B Cells and Antibodies as Targets of Therapeutic Intervention in Neuromyelitis Optica Spectrum Disorders


Abstract
:1. Introduction
2. The Role of B Cells and Antibodies in NMOSD
3. Current and Evolving Therapeutic Strategies in NMOSD
3.1. Immunosuppression
3.1.1. Glucocorticoids
3.1.2. Azathioprine
3.1.3. Cyclophosphamide
3.1.4. Mitoxantrone
3.1.5. Mycophenolate Mofetil
3.1.6. Methotrexate
3.1.7. Intravenous Immunoglobulins
3.2. Anti-CD20 Antibodies
3.2.1. Rituximab
3.2.2. Ocrelizumab
3.2.3. Ofatumumab
3.2.4. Ublituximab
3.2.5. BAT4406F
3.3. Targeting CD19
3.3.1. Inebilizumab
3.3.2. Tandem Chimeric Antigen Receptors (CAR) T Cells Targeting CD19 and CD20
3.4. Brutons Tyrosine Kinase Inhibition
3.4.1. SHR1459
3.5. IL-6 Receptor Antagonism
3.5.1. Tocilizumab
3.5.2. Satralizumab
3.6. Complement Factor Antagonism
3.6.1. Eculizumab
3.6.2. Ravulizumab
3.7. Targeting Pathological AQP4 Antibodies
3.7.1. Aquaporumab
3.7.2. HBM9161
3.8. Other Therapeutic Strategies
3.8.1. Bevacizumab
3.8.2. Bortezomib
3.8.3. Cetirizine
3.8.4. Sivelestat
3.8.5. Telitacicept
3.8.6. C1-Esterase Inhibitor/Cinryze
3.8.7. Tolerogenic Dendritic Cells Loaded with Myelin Peptides
3.8.8. Hematopoietic Stem Cell Transplantation
3.8.9. Mesenchymal Stromal Cells
3.8.10. Dalfampridine
3.8.11. Alpha-1 Antitrypsin
4. Outlook
4.1. Calcineurin Inhibition
4.2. Targeting Eosinophils
4.2.1. Anti-IL5 Antibodies
4.2.2. Anti-IgE Antibodies
4.3. Blocking of Pathogenic Autoantibodies
4.4. Targeting the BAFF/APRIL System
4.5. Microbiota
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| APRIL | A proliferation-inducing ligand |
| AQP4 | Aquaporin 4 |
| BAFF | B cell activating factor |
| BAFFR | BAFF receptor |
| BBB | Blood-brain barrier |
| BCMA | B cell maturation antigen |
| BLyS | B lymphocyte stimulator |
| C1 | Complement factor 1 |
| C5 | Complement factor 5 |
| CAR | Chimeric antigen receptor |
| CD | Cluster of differentiation |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| EDSS | Expanded disability status scale |
| Fc | Fragment crystallizable |
| Ig | Immunoglobulin |
| IL | Interleukin |
| MOG | Myelin oligodendrocyte glycoprotein |
| MS | Multiple sclerosis |
| NMOSD | Neuromyelitis optica spectrum disorders |
| SLE | Systemic lupus erythematosus |
| TACI | Transmembrane activator and calcium-modulator and cyclophilin-ligand activator |
| TNF | Tumor necrosis factor |
References
- Prasad, S.; Chen, J. What you need to know about aqp4, mog, and nmosd. Semin. Neurol. 2019, 39, 718–731. [Google Scholar] [CrossRef]
- Jarius, S.; Wildemann, B. The history of neuromyelitis optica. J. Neuroinflamm. 2013, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Weber, M.S.; Derfuss, T.; Metz, I.; Bruck, W. Defining distinct features of anti-mog antibody associated central nervous system demyelination. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418762083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lana-Peixoto, M.A.; Talim, N. Neuromyelitis optica spectrum disorder and anti-mog syndromes. Biomedicines 2019, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.S.; Derfuss, T.; Bruck, W. Anti-myelin oligodendrocyte glycoprotein antibody-associated central nervous system demyelination-a novel disease entity? JAMA Neurol. 2018, 75, 909–910. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Franciotta, D.; Waters, P.; Zipp, F.; Hohlfeld, R.; Vincent, A.; Wildemann, B. Mechanisms of disease: Aquaporin-4 antibodies in neuromyelitis optica. Nat. Clin. Pract. Neurol. 2008, 4, 202–214. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Kinzel, S.; Weber, M.S. The role of peripheral cns-directed antibodies in promoting inflammatory cns demyelination. Brain Sci. 2017, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Wildemann, B.; Jarius, S.; Orellano, B.; Sasidharan, S.; Weber, M.S.; Stuve, O. Immunopathogenesis of neuromyelitis optica. Adv. Immunol. 2014, 121, 213–242. [Google Scholar] [PubMed]
- Takahashi, T.; Fujihara, K.; Nakashima, I.; Misu, T.; Miyazawa, I.; Nakamura, M.; Watanabe, S.; Shiga, Y.; Kanaoka, C.; Fujimori, J.; et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of nmo: A study on antibody titre. Brain 2007, 130, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausser-Kinzel, S.; Weber, M.S. The role of b cells and antibodies in multiple sclerosis, neuromyelitis optica, and related disorders. Front. Immunol. 2019, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann-Horn, K.; Kinzel, S.; Weber, M.S. Deciphering the role of b cells in multiple sclerosis-towards specific targeting of pathogenic function. Int. J. Mol. Sci. 2017, 18, 2048. [Google Scholar] [CrossRef] [Green Version]
- Kinzel, S.; Lehmann-Horn, K.; Torke, S.; Hausler, D.; Winkler, A.; Stadelmann, C.; Payne, N.; Feldmann, L.; Saiz, A.; Reindl, M.; et al. Myelin-reactive antibodies initiate t cell-mediated cns autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. 2016, 132, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, A.; Bergamaschi, R.; Martinelli, V.; Trojano, M.; Tola, M.R.; Merelli, E.; Mancardi, L.; Gallo, P.; Filippi, M.; Zaffaroni, M.; et al. Clinical characteristics, course and prognosis of relapsing devic’s neuromyelitis optica. J. Neurol. 2004, 251, 47–52. [Google Scholar] [CrossRef]
- Vincent, T.; Saikali, P.; Cayrol, R.; Roth, A.D.; Bar-Or, A.; Prat, A.; Antel, J.P. Functional consequences of neuromyelitis optica-igg astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J. Immunol. 2008, 181, 5730–5737. [Google Scholar] [CrossRef]
