Data-Driven Molecular Dynamics: A Multifaceted Challenge


Abstract
:1. Introduction
2. Learning from Molecular Dynamics Trajectories
2.1. Unsupervised Learning Methods
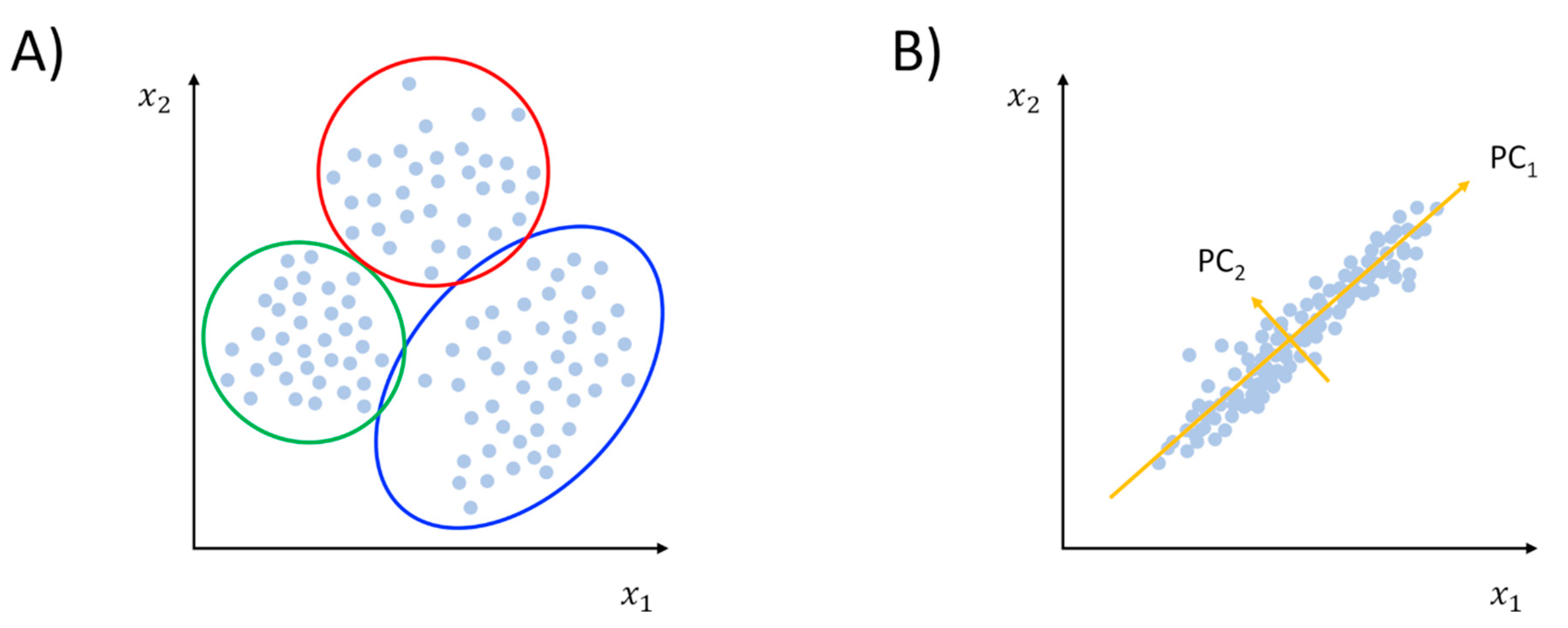
2.1.1. Clustering and PCA: The Grand Old Tools of Trajectory Analysis
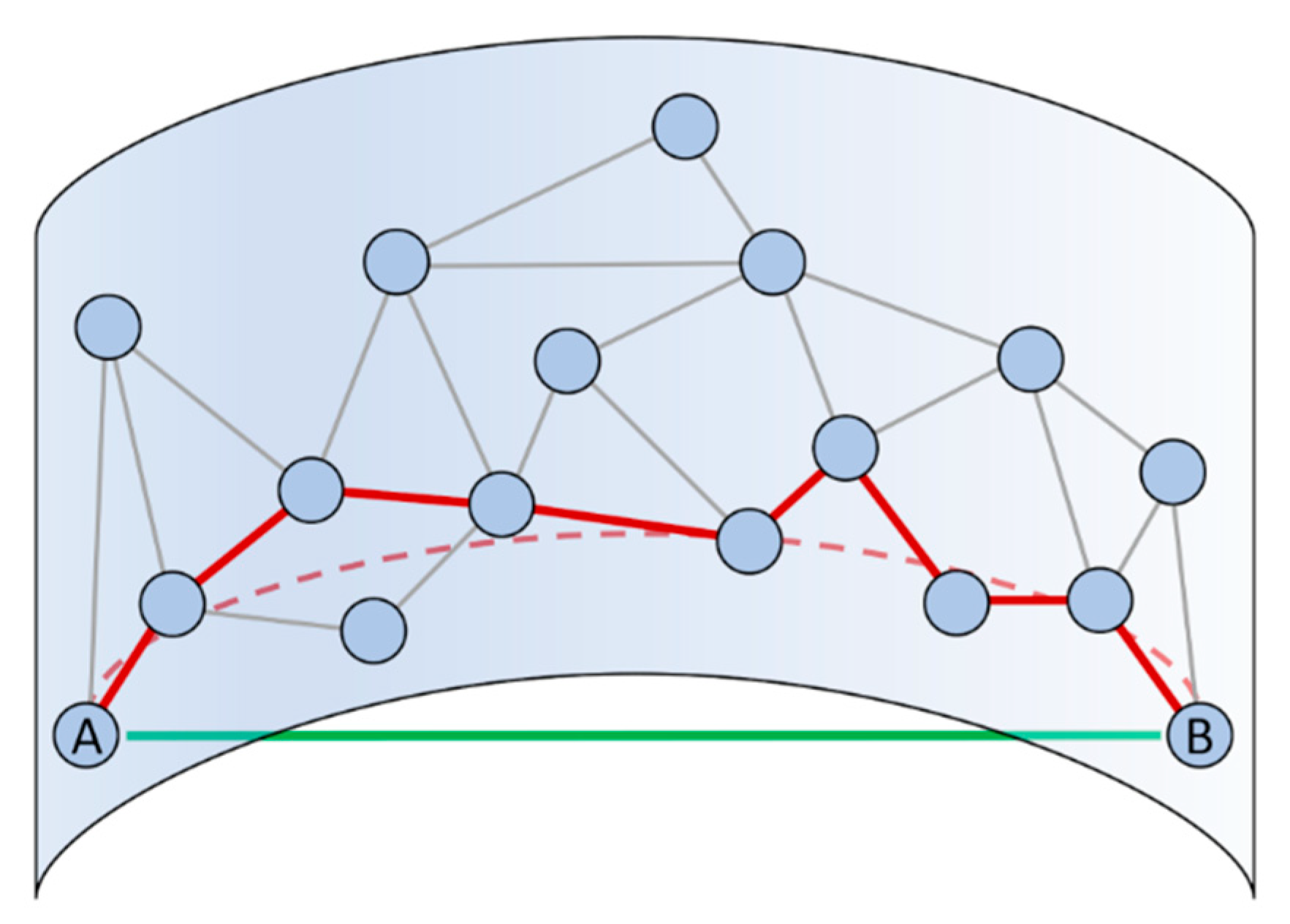
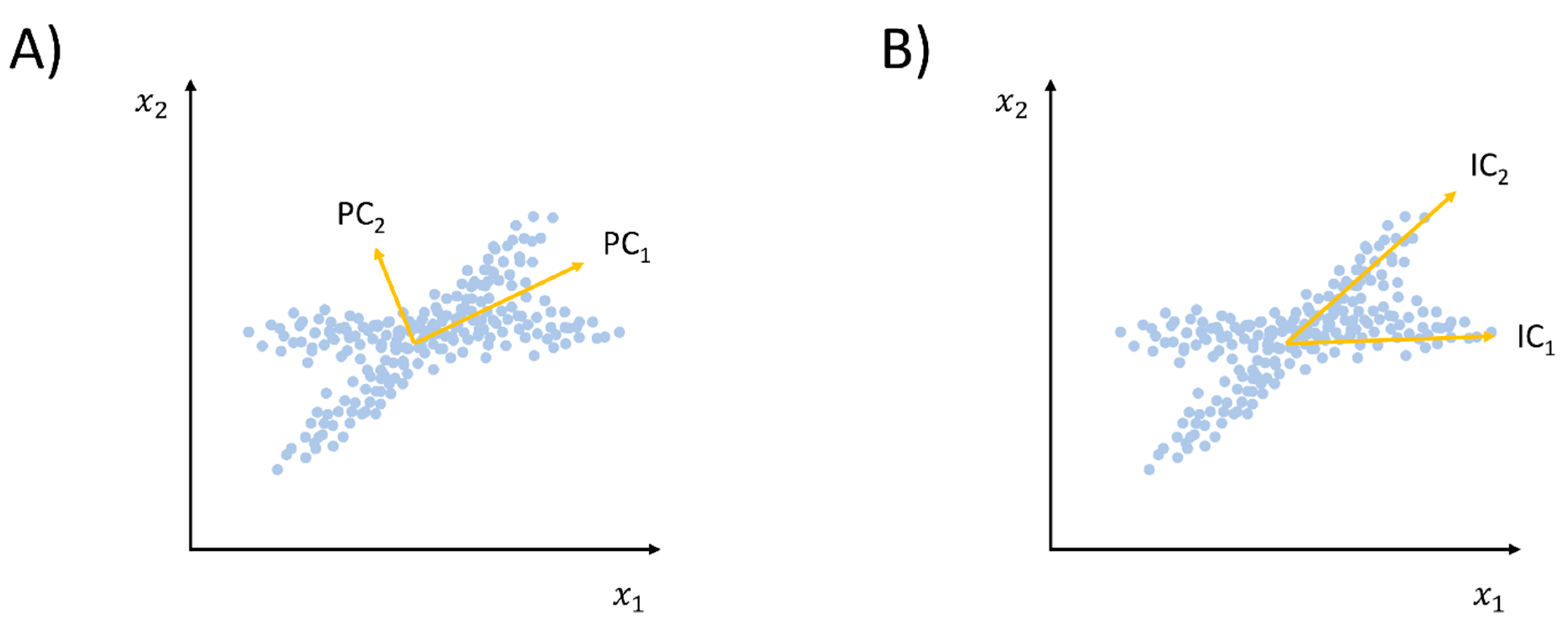
2.1.2. Beyond Linear Dimensionality Reduction
2.1.3. Including Dynamical Information into Geometric Dimensionality Reduction
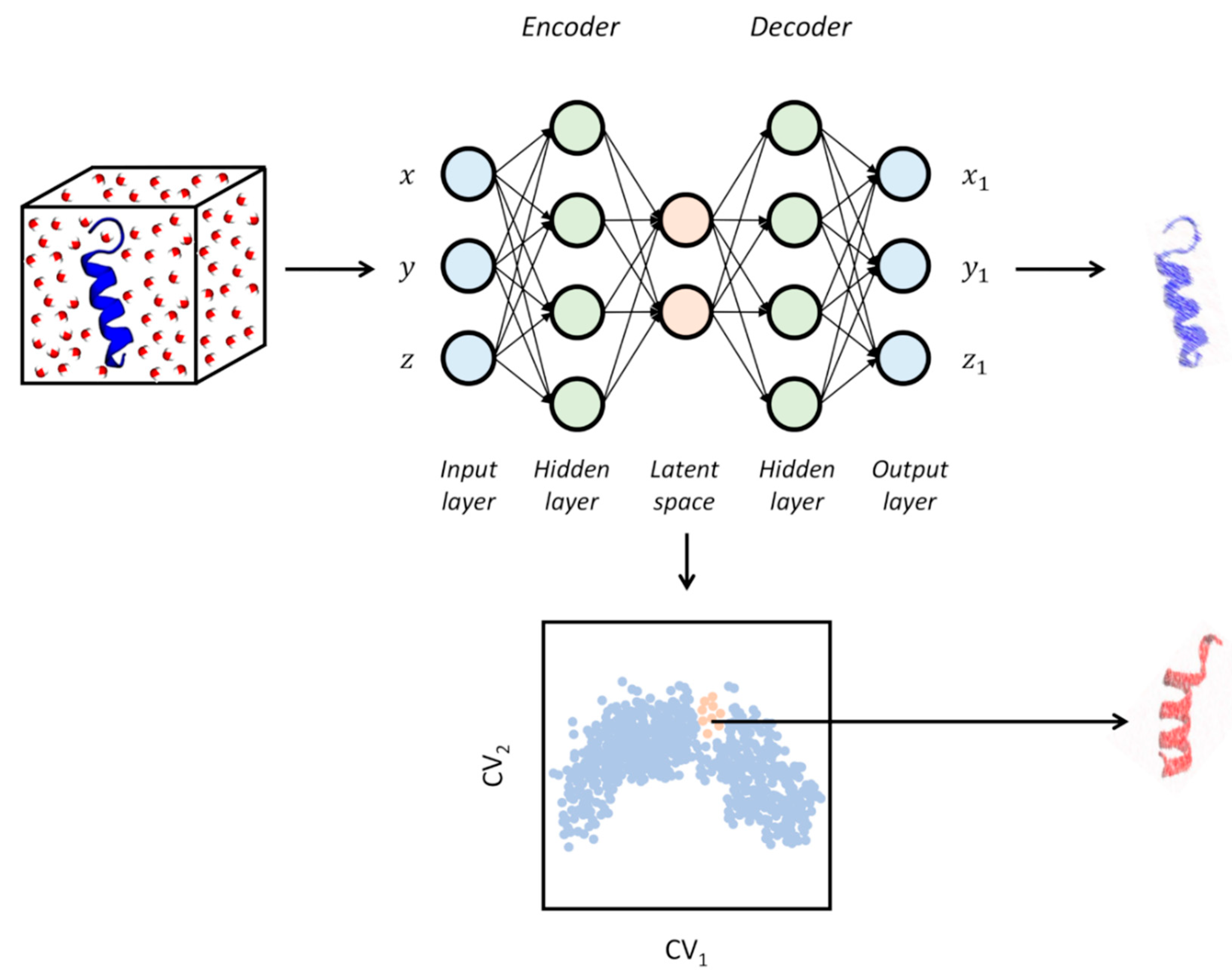
2.1.4. Neural Networks and Deep Learning
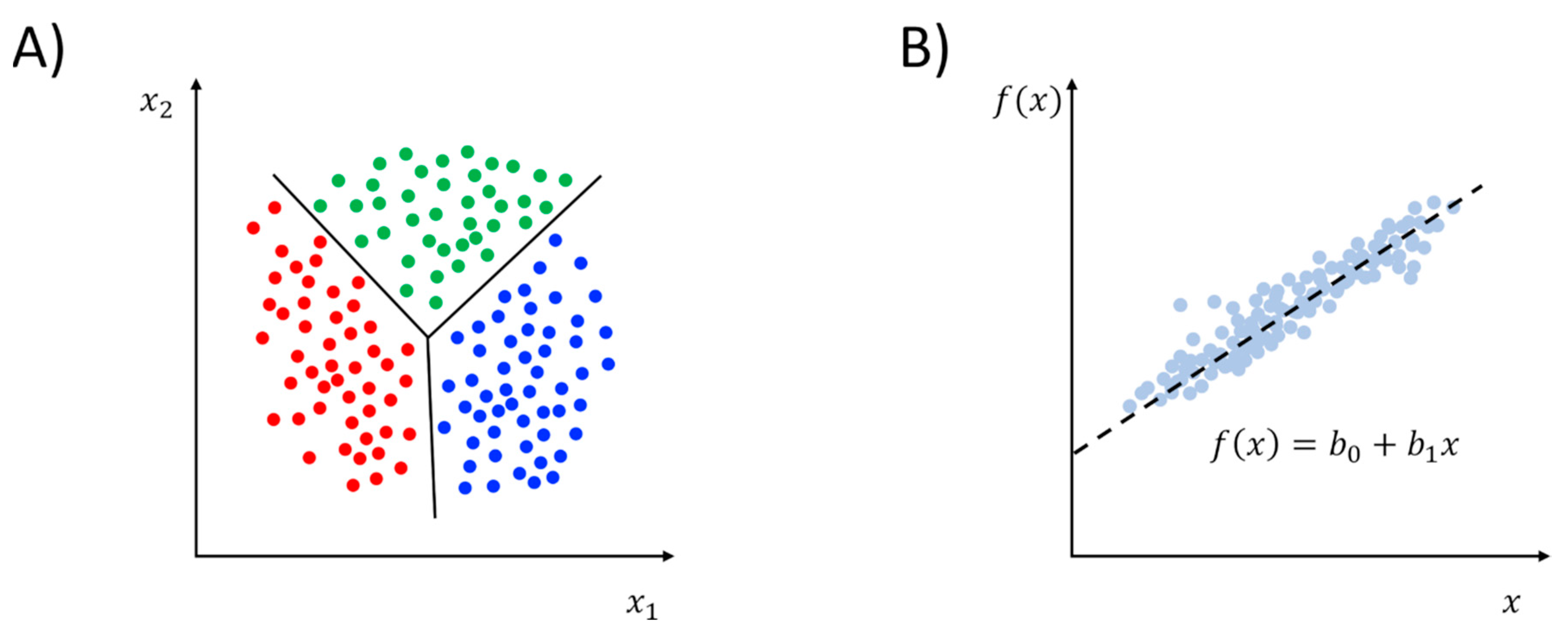
2.2. Supervised Learning Methods
3. Learning from Molecular Dynamics Trajectories and Experimental Data
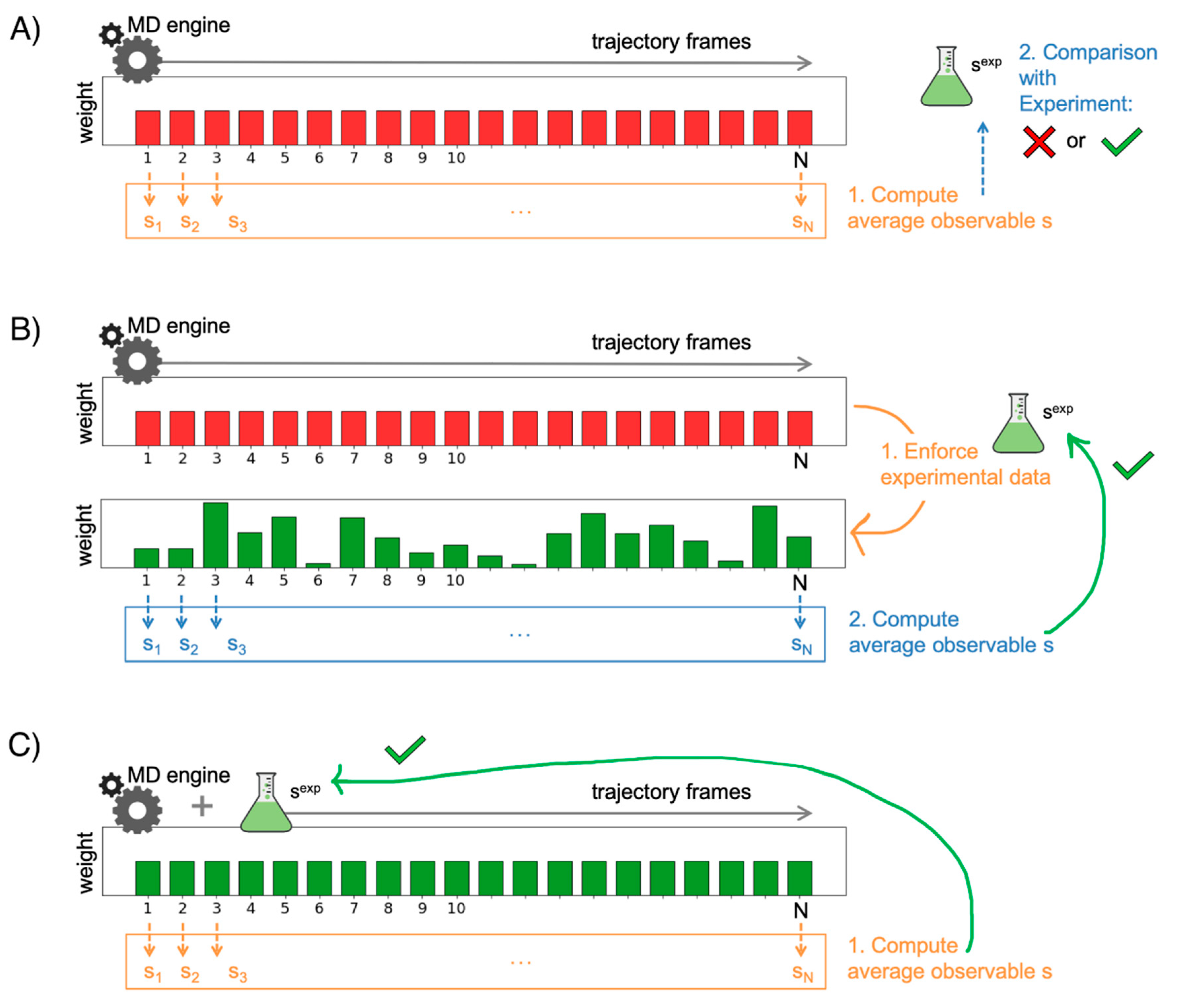
3.1. Validating MD Simulations through Comparison with Experiments
3.2. Improving the Agreement through Ensemble Reweighting
3.3. Enforcing Experimental Information during the Simulations
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hansch, C.; Maloney, P.P.; Fujita, T.; Muir, R.M. Correlation of Biological Activity of Phenoxyacetic Acids with Hammett Substituent Constants and Partition Coefficients. Nature 1962, 194, 178–180. [Google Scholar] [CrossRef]
- Hansch, C.; Fujita, T. p-σ-π Analysis. A Method for the Correlation of Biological Activity and Chemical Structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaduangrat, N.; Lampa, S.; Simeon, S.; Gleeson, M.P.; Spjuth, O.; Nantasenamat, C. Towards reproducible computational drug discovery. J. Cheminform. 2020, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, J. Chemoinformatics: Achievements and Challenges, a Personal View. Molecules 2016, 21, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Hu, Y.; Bajorath, J. Entering the ‘big data’ era in medicinal chemistry: Molecular promiscuity analysis revisited. Future Sci. OA 2017, 3, FSO179. [Google Scholar] [CrossRef] [Green Version]
- Lavecchia, A. Deep learning in drug discovery: Opportunities, challenges and future prospects. Drug Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Gioia, D.; Bertazzo, M.; Recanatini, M.; Masetti, M.; Cavalli, A. Dynamic Docking: A Paradigm Shift in Computational Drug Discovery. Molecules 2017, 22, 2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef] [PubMed]
- Bernetti, M.; Masetti, M.; Rocchia, W.; Cavalli, A. Kinetics of Drug Binding and Residence Time. Annu. Rev. Phys. Chem. 2019, 70, 143–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Borhani, D.W.; Shaw, D.E. The future of molecular dynamics simulations in drug discovery. J. Comput. Aided Mol. Des. 2012, 26, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daidone, I.; Amadei, A. Essential dynamics: Foundation and applications. Wires Comput. Mol. Sci. 2012, 2, 762–770. [Google Scholar] [CrossRef]
- Klein, M.; Sharma, R.; Bohrer, C.H.; Avelis, C.M.; Roberts, E. Biospark: Scalable analysis of large numerical datasets from biological simulations and experiments using Hadoop and Spark. Bioinformatics 2017, 33, 303–305. [Google Scholar] [CrossRef]
- Dean, J.; Ghemawat, S. MapReduce: Simplified data processing on large clusters. Commun. ACM 2008, 51, 107–113. [Google Scholar] [CrossRef]
- Cesari, A.; Reißer, S.; Bussi, G. Using the Maximum Entropy Principle to Combine Simulations and Solution Experiments. Computation 2018, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Fiorin, G.; Klein, M.L.; Hénin, J. Using collective variables to drive molecular dynamics simulations. Mol. Phys. 2013, 111, 3345–3362. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham Iii, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heymann, B.; Tavan, P. Ligand Binding: Molecular Mechanics Calculation of the Streptavidin-Biotin Rupture Force. Science 1996, 271, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isralewitz, B.; Gao, M.; Schulten, K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001, 11, 224–230. [Google Scholar] [CrossRef]
