2A receptors. Life Sci. 2005, 76, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.J.; Rhee, S.Y.; Ryu, J.H.; Cheong, J.H.; Kwon, K.; Yang, S.I.; Park, S.H.; Lee, J.; Kim, H.Y.; Han, S.H.; et al. Activation of adenosine A2A receptor up-regulates BDNF expression in rat primary cortical neurons. Neurochem. Res. 2011, 36, 2259–2269. [Google Scholar] [CrossRef]
- Da Rocha Lapa, F.; da Silva, M.D.; de Almeida Cabrini, D.; Santos, A.R. Anti-inflammatory effects of purine nucleosides, adenosine and inosine, in a mouse model of pleurisy: Evidence for the role of adenosine A2 receptors. Purinergic Signal. 2012, 8, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am. J. Cancer. Res. 2014, 4, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Keddie, J.R.; Poucher, S.M.; Shaw, G.R.; Brooks, R.; Collis, M.G. In vivo characterisation of ZM-241385, a selective adenosine A2A receptor antagonist. Eur. J. Pharmacol. 1996, 301, 107–113. [Google Scholar] [CrossRef]
- Yang, M.; Soohoo, D.; Soelaiman, S.; Kalla, R.; Zablocki, J.; Chu, N.; Leung, K.; Yao, L.; Diamond, I.; Belardinelli, L.; et al. Characterization of the potency, selectivity, and pharmacokinetic profile for six adenosine A2A receptor antagonists. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 375, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kiselgof, E.; Tulshian, D.B.; Arik, L.; Zhang, H.; Fawzi, A. 6-(2-Furanyl)-9H-purin-2-amine derivatives as A2A adenosine antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 2119–2122. [Google Scholar] [CrossRef]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, R.A.; Bertorelli, R.; Varty, G.B.; Lachowicz, J.E.; Forlani, A.; Fredduzzi, S.; Cohen-Williams, M.E.; Higgins, G.A.; Impagnatiello, F.; Nicolussi, E.; et al. Characterization of the potent and highly selective A2A receptor antagonists preladenant and SCH-412348 [7-[2-[4-2,4-difluorophenyl]-1-piperazinyl]ethyl]-2-(2-furanyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine] in rodent models of movement disorders and depression. J. Pharmacol. Exp. Ther. 2009, 330, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Nunez, F.; Taura, J.; Camacho, J.; Lopez-Cano, M.; Fernandez-Duenas, V.; Castro, N.; Castro, J.; Ciruela, F. PBF509, an adenosine A2A receptor antagonist with efficacy in rodent models of movement disorders. Front. Pharmacol. 2018, 9, 1200. [Google Scholar] [CrossRef] [Green Version]
- Chiappori, A.; Williams, C.; Creelan, B.; Tanvetyanon, T.; Gray, J.; Haura, E.; Chen, D.T.; Thapa, R.; Beg, A.; Boyle, T.; et al. Phase I/II study of the A2AR antagonist NIR178 (PBF-509), an oral immunotherapy, in patients (pts) with advanced NSCLC. J. Thorac. Oncol. 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Hauser, R.A.; Olanow, C.W.; Kieburtz, K.D.; Pourcher, E.; Docu-Axelerad, A.; Lew, M.; Kozyolkin, O.; Neale, A.; Resburg, C.; Meya, U.; et al. Tozadenant (SYN115) in patients with Parkinson’s disease who have motor fluctuations on levodopa: A phase 2b, double-blind, randomised trial. Lancet. Neurol. 2014, 13, 767–776. [Google Scholar] [CrossRef]
- Bamford, S.J.; Gillespie, R.J.; Todd, R.S. Triazolo[4,5-d]pyramidine derivatives and their use as purine receptor antagonists. WO2,009,156,737, 30 December 2009. [Google Scholar]
- Schindler, U.; Seitz, L.; Ashok, D.; Piovesan, D.; Tan, J.; DiRenzo, D.; Yin, F.; Leleti, M.; Rosen, B.; Miles, D.; et al. AB928, a dual antagonist of the A2AR and A2BR adenosine receptors, leads to greater immune activation and reduced tumor growth when combined with chemotherapy. Eur. J. Cancer 2018, 92, S14–S15. [Google Scholar] [CrossRef]
- Seitz, L.; Jin, L.; Leleti, M.; Ashok, D.; Jeffrey, J.; Rieger, A.; Tiessen, R.G.; Arold, G.; Tan, J.B.L.; Powers, J.P.; et al. Safety, tolerability, and pharmacology of AB928, a novel dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Investig. New Drugs 2019, 37, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Crosignani, S.; Dickinson, P.; de Matas, M.; Houthuys, E.J.K.H.; Marillier, R.G.; Martinoli, C.; de Henau, O.; Deriessens, G. Thiocarbamate derivatives as A2A inhibitors, pharmaceutical composition thereof and combinations with anticancer agents. WO2,020,053,263, 19 March 2020. [Google Scholar]
- iTeos Therapeutics. Available online: https://www.iteostherapeutics.com/ (accessed on 14 August 2020).