- Soltys, J.; Liu, Y.; Ritchie, A.; Wemlinger, S.; Schaller, K.; Schumann, H.; Owens, G.P.; Bennett, J.L. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J. Clin. Investig. 2019, 129, 2000–2013. [Google Scholar] [CrossRef] [Green Version]
- Mitsdoerffer, M.; Kuchroo, V.; Korn, T. Immunology of neuromyelitis optica: A t cell-b cell collaboration. Ann. N. Y. Acad. Sci. 2013, 1283, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of il-6-producing b cells. J. Exp. Med. 2012, 209, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. Il-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Pitarokoili, K.; Gold, R. Dimethyl fumarate for patients with neuromyelitis optica spectrum disorder. Mult. Scler. 2018, 24, 364–365. [Google Scholar] [CrossRef]
- Weber, M.S.; Hemmer, B. Cooperation of b cells and t cells in the pathogenesis of multiple sclerosis. Results Probl. Cell Differ. 2010, 51, 115–126. [Google Scholar]
- Molnarfi, N.; Schulze-Topphoff, U.; Weber, M.S.; Patarroyo, J.C.; Prod’homme, T.; Varrin-Doyer, M.; Shetty, A.; Linington, C.; Slavin, A.J.; Hidalgo, J.; et al. Mhc class ii-dependent b cell apc function is required for induction of cns autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 2013, 210, 2921–2937. [Google Scholar] [CrossRef]
- Sahraian, M.A.; Moghadasi, A.N.; Azimi, A.R.; Asgari, N.; Akhoundi, F.H.; Abolfazli, R.; Alaie, S.; Ashtari, F.; Ayromlou, H.; Baghbanian, S.M.; et al. Diagnosis and management of neuromyelitis optica spectrum disorder (nmosd) in iran: A consensus guideline and recommendations. Mult. Scler. Relat. Disord. 2017, 18, 144–151. [Google Scholar] [CrossRef]
- Traub, J.; Hausser-Kinzel, S.; Weber, M.S. Differential effects of ms therapeutics on b cells-implications for their use and failure in aqp4-positive nmosd patients. Int. J. Mol. Sci. 2020, 21, 5021. [Google Scholar] [CrossRef]
- Kinzel, S.; Weber, M.S. B cell-directed therapeutics in multiple sclerosis: Rationale and clinical evidence. CNS Drugs 2016, 30, 1137–1148. [Google Scholar] [CrossRef]
- Selmaj, K.; Selmaj, I. Novel emerging treatments for nmosd. Neurol. Neurochir. Pol. 2019, 53, 317–326. [Google Scholar] [CrossRef]
- Abboud, H.; Petrak, A.; Mealy, M.; Sasidharan, S.; Siddique, L.; Levy, M. Treatment of acute relapses in neuromyelitis optica: Steroids alone versus steroids plus plasma exchange. Mult. Scler. 2016, 22, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, C.L.; Wegner, C.; Sprenger, T.; Weber, M.S.; Bruck, W.; Hemmer, B.; Sellner, J. Favourable response to plasma exchange in tumefactive cns demyelination with delayed b-cell response. Mult. Scler. 2012, 18, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.T.; Yu, D.T.; Clements, P.J.; Fowlston, S.; Eisman, J.; Bluestone, R. Effect of corticosteroids on the human immune response: Comparison of one and three daily 1 gm intravenous pulses of methylprednisolone. J. Lab. Clin. Med. 1978, 91, 625–634. [Google Scholar] [PubMed]
- Fauci, A.S.; Dale, D.C.; Balow, J.E. Glucocorticosteroid therapy: Mechanisms of action and clinical considerations. Ann. Intern. Med. 1976, 84, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Olnes, M.J.; Kotliarov, Y.; Biancotto, A.; Cheung, F.; Chen, J.; Shi, R.; Zhou, H.; Wang, E.; Tsang, J.S.; Nussenblatt, R.; et al. Effects of systemically administered hydrocortisone on the human immunome. Sci. Rep. 2016, 6, 23002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slade, J.D.; Hepburn, B. Prednisone-induced alterations of circulating human lymphocyte subsets. J. Lab. Clin. Med. 1983, 101, 479–487. [Google Scholar]
- Lanzillotta, M.; Della-Torre, E.; Milani, R.; Bozzolo, E.; Bozzalla-Cassione, E.; Rovati, L.; Arcidiacono, P.G.; Partelli, S.; Falconi, M.; Ciceri, F.; et al. Effects of glucocorticoids on b-cell subpopulations in patients with igg4-related disease. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S118), 159–166. [Google Scholar]
- Lin, W.; Zhang, P.; Chen, H.; Chen, Y.; Yang, H.; Zheng, W.; Zhang, X.; Zhang, F.; Zhang, W.; Lipsky, P.E. Circulating plasmablasts/plasma cells: A potential biomarker for igg4-related disease. Arthritis Res. Ther. 2017, 19, 25. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; LaForge, K.S.; Sehgal, P.B. On the mechanism for efficient repression of the interleukin-6 promoter by glucocorticoids: Enhancer, tata box, and rna start site (inr motif) occlusion. Mol. Cell. Biol. 1990, 10, 5736–5746. [Google Scholar] [CrossRef] [Green Version]
- Settipane, G.A.; Pudupakkam, R.K.; McGowan, J.H. Corticosteroid effect on immunoglobulins. J. Allergy Clin. Immunol. 1978, 62, 162–166. [Google Scholar] [CrossRef]
- Lack, G.; Ochs, H.D.; Gelfand, E.W. Humoral immunity in steroid-dependent children with asthma and hypogammaglobulinemia. J. Pediatr. 1996, 129, 898–903. [Google Scholar] [CrossRef]
- Kamhieh-Milz, J.; Ghosoun, N.; Sterzer, V.; Salama, A. Effect of glucocorticoid treatment on baff and april expression in patients with immune thrombocytopenia (itp). Clin. Immunol. 2018, 188, 74–80. [Google Scholar] [CrossRef]
- Sellner, J.; Boggild, M.; Clanet, M.; Hintzen, R.Q.; Illes, Z.; Montalban, X.; Du Pasquier, R.A.; Polman, C.H.; Sorensen, P.S.; Hemmer, B. Efns guidelines on diagnosis and management of neuromyelitis optica. Eur. J. Neurol. 2010, 17, 1019–1032. [Google Scholar] [CrossRef]