- Darve, E.; Rodríguez-Gómez, D.; Pohorille, A. Adaptive biasing force method for scalar and vector free energy calculations. J. Chem. Phys. 2008, 128, 144120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562. [Google Scholar] [CrossRef] [Green Version]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [Green Version]
- Abrams, C.; Bussi, G. Enhanced Sampling in Molecular Dynamics Using Metadynamics, Replica-Exchange, and Temperature-Acceleration. Entropy 2014, 16, 163–199. [Google Scholar] [CrossRef] [Green Version]
- Decherchi, S.; Masetti, M.; Vyalov, I.; Rocchia, W. Implicit solvent methods for free energy estimation. Eur. J. Med. Chem. 2015, 91, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrdanz, M.A.; Zheng, W.; Clementi, C. Discovering Mountain Passes via Torchlight: Methods for the Definition of Reaction Coordinates and Pathways in Complex Macromolecular Reactions. Annu. Rev. Phys. Chem. 2013, 64, 295–316. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, D.M.; Chong, L.T. Weighted Ensemble Simulation: Review of Methodology, Applications, and Software. Annu. Rev. Biophys. 2017, 46, 43–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betz, R.M.; Dror, R.O. How Effectively Can Adaptive Sampling Methods Capture Spontaneous Ligand Binding? J. Chem. Theory Comput. 2019, 15, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.L. Machine learning and data science in soft materials engineering. J. Phys. Condens. Matter 2017, 30, 043002. [Google Scholar] [CrossRef] [PubMed]
- Ceriotti, M. Unsupervised machine learning in atomistic simulations, between predictions and understanding. J. Chem. Phys. 2019, 150, 150901. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef]
- Bottegoni, G.; Rocchia, W.; Cavalli, A. Application of Conformational Clustering in Protein–Ligand Docking. In Computational Drug Discovery and Design; Baron, R., Ed.; Springer: New York, NY, USA, 2012; pp. 169–186. [Google Scholar]
- Tribello, G.A.; Ceriotti, M.; Parrinello, M. A self-learning algorithm for biased molecular dynamics. Proc. Natl. Acad. Sci. USA 2010, 107, 17509. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Gasparotto, P. Using Dimensionality Reduction to Analyze Protein Trajectories. Front. Mol. Biosci. 2019, 6, 46. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; de Groot, B.L.; van Aalten, D.M.F.; Berendsen, H.J.C. An Efficient Method for Sampling the Essential Subspace of Proteins. J. Biomol. Struct. Dyn. 1996, 13, 615–625. [Google Scholar] [CrossRef]
- Spiwok, V.; Lipovová, P.; Králová, B. Metadynamics in Essential Coordinates: Free Energy Simulation of Conformational Changes. J. Phys. Chem. B 2007, 111, 3073–3076. [Google Scholar] [CrossRef] [PubMed]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glykos, N.M. Software news and updates carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Gowers, R.; Linke, M.; Barnoud, J.; Reddy, T.; Melo, M.; Seyler, S.; Domański, J.; Dotson, D.; Buchoux, S.; Kenney, I.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the Python in Science Conference 2016, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar] [CrossRef] [Green Version]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.; Nguyen, P.H.; Stock, G. Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins Struct. Funct. Bioinform. 2005, 58, 45–52. [Google Scholar] [CrossRef]