- Dore, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A2A receptor in complex with ZM-241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Rucktooa, P.; Cheng, R.K.Y.; Segala, E.; Geng, T.; Errey, J.C.; Brown, G.A.; Cooke, R.M.; Marshall, F.H.; Dore, A.S. Towards high throughput GPCR crystallography: In meso soaking of adenosine A2A receptor crystals. Sci. Rep. 2018, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Congreve, M.; Andrews, S.P.; Dore, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef]
- Guo, D.; Xia, L.; van Veldhoven, J.P.; Hazeu, M.; Mocking, T.; Brussee, J.; Ijzerman, A.P.; Heitman, L.H. Binding kinetics of ZM-241385 derivatives at the human adenosine A2A receptor. Chem. Med. Chem. 2014, 9, 752–761. [Google Scholar] [CrossRef]
- Sun, B.; Bachhawat, P.; Chu, M.L.; Wood, M.; Ceska, T.; Sands, Z.A.; Mercier, J.; Lebon, F.; Kobilka, T.S.; Kobilka, B.K. Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc. Natl. Acad. Sci. USA 2017, 114, 2066–2071. [Google Scholar] [CrossRef] [Green Version]
- Igonet, S.; Raingeval, C.; Cecon, E.; Pucic-Bakovic, M.; Lauc, G.; Cala, O.; Baranowski, M.; Perez, J.; Jockers, R.; Krimm, I.; et al. Enabling STD-NMR fragment screening using stabilized native GPCR: A case study of adenosine receptor. Sci. Rep. 2018, 8, 8142. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | Ki (nM) | References | |||
|---|---|---|---|---|---|
| A1AR | A2AAR | A2BAR | A3AR | ||
| KW-6002 | 9600 | 12 | 1800 | >3000 | [44] |
| ZM-241385 | 255 | 0.8 | 50 | >10,000 | [45,46] |
| SCH-58261 | 287 | 0.6 | 5011 | >10,000 | [47] |
| MK-3814 | >1000 | 1.1 | >1700 | >1000 | [48] |
| PBF-509 | 2500 | 12 | 1000 | 5000 | [18] |
| SYN-115 | 1350 | 5 | 700 | 1570 | [49] |
| AZD-4635 | 160 | 1.7 | 64 | >10,000 | [50] |
| V-2006 | 68 | 1.3 | 63 | 1005 | [51] |
| CPI-444 | 192 | 3.54 | 1528 | 2455 | [19] |
| AB-928 | 64 | 1.5 | 2.0 | 489 | [42] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Yan, W.; Duan, W.; Wüthrich, K.; Cheng, J. Tumor Immunotherapy Using A2A Adenosine Receptor Antagonists. Pharmaceuticals 2020, 13, 237. https://doi.org/10.3390/ph13090237
Zhang J, Yan W, Duan W, Wüthrich K, Cheng J. Tumor Immunotherapy Using A2A Adenosine Receptor Antagonists. Pharmaceuticals. 2020; 13(9):237. https://doi.org/10.3390/ph13090237
Chicago/Turabian StyleZhang, Jinfeng, Wenzhong Yan, Wenwen Duan, Kurt Wüthrich, and Jianjun Cheng. 2020. "Tumor Immunotherapy Using A2A Adenosine Receptor Antagonists" Pharmaceuticals 13, no. 9: 237. https://doi.org/10.3390/ph13090237
APA StyleZhang, J., Yan, W., Duan, W., Wüthrich, K., & Cheng, J. (2020). Tumor Immunotherapy Using A2A Adenosine Receptor Antagonists. Pharmaceuticals, 13(9), 237. https://doi.org/10.3390/ph13090237