- Kimbrough, D.J.; Fujihara, K.; Jacob, A.; Lana-Peixoto, M.A.; Leite, M.I.; Levy, M.; Marignier, R.; Nakashima, I.; Palace, J.; de Seze, J.; et al. Treatment of neuromyelitis optica: Review and recommendations. Mult. Scler. Relat. Disord. 2012, 1, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Elsone, L.; Kitley, J.; Luppe, S.; Lythgoe, D.; Mutch, K.; Jacob, S.; Brown, R.; Moss, K.; McNeillis, B.; Goh, Y.Y.; et al. Long-term efficacy, tolerability and retention rate of azathioprine in 103 aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder patients: A multicentre retrospective observational study from the UK. Mult. Scler. 2014, 20, 1533–1540. [Google Scholar] [CrossRef]
- Evans, W.E. Pharmacogenetics of thiopurine s-methyltransferase and thiopurine therapy. Ther. Drug Monit. 2004, 26, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Maltzman, J.S.; Koretzky, G.A. Azathioprine: Old drug, new actions. J. Clin. Investig. 2003, 111, 1122–1124. [Google Scholar] [CrossRef]
- Leandro, M.J.; Cambridge, G.; Edwards, J.C.; Ehrenstein, M.R.; Isenberg, D.A. B-cell depletion in the treatment of patients with systemic lupus erythematosus: A longitudinal analysis of 24 patients. Rheumatology 2005, 44, 1542–1545. [Google Scholar] [CrossRef] [Green Version]
- Hayry, P.; von Willebrand, E.; Ahonen, J.; Eklund, B.; Salmela, K.; Hockerstedt, K.; Pettersson, E.; Koskimies, S. Effects of cyclosporine, azathioprine, and steroids on the renal transplant, on the cytologic patterns of intragraft inflammation, and on concomitant rejection-associated changes in recipient blood. Transplant. Proc. 1988, 20, 153–162. [Google Scholar]
- Bottomley, M.J.; Chen, M.; Fuggle, S.; Harden, P.N.; Wood, K.J. Application of operational tolerance signatures are limited by variability and type of immunosuppression in renal transplant recipients: A cross-sectional study. Transplant. Direct 2017, 3, e125. [Google Scholar] [CrossRef]
- Thiel, J.; Salzer, U.; Hassler, F.; Effelsberg, N.M.; Hentze, C.; Sic, H.; Bartsch, M.; Miehle, N.; Peter, H.H.; Warnatz, K.; et al. B cell homeostasis is disturbed by immunosuppressive therapies in patients with anca-associated vasculitides. Autoimmunity 2013, 46, 429–438. [Google Scholar] [CrossRef]
- Roekevisch, E.; Szegedi, K.; Hack, D.P.; Schram, M.E.; Res, P.; Bos, J.D.; Leeflang, M.M.G.; Luiten, R.M.; Kezic, S.; Spuls, P.I.; et al. Effect of immunosuppressive treatment on biomarkers in adult atopic dermatitis patients. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Sokollik, C.; McLin, V.A.; Vergani, D.; Terziroli Beretta-Piccoli, B.; Mieli-Vergani, G. Juvenile autoimmune hepatitis: A comprehensive review. J. Autoimmun. 2018, 95, 69–76. [Google Scholar] [CrossRef]
- Hernandez-Breijo, B.; Gomez, A.; Soukka, S.; Johansson, P.; Parodis, I. Antimalarial agents diminish while methotrexate, azathioprine and mycophenolic acid increase baff levels in systemic lupus erythematosus. Autoimmun. Rev. 2019, 18, 102372. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Stuve, O. Cyclophosphamide in multiple sclerosis: Scientific rationale, history and novel treatment paradigms. Ther. Adv. Neurol. Disord. 2009, 2, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Yaguchi, H.; Sakushima, K.; Takahashi, I.; Nishimura, H.; Yashima-Yamada, M.; Nakamura, M.; Tsuzaka, K.; Maruo, Y.; Takahashi, T.; Yabe, I.; et al. Efficacy of intravenous cyclophosphamide therapy for neuromyelitis optica spectrum disorder. Intern. Med. 2013, 52, 969–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, C.C.; To, C.H.; Mak, A.; Poon, W.L. Immunoablative cyclophosphamide for refractory lupus-related neuromyelitis optica. J. Rheumatol. 2008, 35, 172–174. [Google Scholar]
- Torres, J.; Pruitt, A.; Balcer, L.; Galetta, S.; Markowitz, C.; Dahodwala, N. Analysis of the treatment of neuromyelitis optica. J. Neurol. Sci. 2015, 351, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Saida, T. [treatment of nmo]. Rinsho Shinkeigaku 2009, 49, 902–905. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.G.; Tilby, M.J. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 1992, 6, 163–173. [Google Scholar] [CrossRef]
- Brodsky, R.A. High-dose cyclophosphamide for autoimmunity and alloimmunity. Immunol. Res. 2010, 47, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.P.; Cupps, T.R.; Whalen, G.; Fauci, A.S. Selective effects of cyclophosphamide therapy on activation, proliferation, and differentiation of human b cells. J. Clin. Investig. 1987, 79, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Fassbinder, T.; Saunders, U.; Mickholz, E.; Jung, E.; Becker, H.; Schluter, B.; Jacobi, A.M. Differential effects of cyclophosphamide and mycophenolate mofetil on cellular and serological parameters in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorner, T.; Jacobi, A.M.; Lipsky, P.E. B cells in autoimmunity. Arthritis Res. Ther. 2009, 11, 247. [Google Scholar] [CrossRef] [Green Version]
- Moschella, F.; Torelli, G.F.; Valentini, M.; Urbani, F.; Buccione, C.; Petrucci, M.T.; Natalino, F.; Belardelli, F.; Foa, R.; Proietti, E. Cyclophosphamide induces a type i interferon-associated sterile inflammatory response signature in cancer patients’ blood cells: Implications for cancer chemoimmunotherapy. Clin. Cancer Res. 2013, 19, 4249–4261. [Google Scholar] [CrossRef] [Green Version]
- Trebst, C.; Jarius, S.; Berthele, A.; Paul, F.; Schippling, S.; Wildemann, B.; Borisow, N.; Kleiter, I.; Aktas, O.; Kumpfel, T.; et al. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the neuromyelitis optica study group (nemos). J. Neurol. 2014, 261, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Weinstock-Guttman, B.; Ramanathan, M.; Lincoff, N.; Napoli, S.Q.; Sharma, J.; Feichter, J.; Bakshi, R. Study of mitoxantrone for the treatment of recurrent neuromyelitis optica (devic disease). Arch. Neurol. 2006, 63, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kim, W.; Park, M.S.; Sohn, E.H.; Li, X.F.; Kim, H.J. Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch. Neurol. 2011, 68, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Cabre, P.; Olindo, S.; Marignier, R.; Jeannin, S.; Merle, H.; Smadja, D.; Aegis of French National Observatory of Multiple, S. Efficacy of mitoxantrone in neuromyelitis optica spectrum: Clinical and neuroradiological study. J. Neurol. Neurosurg. Psychiatry 2013, 84, 511–516. [Google Scholar] [CrossRef]