- Altis, A.; Nguyen, P.H.; Hegger, R.; Stock, G. Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 2007, 126, 244111. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, M.; Decherchi, S.; De Simone, A.; Recanatini, M.; Cavalli, A.; Bottegoni, G. Multi-target dopamine D3 receptor modulators: Actionable knowledge for drug design from molecular dynamics and machine learning. Eur. J. Med. Chem. 2020, 188, 111975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, O.M. Geometric versus topological clustering: An insight into conformation mapping. Proteins Struct. Funct. Bioinform. 1997, 27, 213–226. [Google Scholar] [CrossRef]
- Troyer, J.M.; Cohen, F.E. Protein conformational landscapes: Energy minimization and clustering of a long molecular dynamics trajectory. Proteins Struct. Funct. Bioinform. 1995, 23, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Pisani, P.; Caporuscio, F.; Carlino, L.; Rastelli, G. Molecular Dynamics Simulations and Classical Multidimensional Scaling Unveil New Metastable States in the Conformational Landscape of CDK2. PLoS ONE 2016, 11, e0154066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniou, D.; Schwartz, S.D. Toward Identification of the Reaction Coordinate Directly from the Transition State Ensemble Using the Kernel PCA Method. J. Phys. Chem. B 2011, 115, 2465–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freddolino, P.L.; Schulten, K. Common Structural Transitions in Explicit-Solvent Simulations of Villin Headpiece Folding. Biophys. J. 2009, 97, 2338–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, A.; Freddolino, P.L.; Schulten, K. Going beyond Clustering in MD Trajectory Analysis: An Application to Villin Headpiece Folding. PLoS ONE 2010, 5, e9890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceriotti, M.; Tribello, G.A.; Parrinello, M. Simplifying the representation of complex free-energy landscapes using sketch-map. Proc. Natl. Acad. Sci. USA 2011, 108, 13023. [Google Scholar] [CrossRef] [Green Version]
- Ceriotti, M.; Tribello, G.A.; Parrinello, M. Demonstrating the Transferability and the Descriptive Power of Sketch-Map. J. Chem. Theory Comput. 2013, 9, 1521–1532. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Ceriotti, M.; Parrinello, M. Using sketch-map coordinates to analyze and bias molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2012, 109, 5196. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, L.; Ardèvol, A.; Parrinello, M.; Lutz, H.; Lu, H.; Weidner, T.; Corni, S. The interaction with gold suppresses fiber-like conformations of the amyloid β (16–22) peptide. Nanoscale 2016, 8, 8737–8748. [Google Scholar] [CrossRef] [Green Version]
- Tenenbaum, J.B.; Silva, V.d.; Langford, J.C. A Global Geometric Framework for Nonlinear Dimensionality Reduction. Science 2000, 290, 2319. [Google Scholar] [CrossRef]
- Das, P.; Moll, M.; Stamati, H.; Kavraki, L.E.; Clementi, C. Low-dimensional, free-energy landscapes of protein-folding reactions by nonlinear dimensionality reduction. Proc. Natl. Acad. Sci. USA 2006, 103, 9885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamati, H.; Clementi, C.; Kavraki, L.E. Application of nonlinear dimensionality reduction to characterize the conformational landscape of small peptides. Proteins Struct. Funct. Bioinform. 2010, 78, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiwok, V.; Králová, B. Metadynamics in the conformational space nonlinearly dimensionally reduced by Isomap. J. Chem. Phys. 2011, 135, 224504. [Google Scholar] [CrossRef] [PubMed]
- Branduardi, D.; Gervasio, F.L.; Parrinello, M. From A to B in free energy space. J. Chem. Phys. 2007, 126, 054103. [Google Scholar] [CrossRef]
- Bonomi, M.; Bussi, G.; Camilloni, C.; Tribello, G.A.; Banáš, P.; Barducci, A.; Bernetti, M.; Bolhuis, P.G.; Bottaro, S.; Branduardi, D.; et al. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. [Google Scholar] [CrossRef] [Green Version]
- Hashemian, B.; Millán, D.; Arroyo, M. Modeling and enhanced sampling of molecular systems with smooth and nonlinear data-driven collective variables. J. Chem. Phys. 2013, 139, 214101. [Google Scholar] [CrossRef] [Green Version]
- Schuetz, D.A.; Bernetti, M.; Bertazzo, M.; Musil, D.; Eggenweiler, H.-M.; Recanatini, M.; Masetti, M.; Ecker, G.F.; Cavalli, A. Predicting Residence Time and Drug Unbinding Pathway through Scaled Molecular Dynamics. J. Chem. Inf. Model. 2019, 59, 535–549. [Google Scholar] [CrossRef]
- Lange, O.F.; Grubmüller, H. Generalized correlation for biomolecular dynamics. Proteins Struct. Funct. Bioinform. 2006, 62, 1053–1061. [Google Scholar] [CrossRef] [Green Version]
- Lange, O.F.; Grubmüller, H. Full correlation analysis of conformational protein dynamics. Proteins Struct. Funct. Bioinform. 2008, 70, 1294–1312. [Google Scholar] [CrossRef]
- Masetti, M.; Falchi, F.; Recanatini, M. Protein Dynamics of the HIF-2 alpha PAS-B Domain upon Heterodimerization and Ligand Binding. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Ferguson, A.L.; Panagiotopoulos, A.Z.; Debenedetti, P.G.; Kevrekidis, I.G. Systematic determination of order parameters for chain dynamics using diffusion maps. Proc. Natl. Acad. Sci. USA 2010, 107, 13597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrdanz, M.A.; Zheng, W.; Maggioni, M.; Clementi, C. Determination of reaction coordinates via locally scaled diffusion map. J. Chem. Phys. 2011, 134, 124116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Rohrdanz, M.A.; Clementi, C. Rapid Exploration of Configuration Space with Diffusion-Map-Directed Molecular Dynamics. J. Phys. Chem. B 2013, 117, 12769–12776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preto, J.; Clementi, C. Fast recovery of free energy landscapes via diffusion-map-directed molecular dynamics. Phys. Chem. Chem. Phys. 2014, 16, 19181–19191. [Google Scholar] [CrossRef]
- Chiavazzo, E.; Covino, R.; Coifman, R.R.; Gear, C.W.; Georgiou, A.S.; Hummer, G.; Kevrekidis, I.G. Intrinsic map dynamics exploration for uncharted effective free-energy landscapes. Proc. Natl. Acad. Sci. USA 2017, 114, E5494. [Google Scholar] [CrossRef] [Green Version]
- Naritomi, Y.; Fuchigami, S. Slow dynamics in protein fluctuations revealed by time-structure based independent component analysis: The case of domain motions. J. Chem. Phys. 2011, 134, 065101. [Google Scholar] [CrossRef]
- Husic, B.E.; Pande, V.S. Markov State Models: From an Art to a Science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef]
- Chodera, J.D.; Noé, F. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014, 25, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Schwantes, C.R.; McGibbon, R.T.; Pande, V.S. Perspective: Markov models for long-timescale biomolecular dynamics. J. Chem. Phys. 2014, 141, 090901. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Cao, S.; Zhu, L.; Huang, X. Constructing Markov State Models to elucidate the functional conformational changes of complex biomolecules. Wires Comput. Mol. Sci. 2018, 8, e1343. [Google Scholar] [CrossRef]
- Pérez-Hernández, G.; Paul, F.; Giorgino, T.; De Fabritiis, G.; Noé, F. Identification of slow molecular order parameters for Markov model construction. J. Chem. Phys. 2013, 139, 015102. [Google Scholar] [CrossRef] [PubMed]