- Mazerski, J.; Martelli, S.; Borowski, E. The geometry of intercalation complex of antitumor mitoxantrone and ametantrone with DNA: Molecular dynamics simulations. Acta Biochim. Pol. 1998, 45, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, O.; Wiendl, H.; Kieseier, B.C.; Archelos, J.J.; Hemmer, B.; Stuve, O.; Hartung, H.P. Multiple sclerosis: Mitoxantrone promotes differential effects on immunocompetent cells in vitro. J. Neuroimmunol. 2005, 168, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Chanvillard, C.; Millward, J.M.; Lozano, M.; Hamann, I.; Paul, F.; Zipp, F.; Dorr, J.; Infante-Duarte, C. Mitoxantrone induces natural killer cell maturation in patients with secondary progressive multiple sclerosis. PLoS ONE 2012, 7, e39625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, J.M.; DeJoy, S.Q.; Gibbons, J.J., Jr. Selective immunomodulation by the antineoplastic agent mitoxantrone. I. Suppression of b lymphocyte function. J. Immunol. 1986, 137, 727–732. [Google Scholar]
- Fox, E.J. Mechanism of action of mitoxantrone. Neurology 2004, 63, S15–S18. [Google Scholar] [CrossRef] [PubMed]
- Kannel, K.; Alnek, K.; Vahter, L.; Gross-Paju, K.; Uibo, R.; Kisand, K.V. Changes in blood b cell-activating factor (baff) levels in multiple sclerosis: A sign of treatment outcome. PLoS ONE 2015, 10, e0143393. [Google Scholar] [CrossRef]
- Montcuquet, A.; Collongues, N.; Papeix, C.; Zephir, H.; Audoin, B.; Laplaud, D.; Bourre, B.; Brochet, B.; Camdessanche, J.P.; Labauge, P.; et al. Effectiveness of mycophenolate mofetil as first-line therapy in aqp4-igg, mog-igg, and seronegative neuromyelitis optica spectrum disorders. Mult. Scler. 2017, 23, 1377–1384. [Google Scholar] [CrossRef]
- Huh, S.Y.; Kim, S.H.; Hyun, J.W.; Joung, A.R.; Park, M.S.; Kim, B.J.; Kim, H.J. Mycophenolate mofetil in the treatment of neuromyelitis optica spectrum disorder. JAMA Neurol. 2014, 71, 1372–1378. [Google Scholar] [CrossRef]
- Walkiewicz-Pielaszek, K.; Swacha, M.; Bullo-Piontecka, B.; Rutkowski, B.; Olesinska, M. Mycophenolate mofetil--20 years of experience in treatment of rheumatic diseases. Postepy Hig. Med. Dosw. Online 2015, 69, 176–187. [Google Scholar]
- Allison, A.C.; Eugui, E.M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000, 47, 85–118. [Google Scholar] [CrossRef]
- Karnell, J.L.; Karnell, F.G., 3rd; Stephens, G.L.; Rajan, B.; Morehouse, C.; Li, Y.; Swerdlow, B.; Wilson, M.; Goldbach-Mansky, R.; Groves, C.; et al. Mycophenolic acid differentially impacts b cell function depending on the stage of differentiation. J. Immunol. 2011, 187, 3603–3612. [Google Scholar] [CrossRef] [Green Version]
- Eickenberg, S.; Mickholz, E.; Jung, E.; Nofer, J.R.; Pavenstadt, H.J.; Jacobi, A.M. Mycophenolic acid counteracts b cell proliferation and plasmablast formation in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2012, 14, R110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, M.S.; Kim, E.Y.; Park, H.J.; Chang, C.Y.; Park, K.S.; Jung, D.Y.; Kwon, C.H.; Joh, J.W.; Kim, S.J. Mycophenolate mofetil promotes down-regulation of expanded b cells and production of tnf-alpha in an experimental murine model of colitis. Cytokine 2008, 44, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kitley, J.; Elsone, L.; George, J.; Waters, P.; Woodhall, M.; Vincent, A.; Jacob, A.; Leite, M.I.; Palace, J. Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.S.; Malhotra, K.; Scott, T. Treatment of neuromyelitis optica/neuromyelitis optica spectrum disorders with methotrexate. BMC Neurol. 2014, 14, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beh, S.C.; Kildebeck, E.; Narayan, R.; Desena, A.; Schell, D.; Rowe, E.S.; Rowe, V.; Burns, D.; Whitworth, L.; Frohman, T.C.; et al. High-dose methotrexate with leucovorin rescue: For monumentally severe cns inflammatory syndromes. J. Neurol. Sci. 2017, 372, 187–195. [Google Scholar] [CrossRef]
- Rajagopalan, P.T.; Zhang, Z.; McCourt, L.; Dwyer, M.; Benkovic, S.J.; Hammes, G.G. Interaction of dihydrofolate reductase with methotrexate: Ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 13481–13486. [Google Scholar] [CrossRef] [Green Version]
- Fortea-Gordo, P.; Villalba, A.; Nuno, L.; Santos-Bornez, M.J.; Peiteado, D.; Monjo, I.; Puig-Kroger, A.; Sanchez-Mateos, P.; Martin-Mola, E.; Balsa, A.; et al. Circulating cd19+cd24hicd38hi regulatory b cells as biomarkers of response to methotrexate in early rheumatoid arthritis. Rheumatology 2020, 59, 3081–3091. [Google Scholar] [CrossRef]
- Aggarwal, A.; Misra, R. Methotrexate inhibits interleukin-6 production in patients with juvenile rheumatoid arthritis. Rheumatol. Int. 2003, 23, 134–137. [Google Scholar] [CrossRef]
- Hughes, R.A.; Donofrio, P.; Bril, V.; Dalakas, M.C.; Deng, C.; Hanna, K.; Hartung, H.P.; Latov, N.; Merkies, I.S.; van Doorn, P.A.; et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ice study): A randomised placebo-controlled trial. Lancet Neurol. 2008, 7, 136–144. [Google Scholar] [CrossRef]
- Magraner, M.J.; Coret, F.; Casanova, B. The effect of intravenous immunoglobulin on neuromyelitis optica. Neurologia 2013, 28, 65–72. [Google Scholar] [CrossRef]