- Schwantes, C.R.; Pande, V.S. Improvements in Markov State Model Construction Reveal Many Non-Native Interactions in the Folding of NTL9. J. Chem. Theory Comput. 2013, 9, 2000–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- M. Sultan, M.; Pande, V.S. tICA-Metadynamics: Accelerating Metadynamics by Using Kinetically Selected Collective Variables. J. Chem. Theory Comput. 2017, 13, 2440–2447. [Google Scholar] [CrossRef]
- Hinton, G.E.; Salakhutdinov, R.R. Reducing the Dimensionality of Data with Neural Networks. Science 2006, 313, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehmeyer, C.; Noé, F. Time-lagged autoencoders: Deep learning of slow collective variables for molecular kinetics. J. Chem. Phys. 2018, 148, 241703. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.X.; Wayment-Steele, H.K.; Sultan, M.M.; Husic, B.E.; Pande, V.S. Variational encoding of complex dynamics. Phys. Rev. E 2018, 97, 062412. [Google Scholar] [CrossRef] [Green Version]
- Mardt, A.; Pasquali, L.; Wu, H.; Noé, F. VAMPnets for deep learning of molecular kinetics. Nat. Commun. 2018, 9, 5. [Google Scholar] [CrossRef]
- Chen, W.; Ferguson, A.L. Molecular enhanced sampling with autoencoders: On-the-fly collective variable discovery and accelerated free energy landscape exploration. J. Comput. Chem. 2018, 39, 2079–2102. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Tan, A.R.; Ferguson, A.L. Collective variable discovery and enhanced sampling using autoencoders: Innovations in network architecture and error function design. J. Chem. Phys. 2018, 149, 072312. [Google Scholar] [CrossRef]
- Ribeiro, J.M.L.; Bravo, P.; Wang, Y.; Tiwary, P. Reweighted autoencoded variational Bayes for enhanced sampling (RAVE). J. Chem. Phys. 2018, 149, 072301. [Google Scholar] [CrossRef] [Green Version]
- Lemke, T.; Peter, C. EncoderMap: Dimensionality Reduction and Generation of Molecule Conformations. J. Chem. Theory Comput. 2019, 15, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Lemke, T.; Berg, A.; Jain, A.; Peter, C. EncoderMap(II): Visualizing Important Molecular Motions with Improved Generation of Protein Conformations. J. Chem. Inf. Model. 2019, 59, 4550–4560. [Google Scholar] [CrossRef] [PubMed]
- Trapl, D.; Horvacanin, I.; Mareska, V.; Ozcelik, F.; Unal, G.; Spiwok, V. Anncolvar: Approximation of Complex Collective Variables by Artificial Neural Networks for Analysis and Biasing of Molecular Simulations. Front. Mol. Biosci. 2019, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.M.; Wayment-Steele, H.K.; Pande, V.S. Transferable Neural Networks for Enhanced Sampling of Protein Dynamics. J. Chem. Theory Comput. 2018, 14, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; de Groot, B.L. Detection of Functional Modes in Protein Dynamics. PLoS Comput. Biol. 2009, 5, e1000480. [Google Scholar] [CrossRef] [Green Version]
- Krivobokova, T.; Briones, R.; Hub, J.S.; Munk, A.; de Groot, B.L. Partial Least-Squares Functional Mode Analysis: Application to the Membrane Proteins AQP1, Aqy1, and CLC-ec1. Biophys. J. 2012, 103, 786–796. [Google Scholar] [CrossRef] [Green Version]
- Kopec, W.; Rothberg, B.S.; de Groot, B.L. Molecular mechanism of a potassium channel gating through activation gate-selectivity filter coupling. Nat. Commun. 2019, 10, 5366. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.H.; de Groot, B.L. Ubiquitin Dynamics in Complexes Reveal Molecular Recognition Mechanisms Beyond Induced Fit and Conformational Selection. PLoS Comput. Biol. 2012, 8, e1002704. [Google Scholar] [CrossRef]
- Sakuraba, S.; Kono, H. Spotting the difference in molecular dynamics simulations of biomolecules. J. Chem. Phys. 2016, 145, 074116. [Google Scholar] [CrossRef] [Green Version]
- Mendels, D.; Piccini, G.; Parrinello, M. Collective Variables from Local Fluctuations. J. Phys. Chem. Lett. 2018, 9, 2776–2781. [Google Scholar] [CrossRef]
- Piccini, G.; Mendels, D.; Parrinello, M. Metadynamics with Discriminants: A Tool for Understanding Chemistry. J. Chem. Theory Comput. 2018, 14, 5040–5044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendels, D.; Piccini, G.; Brotzakis, Z.F.; Yang, Y.I.; Parrinello, M. Folding a small protein using harmonic linear discriminant analysis. J. Chem. Phys. 2018, 149, 194113. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.M.; Pande, V.S. Automated design of collective variables using supervised machine learning. J. Chem. Phys. 2018, 149, 094106. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, A.B.; Sali, A.; Wilson, I.A. Integrative Structural Biology. Science 2013, 339, 913. [Google Scholar] [CrossRef] [Green Version]
- Bottaro, S.; Lindorff-Larsen, K. Biophysical experiments and biomolecular simulations: A perfect match? Science 2018, 361, 355. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, M.; Heller, G.T.; Camilloni, C.; Vendruscolo, M. Principles of protein structural ensemble determination. Curr. Opin. Struct. Biol. 2017, 42, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]
- Allnér, O.; Nilsson, L.; Villa, A. Magnesium Ion–Water Coordination and Exchange in Biomolecular Simulations. J. Chem. Theory Comput. 2012, 8, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Ibragimova, G.T.; Wade, R.C. Importance of Explicit Salt Ions for Protein Stability in Molecular Dynamics Simulation. Biophys. J. 1998, 74, 2906–2911. [Google Scholar] [CrossRef] [Green Version]
- Ross, G.A.; Rustenburg, A.S.; Grinaway, P.B.; Fass, J.; Chodera, J.D. Biomolecular Simulations under Realistic Macroscopic Salt Conditions. J. Phys. Chem. B 2018, 122, 5466–5486. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A. Chemical shifts in biomolecules. Curr. Opin. Struct. Biol. 2013, 23, 172–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolman, J.R.; Ruan, K. NMR Residual Dipolar Couplings as Probes of Biomolecular Dynamics. Chem. Rev. 2006, 106, 1720–1736. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural Characterization of Flexible Proteins Using Small-Angle X-ray Scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G. DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piston, D.W.; Kremers, G.-J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef]
- Jaynes, E.T. Information Theory and Statistical Mechanics. Phys. Rev. 1957, 106, 620–630. [Google Scholar] [CrossRef]
- Caticha, A. Relative Entropy and Inductive Inference. Aip Conf. Proc. 2004, 707, 75–96. [Google Scholar] [CrossRef]
- Boomsma, W.; Ferkinghoff-Borg, J.; Lindorff-Larsen, K. Combining Experiments and Simulations Using the Maximum Entropy Principle. PLoS Comput. Biol. 2014, 10, e1003406. [Google Scholar] [CrossRef] [Green Version]
- Bottaro, S.; Bussi, G.; Kennedy, S.D.; Turner, D.H.; Lindorff-Larsen, K. Conformational ensembles of RNA oligonucleotides from integrating NMR and molecular simulations. Sci. Adv. 2018, 4, eaar8521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Różycki, B.; Kim, Y.C.; Hummer, G. SAXS Ensemble Refinement of ESCRT-III CHMP3 Conformational Transitions. Structure 2011, 19, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martinez, M.; Crehuet, R. Application of the maximum entropy principle to determine ensembles of intrinsically disordered proteins from residual dipolar couplings. Phys. Chem. Chem. Phys. 2014, 16, 26030–26039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podbevšek, P.; Fasolo, F.; Bon, C.; Cimatti, L.; Reißer, S.; Carninci, P.; Bussi, G.; Zucchelli, S.; Plavec, J.; Gustincich, S. Structural determinants of the SINE B2 element embedded in the long non-coding RNA activator of translation AS Uchl1. Sci. Rep. 2018, 8, 3189. [Google Scholar] [CrossRef] [Green Version]