- Viswanathan, S.; Wong, A.H.; Quek, A.M.; Yuki, N. Intravenous immunoglobulin may reduce relapse frequency in neuromyelitis optica. J. Neuroimmunol. 2015, 282, 92–96. [Google Scholar] [CrossRef]
- Li, X.; Tian, D.C.; Fan, M.; Xiu, Y.; Wang, X.; Li, T.; Jia, D.; Xu, W.; Song, T.; Shi, F.D.; et al. Intravenous immunoglobulin for acute attacks in neuromyelitis optica spectrum disorders (nmosd). Mult. Scler. Relat. Disord. 2020, 44, 102325. [Google Scholar] [CrossRef]
- Koffman, B.M.; Dalakas, M.C. Effect of high-dose intravenous immunoglobulin on serum chemistry, hematology, and lymphocyte subpopulations: Assessments based on controlled treatment trials in patients with neurological diseases. Muscle Nerve 1997, 20, 1102–1107. [Google Scholar] [CrossRef]
- Brem, M.D.; Jacobs, B.C.; van Rijs, W.; Fokkink, W.J.R.; Tio-Gillen, A.P.; Walgaard, C.; van Doorn, P.A.; Ijspeert, H.; van der Burg, M.; Huizinga, R. Ivig-induced plasmablasts in patients with guillain-barre syndrome. Ann. Clin. Transl. Neurol. 2019, 6, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Rigal, D.; Vermot-Desroches, C.; Heitz, S.; Bernaud, J.; Alfonsi, F.; Monier, J.C. Effects of intravenous immunoglobulins (ivig) on peripheral blood b, nk, and t cell subpopulations in women with recurrent spontaneous abortions: Specific effects on lfa-1 and cd56 molecules. Clin. Immunol. Immunopathol. 1994, 71, 309–314. [Google Scholar] [CrossRef]
- de Grandmont, M.J.; Racine, C.; Roy, A.; Lemieux, R.; Neron, S. Intravenous immunoglobulins induce the in vitro differentiation of human b lymphocytes and the secretion of igg. Blood 2003, 101, 3065–3073. [Google Scholar] [CrossRef] [Green Version]
- Kessel, A.; Peri, R.; Haj, T.; Snir, A.; Slobodin, G.; Sabo, E.; Rosner, I.; Shoenfeld, Y.; Toubi, E. Ivig attenuates tlr-9 activation in b cells from sle patients. J. Clin. Immunol. 2011, 31, 30–38. [Google Scholar] [CrossRef]
- Seite, J.F.; Guerrier, T.; Cornec, D.; Jamin, C.; Youinou, P.; Hillion, S. Tlr9 responses of b cells are repressed by intravenous immunoglobulin through the recruitment of phosphatase. J. Autoimmun. 2011, 37, 190–197. [Google Scholar] [CrossRef]
- Toyoda, M.; Zhang, X.M.; Petrosian, A.; Wachs, K.; Moudgil, A.; Jordan, S.C. Inhibition of allospecific responses in the mixed lymphocyte reaction by pooled human gamma-globulin. Transpl. Immunol. 1994, 2, 337–341. [Google Scholar] [CrossRef]
- Paquin Proulx, D.; Aubin, E.; Lemieux, R.; Bazin, R. Inhibition of b cell-mediated antigen presentation by intravenous immunoglobulins (ivig). Clin. Immunol. 2010, 135, 422–429. [Google Scholar] [CrossRef]
- Kabuto, M.; Fujimoto, N.; Tanaka, T. Increase of interleukin-10-producing b cells associated with long-term remission after i.V. Immunoglobulin treatment for pemphigus. J. Dermatol. 2016, 43, 815–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, N.; Mori, M.; Kobayashi, Y.; Miyamae, T.; Imagawa, T.; Okuyama, T.; Kurozumi, H.; Yokota, S. Intravenous gamma-globulin therapy improves hypercytokinemia in the acute phase of kawasaki disease. Mod. Rheumatol. 2004, 14, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Ozawa, T.; Mushiake, K.; Motoyoshi, F.; Kameyama, T.; Kasahara, K.; Kaneko, H.; Yamashina, M.; Kato, Y.; Orii, T. Suppression of immunoglobulin production of lymphocytes by intravenous immunoglobulin. J. Clin. Immunol. 1991, 11, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Mazer, B.D.; Gelfand, E.W. An open-label study of high-dose intravenous immunoglobulin in severe childhood asthma. J. Allergy Clin. Immunol. 1991, 87, 976–983. [Google Scholar] [CrossRef]
- Le Pottier, L.; Bendaoud, B.; Dueymes, M.; Daridon, C.; Youinou, P.; Shoenfeld, Y.; Pers, J.O. Baff, a new target for intravenous immunoglobulin in autoimmunity and cancer. J. Clin. Immunol. 2007, 27, 257–265. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Asavapanumas, N.; Phuan, P.W.; Verkman, A.S. Potential therapeutic benefit of c1-esterase inhibitor in neuromyelitis optica evaluated in vitro and in an experimental rat model. PLoS ONE 2014, 9, e106824. [Google Scholar] [CrossRef]
- Mealy, M.A.; Shin, K.; John, G.; Levy, M. Bevacizumab is safe in acute relapses of neuromyelitis optica. Clin. Exp. Neuroimmunol. 2015, 6, 413–418. [Google Scholar] [CrossRef]
- Mealy, M.A.; Levy, M. A pilot safety study of ublituximab, a monoclonal antibody against cd20, in acute relapses of neuromyelitis optica spectrum disorder. Medicine 2019, 98, e15944. [Google Scholar] [CrossRef]
- Florez-Grau, G.; Zubizarreta, I.; Cabezon, R.; Villoslada, P.; Benitez-Ribas, D. Tolerogenic dendritic cells as a promising antigen-specific therapy in the treatment of multiple sclerosis and neuromyelitis optica from preclinical to clinical trials. Front. Immunol. 2018, 9, 1169. [Google Scholar] [CrossRef] [Green Version]
- Burt, R.K.; Balabanov, R.; Han, X.; Burns, C.; Gastala, J.; Jovanovic, B.; Helenowski, I.; Jitprapaikulsan, J.; Fryer, J.P.; Pittock, S.J. Autologous nonmyeloablative hematopoietic stem cell transplantation for neuromyelitis optica. Neurology 2019, 93, e1732–e1741. [Google Scholar] [CrossRef]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Katz Sand, I.; Fabian, M.T.; Telford, R.; Kraus, T.A.; Chehade, M.; Masilamani, M.; Moran, T.; Farrell, C.; Ebel, S.; Cook, L.J.; et al. Open-label, add-on trial of cetirizine for neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, K.; Wymbs, N.F.; Huang, H.; Mealy, M.A.; Pardo, C.A.; Zackowski, K.; Levy, M. Randomized, placebo-controlled crossover study of dalfampridine extended-release in transverse myelitis. Mult. Scler. J. Exp. Transl. Clin. 2017, 3, 2055217317740145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tian, D.C.; Yang, C.S.; Han, B.; Wang, J.; Yang, L.; Shi, F.D. Safety and efficacy of bortezomib in patients with highly relapsing neuromyelitis optica spectrum disorder. JAMA Neurol. 2017, 74, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Fujihara, K.; Paul, F.; Cutter, G.R.; Marignier, R.; et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (n-momentum): A double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019, 394, 1352–1363. [Google Scholar] [CrossRef]