- Leung, H.T.A.; Bignucolo, O.; Aregger, R.; Dames, S.A.; Mazur, A.; Bernèche, S.; Grzesiek, S. A Rigorous and Efficient Method To Reweight Very Large Conformational Ensembles Using Average Experimental Data and To Determine Their Relative Information Content. J. Chem. Theory Comput. 2016, 12, 383–394. [Google Scholar] [CrossRef]
- Bradshaw, R.T.; Marinelli, F.; Faraldo-Gómez, J.D.; Forrest, L.R. Interpretation of HDX Data by Maximum-Entropy Reweighting of Simulated Structural Ensembles. Biophys. J. 2020, 118, 1649–1664. [Google Scholar] [CrossRef]
- Shen, T.; Hamelberg, D. A statistical analysis of the precision of reweighting-based simulations. J. Chem. Phys. 2008, 129, 034103. [Google Scholar] [CrossRef]
- Rangan, R.; Bonomi, M.; Heller, G.T.; Cesari, A.; Bussi, G.; Vendruscolo, M. Determination of Structural Ensembles of Proteins: Restraining vs Reweighting. J. Chem. Theory Comput. 2018, 14, 6632–6641. [Google Scholar] [CrossRef]
- Pitera, J.W.; Chodera, J.D. On the Use of Experimental Observations to Bias Simulated Ensembles. J. Chem. Theory Comput. 2012, 8, 3445–3451. [Google Scholar] [CrossRef]
- Cesari, A.; Gil-Ley, A.; Bussi, G. Combining Simulations and Solution Experiments as a Paradigm for RNA Force Field Refinement. J. Chem. Theory Comput. 2016, 12, 6192–6200. [Google Scholar] [CrossRef] [Green Version]
- Reißer, S.; Zucchelli, S.; Gustincich, S.; Bussi, G. Conformational ensembles of an RNA hairpin using molecular dynamics and sparse NMR data. Nucleic Acids Res. 2019, 48, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Fennen, J.; Torda, A.E.; van Gunsteren, W.F. Structure refinement with molecular dynamics and a Boltzmann-weighted ensemble. J. Biomol. NMR 1995, 6, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Vendruscolo, M. Determination of Protein Structures Consistent with NMR Order Parameters. J. Am. Chem. Soc. 2004, 126, 8090–8091. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Best, R.B.; DePristo, M.A.; Dobson, C.M.; Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Camilloni, C.; Vendruscolo, M. Molecular dynamics simulations with replica-averaged structural restraints generate structural ensembles according to the maximum entropy principle. J. Chem. Phys. 2013, 138, 094112. [Google Scholar] [CrossRef]
- Roux, B.; Weare, J. On the statistical equivalence of restrained-ensemble simulations with the maximum entropy method. J. Chem. Phys. 2013, 138, 084107. [Google Scholar] [CrossRef] [Green Version]
- Hermann, M.R.; Hub, J.S. SAXS-Restrained Ensemble Simulations of Intrinsically Disordered Proteins with Commitment to the Principle of Maximum Entropy. J. Chem. Theory Comput. 2019, 15, 5103–5115. [Google Scholar] [CrossRef]
- Bonomi, M.; Camilloni, C.; Cavalli, A.; Vendruscolo, M. Metainference: A Bayesian inference method for heterogeneous systems. Sci. Adv. 2016, 2, e1501177. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, M.; Camilloni, C.; Vendruscolo, M. Metadynamic metainference: Enhanced sampling of the metainference ensemble using metadynamics. Sci. Rep. 2016, 6, 31232. [Google Scholar] [CrossRef]
- Heller, G.T.; Aprile, F.A.; Bonomi, M.; Camilloni, C.; De Simone, A.; Vendruscolo, M. Sequence Specificity in the Entropy-Driven Binding of a Small Molecule and a Disordered Peptide. J. Mol. Biol. 2017, 429, 2772–2779. [Google Scholar] [CrossRef]
- Hultqvist, G.; Åberg, E.; Camilloni, C.; Sundell, G.N.; Andersson, E.; Dogan, J.; Chi, C.N.; Vendruscolo, M.; Jemth, P. Emergence and evolution of an interaction between intrinsically disordered proteins. eLife 2017, 6, e16059. [Google Scholar] [CrossRef] [PubMed]
- Buckle, E.L.; Prakash, A.; Bonomi, M.; Sampath, J.; Pfaendtner, J.; Drobny, G.P. Solid-State NMR and MD Study of the Structure of the Statherin Mutant SNa15 on Mineral Surfaces. J. Am. Chem. Soc. 2019, 141, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Hora, M.; Kazman, P.; Göbl, C.; Camilloni, C.; Reif, B.; Buchner, J. The Antibody Light-Chain Linker Regulates Domain Orientation and Amyloidogenicity. J. Mol. Biol. 2018, 430, 4925–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonomi, M.; Pellarin, R.; Vendruscolo, M. Simultaneous Determination of Protein Structure and Dynamics Using Cryo-Electron Microscopy. Biophys. J. 2018, 114, 1604–1613. [Google Scholar] [CrossRef]
- Vahidi, S.; Ripstein, Z.A.; Bonomi, M.; Yuwen, T.; Mabanglo, M.F.; Juravsky, J.B.; Rizzolo, K.; Velyvis, A.; Houry, W.A.; Vendruscolo, M.; et al. Reversible inhibition of the ClpP protease via an N-terminal conformational switch. Proc. Natl. Acad. Sci. USA 2018, 115, E6447. [Google Scholar] [CrossRef] [Green Version]
- Paissoni, C.; Jussupow, A.; Camilloni, C. Determination of Protein Structural Ensembles by Hybrid-Resolution SAXS Restrained Molecular Dynamics. J. Chem. Theory Comput. 2020, 16, 2825–2834. [Google Scholar] [CrossRef]
- Paissoni, C.; Jussupow, A.; Camilloni, C. Martini bead form factors for nucleic acids and their application in the refinement of protein-nucleic acid complexes against SAXS data. J. Appl. Crystallogr. 2019, 52, 394–402. [Google Scholar] [CrossRef]
- Kooshapur, H.; Choudhury, N.R.; Simon, B.; Mühlbauer, M.; Jussupow, A.; Fernandez, N.; Jones, A.N.; Dallmann, A.; Gabel, F.; Camilloni, C.; et al. Structural basis for terminal loop recognition and stimulation of pri-miRNA-18a processing by hnRNP A1. Nat. Commun. 2018, 9, 2479. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Learning Task | Method |
|---|---|---|
| Supervised Learning | Regression | Linear regression 1 Non-linear regression Support vector regression (SVR) Artificial neural network (ANN) |
| Classification | Logistic regression (LR) 1 Linear discriminant analysis (LDA) 1 Support vector machines (SVR) k-nearest neighbor (kNN) Decision trees/random forests Artificial neural network (ANN) | |
| Unsupervised Learning | Clustering | Hierarchical agglomerative/divisive k-means/-medoid Gaussian mixture models (GMM) 1 Density-based (DBSCAN) Self-organizing maps (SOM) |
| Dimensionality Reduction | Principal component analysis (PCA) 1 Kernel-PCA (kPCA) 1 Independent component analysis (ICA) 1 Multidimensional scaling (MDS) 1 Isometric feature mapping (IsoMap) 1 Locally linear embedding (LLE) Diffusion maps (dMaps) 1 Artificial neural network (ANN) 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernetti, M.; Bertazzo, M.; Masetti, M. Data-Driven Molecular Dynamics: A Multifaceted Challenge. Pharmaceuticals 2020, 13, 253. https://doi.org/10.3390/ph13090253
Bernetti M, Bertazzo M, Masetti M. Data-Driven Molecular Dynamics: A Multifaceted Challenge. Pharmaceuticals. 2020; 13(9):253. https://doi.org/10.3390/ph13090253
Chicago/Turabian StyleBernetti, Mattia, Martina Bertazzo, and Matteo Masetti. 2020. "Data-Driven Molecular Dynamics: A Multifaceted Challenge" Pharmaceuticals 13, no. 9: 253. https://doi.org/10.3390/ph13090253
APA StyleBernetti, M., Bertazzo, M., & Masetti, M. (2020). Data-Driven Molecular Dynamics: A Multifaceted Challenge. Pharmaceuticals, 13(9), 253. https://doi.org/10.3390/ph13090253