- Nikoo, Z.; Badihian, S.; Shaygannejad, V.; Asgari, N.; Ashtari, F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: A randomized clinical trial. J. Neurol. 2017, 264, 2003–2009. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Qiu, W.; Ma, H.; Zhang, X.; Zhu, Z.; Yang, C.S.; Jia, D.; Zhang, T.X.; Yuan, M.; et al. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (tango): An open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020, 19, 391–401. [Google Scholar] [CrossRef]
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.P.; Saiz, A.; Yamazaki, H.; et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N. Engl. J. Med. 2019, 381, 2114–2124. [Google Scholar] [CrossRef]
- Traboulsee, A.; Greenberg, B.M.; Bennett, J.L.; Szczechowski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Wright, P.; et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: A randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020, 19, 402–412. [Google Scholar] [CrossRef]
- Absoud, M.; Brex, P.; Ciccarelli, O.; Diribe, O.; Giovannoni, G.; Hellier, J.; Howe, R.; Holland, R.; Kelly, J.; McCrone, P.; et al. A multicentre randomised controlled trial of intravenous immunoglobulin compared with standard therapy for the treatment of transverse myelitis in adults and children (strive). Health Technol. Assess. 2017, 21, 1–50. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Wang, J.; Zhou, Y.; Yang, H.; Wang, Z.; Yan, Z.; Long, Y.; Yin, J.; Feng, H.; Li, C.; et al. Low-dose mycophenolate mofetil for treatment of neuromyelitis optica spectrum disorders: A prospective multicenter study in south china. Front. Immunol. 2018, 9, 2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinley, M.P.; Moss, B.P.; Cohen, J.A. Safety of monoclonal antibodies for the treatment of multiple sclerosis. Expert Opin. Drug Saf. 2017, 16, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Cabre, P.; Mejdoubi, M.; Jeannin, S.; Merle, H.; Plumelle, Y.; Cavillon, G.; Smadja, D.; Marignier, R.; Francophone Society of Multiple, S.; investigators, O. Treatment of neuromyelitis optica with rituximab: A 2-year prospective multicenter study. J. Neurol. 2018, 265, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Alldredge, B.; Jordan, A.; Imitola, J.; Racke, M.K. Safety and efficacy of rituximab: Experience of a single multiple sclerosis center. Clin. Neuropharmacol. 2018, 41, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chai, B.; Gu, C.; Wu, R.; Dong, T.; Yao, Y.; Zhang, Y. Effectiveness of rituximab in neuromyelitis optica: A meta-analysis. BMC Neurol. 2019, 19, 36. [Google Scholar] [CrossRef] [Green Version]
- Ellrichmann, G.; Bolz, J.; Peschke, M.; Duscha, A.; Hellwig, K.; Lee, D.H.; Linker, R.A.; Gold, R.; Haghikia, A. Peripheral cd19(+) b-cell counts and infusion intervals as a surrogate for long-term b-cell depleting therapy in multiple sclerosis and neuromyelitis optica/neuromyelitis optica spectrum disorders. J. Neurol. 2019, 266, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Milo, R. Therapies for multiple sclerosis targeting b cells. Croat. Med. J. 2019, 60, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Jakimovski, D.; Weinstock-Guttman, B.; Ramanathan, M.; Kolb, C.; Hojnacki, D.; Minagar, A.; Zivadinov, R. Ocrelizumab: A b-cell depleting therapy for multiple sclerosis. Expert Opin. Biol. Ther. 2017, 17, 1163–1172. [Google Scholar] [CrossRef]
- Nissimov, N.; Hajiyeva, Z.; Torke, S.; Grondey, K.; Bruck, W.; Hausser-Kinzel, S.; Weber, M.S. B cells reappear less mature and more activated after their anti-cd20-mediated depletion in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 25690–25699. [Google Scholar] [CrossRef]
- Hausler, D.; Hausser-Kinzel, S.; Feldmann, L.; Torke, S.; Lepennetier, G.; Bernard, C.C.A.; Zamvil, S.S.; Bruck, W.; Lehmann-Horn, K.; Weber, M.S. Functional characterization of reappearing b cells after anti-cd20 treatment of cns autoimmune disease. Proc. Natl. Acad. Sci. USA 2018, 115, 9773–9778. [Google Scholar] [CrossRef] [Green Version]
- Vallerskog, T.; Heimburger, M.; Gunnarsson, I.; Zhou, W.; Wahren-Herlenius, M.; Trollmo, C.; Malmstrom, V. Differential effects on baff and april levels in rituximab-treated patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenstein, M.R.; Wing, C. The baffling effects of rituximab in lupus: Danger ahead? Nat. Rev. Rheumatol. 2016, 12, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ning, Q.; Jin, K.; Xie, J.; Ye, J. Does rituximab improve clinical outcomes of patients with thyroid-associated ophthalmopathy? A systematic review and meta-analysis. BMC Ophthalmol. 2018, 18, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann-Horn, K.; Schleich, E.; Hertzenberg, D.; Hapfelmeier, A.; Kumpfel, T.; von Bubnoff, N.; Hohlfeld, R.; Berthele, A.; Hemmer, B.; Weber, M.S. Anti-cd20 b-cell depletion enhances monocyte reactivity in neuroimmunological disorders. J. Neuroinflamm. 2011, 8, 146. [Google Scholar] [CrossRef] [Green Version]
- Dorner, T.; Burmester, G.R. New approaches of b-cell-directed therapy: Beyond rituximab. Curr. Opin. Rheumatol. 2008, 20, 263–268. [Google Scholar] [CrossRef]
- Soe, Z.N.; Allsup, D. The use of ofatumumab in the treatment of b-cell malignancies. Future Oncol. 2017, 13, 2611–2628. [Google Scholar] [CrossRef]
- Bar-Or, A.; Grove, R.A.; Austin, D.J.; Tolson, J.M.; VanMeter, S.A.; Lewis, E.W.; Derosier, F.J.; Lopez, M.C.; Kavanagh, S.T.; Miller, A.E.; et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: The mirror study. Neurology 2018, 90, e1805–e1814. [Google Scholar] [CrossRef]
- Kurrasch, R.; Brown, J.C.; Chu, M.; Craigen, J.; Overend, P.; Patel, B.; Wolfe, S.; Chang, D.J. Subcutaneously administered ofatumumab in rheumatoid arthritis: A phase i/ii study of safety, tolerability, pharmacokinetics, and pharmacodynamics. J. Rheumatol. 2013, 40, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.A.; Hummert, M.W.; et al. Mog-igg in nmo and related disorders: A multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J. Neuroinflamm. 2016, 13, 280. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.; Lovett-Racke, A.E.; Gormley, M.; Liu, Y.; Petracca, M.; Cocozza, S.; Shubin, R.; Wray, S.; Weiss, M.S.; Bosco, J.A.; et al. A phase 2 multicenter study of ublituximab, a novel glycoengineered anti-cd20 monoclonal antibody, in patients with relapsing forms of multiple sclerosis. Mult. Scler. 2020. [Google Scholar] [CrossRef]
- Chen, D.; Gallagher, S.; Monson, N.L.; Herbst, R.; Wang, Y. Inebilizumab, a b cell-depleting anti-cd19 antibody for the treatment of autoimmune neurological diseases: Insights from preclinical studies. J. Clin. Med. 2016, 5, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, J.E. Inebilizumab: First approval. Drugs 2020, 80, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; Bargou, R.C.; Nagorsen, D.; Friberg, G.R.; Baeuerle, P.A.; Forman, S.J. Redirecting t cells to eradicate b-cell acute lymphoblastic leukemia: Bispecific t-cell engagers and chimeric antigen receptors. Leukemia 2017, 31, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Xu, Q.; Pu, C.; Zhu, K.; Lu, C.; Jiang, Y.; Xiao, L.; Han, Y.; Lu, L. Therapeutic efficacy of anti-cd19 car-t cells in a mouse model of systemic lupus erythematosus. Cell. Mol. Immunol. 2020. [CrossRef]
- Corneth, O.B.J.; Klein Wolterink, R.G.J.; Hendriks, R.W. Btk signaling in b cell differentiation and autoimmunity. Curr. Top. Microbiol. Immunol. 2016, 393, 67–105. [Google Scholar] [PubMed]
- Deeks, E.D. Ibrutinib: A review in chronic lymphocytic leukaemia. Drugs 2017, 77, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Dhillon, S. Acalabrutinib: First global approval. Drugs 2018, 78, 139–145. [Google Scholar] [CrossRef]
- Syed, Y.Y. Zanubrutinib: First approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-controlled trial of an oral btk inhibitor in multiple sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef]
- Ayzenberg, I.; Kleiter, I.; Schroder, A.; Hellwig, K.; Chan, A.; Yamamura, T.; Gold, R. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-cd20 therapy. JAMA Neurol. 2013, 70, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Uchida, T.; Mori, M.; Uzawa, A.; Masuda, H.; Muto, M.; Ohtani, R.; Kuwabara, S. Increased cerebrospinal fluid metalloproteinase-2 and interleukin-6 are associated with albumin quotient in neuromyelitis optica: Their possible role on blood-brain barrier disruption. Mult. Scler. 2017, 23, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Naka, T.; Kishimoto, T. Il-6-dependent and -independent pathways in the development of interleukin 17-producing t helper cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12099–12104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, M.; Matsuoka, T.; Miyamoto, K.; Kusunoki, S.; Okamoto, T.; Murata, M.; Miyake, S.; Aranami, T.; Yamamura, T. Efficacy of the anti-il-6 receptor antibody tocilizumab in neuromyelitis optica: A pilot study. Neurology 2014, 82, 1302–1306. [Google Scholar] [CrossRef] [Green Version]
- Ringelstein, M.; Ayzenberg, I.; Harmel, J.; Lauenstein, A.S.; Lensch, E.; Stogbauer, F.; Hellwig, K.; Ellrichmann, G.; Stettner, M.; Chan, A.; et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015, 72, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Ectrims 2019 committees. Mult. Scler. 2019, 25, 1–2. [CrossRef]
- Moura, R.A.; Quaresma, C.; Vieira, A.R.; Goncalves, M.J.; Polido-Pereira, J.; Romao, V.C.; Martins, N.; Canhao, H.; Fonseca, J.E. B-cell phenotype and igd-cd27- memory b cells are affected by tnf-inhibitors and tocilizumab treatment in rheumatoid arthritis. PLoS ONE 2017, 12, e0182927. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xiao, D.M.; Qin, W.; Xie, B.H.; Wang, T.H.; Huang, H.; Zhao, B.J.; Han, X.; Sun, Q.Q.; Wu, X.D.; et al. The clinical value of hematological markers in rheumatoid arthritis patients treated with tocilizumab. J. Clin. Lab. Anal. 2019, 33, e22862. [Google Scholar] [CrossRef]
- Roll, P.; Muhammad, K.; Schumann, M.; Kleinert, S.; Einsele, H.; Dorner, T.; Tony, H.P. In vivo effects of the anti-interleukin-6 receptor inhibitor tocilizumab on the b cell compartment. Arthritis Rheum. 2011, 63, 1255–1264. [Google Scholar] [CrossRef]
- Choi, I.A.; Lee, S.J.; Park, W.; Park, S.H.; Shim, S.C.; Baek, H.J.; Yoo, D.H.; Kim, H.A.; Lee, S.K.; Lee, Y.J.; et al. Effects of tocilizumab therapy on serum interleukin-33 and interleukin-6 levels in patients with rheumatoid arthritis. Arch. Rheumatol. 2018, 33, 389–394. [Google Scholar] [CrossRef]
- Igawa, T.; Ishii, S.; Tachibana, T.; Maeda, A.; Higuchi, Y.; Shimaoka, S.; Moriyama, C.; Watanabe, T.; Takubo, R.; Doi, Y.; et al. Antibody recycling by engineered ph-dependent antigen binding improves the duration of antigen neutralization. Nat. Biotechnol. 2010, 28, 1203–1207. [Google Scholar] [CrossRef]
- Heo, Y.A. Satralizumab: First approval. Drugs 2020, 80, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Brachet, G.; Bourquard, T.; Gallay, N.; Reiter, E.; Gouilleux-Gruart, V.; Poupon, A.; Watier, H. Eculizumab epitope on complement c5: Progress towards a better understanding of the mechanism of action. Mol. Immunol. 2016, 77, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Eculizumab: A review in generalized myasthenia gravis. Drugs 2018, 78, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, J.E. Eculizumab: A review in neuromyelitis optica spectrum disorder. Drugs 2020, 80, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, F.; Murphy, O.; Pardo, S.; Levy, M. Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin. Investig. Drugs 2018, 27, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Weitz, I.C.; Razavi, P.; Rochanda, L.; Zwicker, J.; Furie, B.; Manly, D.; Mackman, N.; Green, R.; Liebman, H.A. Eculizumab therapy results in rapid and sustained decreases in markers of thrombin generation and inflammation in patients with pnh independent of its effects on hemolysis and microparticle formation. Thromb. Res. 2012, 130, 361–368. [Google Scholar] [CrossRef]
- Alfinito, F.; Ruggiero, G.; Sica, M.; Udhayachandran, A.; Rubino, V.; Della Pepa, R.; Palatucci, A.T.; Annunziatella, M.; Notaro, R.; Risitano, A.M.; et al. Eculizumab treatment modifies the immune profile of pnh patients. Immunobiology 2012, 217, 698–703. [Google Scholar] [CrossRef]
- Sheridan, D.; Yu, Z.X.; Zhang, Y.; Patel, R.; Sun, F.; Lasaro, M.A.; Bouchard, K.; Andrien, B.; Marozsan, A.; Wang, Y.; et al. Design and preclinical characterization of alxn1210: A novel anti-c5 antibody with extended duration of action. PLoS ONE 2018, 13, e0195909. [Google Scholar] [CrossRef] [Green Version]
- Ravulizumab. In Drugs and Lactation Database (Lactmed); National Library of Medicine (US): Bethesda, MD, USA, 2006.
- Verkman, A.S.; Phuan, P.W.; Asavapanumas, N.; Tradtrantip, L. Biology of aqp4 and anti-aqp4 antibody: Therapeutic implications for nmo. Brain Pathol. 2013, 23, 684–695. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.S.; Smith, A.J.; Phuan, P.W.; Tradtrantip, L.; Anderson, M.O. The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin. Ther. Targets 2017, 21, 1161–1170. [Google Scholar] [CrossRef]
- Duan, T.; Tradtrantip, L.; Phuan, P.W.; Bennett, J.L.; Verkman, A.S. Affinity-matured ‘aquaporumab’ anti-aquaporin-4 antibody for therapy of seropositive neuromyelitis optica spectrum disorders. Neuropharmacology 2020, 162, 107827. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M.; Rossiter, D.; Fehmi, J.; Stevens, J.C.; Renowden, S.A.; Cohen, N.; Bailey, C.; Scolding, N.J. Tumefactive demyelination presenting during bevacizumab treatment. BMJ Case Rep. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker-Caulfield, M.E.; Guo, Y.; Johnson, R.K.; McCarthy, C.B.; Fitz-Gibbon, P.D.; Lucchinetti, C.F.; Howe, C.L. Nfkappab signaling drives pro-granulocytic astroglial responses to neuromyelitis optica patient igg. J. Neuroinflamm. 2015, 12, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H. Bortezomib for neuromyelitis optica spectrum disorder: A new therapeutic option for the more severe forms? JAMA Neurol. 2018, 75, 128–129. [Google Scholar] [CrossRef]
- Zhang, H.; Verkman, A.S. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J. Clin. Investig. 2013, 123, 2306–2316. [Google Scholar] [CrossRef]
- Muroishi, T.; Sakai, K.; Yanase, D.; Ikeda, Y.; Machiya, T.; Kato-Motozaki, Y.; Samuraki, M.; Yamada, M. Serum anti-aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder presenting as acute eosinophilic encephalomyelitis. J. Clin. Neurosci. 2018, 48, 93–94. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; MacDonald, C.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin g-induced damage in mouse brain. Ann. Neurol. 2012, 71, 323–333. [Google Scholar] [CrossRef]
- Herges, K.; de Jong, B.A.; Kolkowitz, I.; Dunn, C.; Mandelbaum, G.; Ko, R.M.; Maini, A.; Han, M.H.; Killestein, J.; Polman, C.; et al. Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult. Scler. 2012, 18, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Paul, F.; Franciotta, D.; Ruprecht, K.; Ringelstein, M.; Bergamaschi, R.; Rommer, P.; Kleiter, I.; Stich, O.; Reuss, R.; et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: Results from 211 lumbar punctures. J. Neurol. Sci. 2011, 306, 82–90. [Google Scholar] [CrossRef]
- Vincent, F.B.; Morand, E.F.; Schneider, P.; Mackay, F. The baff/april system in sle pathogenesis. Nat. Rev. Rheumatol. 2014, 10, 365–373. [Google Scholar] [CrossRef]
- Okada, K.; Matsushita, T.; Kira, J.; Tsuji, S. B-cell activating factor of the tnf family is upregulated in neuromyelitis optica. Neurology 2010, 74, 177–178. [Google Scholar] [CrossRef] [PubMed]
- 2019 acr/arp annual meeting abstract supplement. Arthritis Rheumatol. 2019, 71 (Suppl. S10), 1–5362. [CrossRef] [PubMed]
- Chamberlain, J.L.; Huda, S.; Whittam, D.H.; Matiello, M.; Morgan, B.P.; Jacob, A. Role of complement and potential of complement inhibitors in myasthenia gravis and neuromyelitis optica spectrum disorders: A brief review. J. Neurol. 2019. [Google Scholar] [CrossRef]
- Levy, M.; Mealy, M.A. Purified human c1-esterase inhibitor is safe in acute relapses of neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- Hakobyan, S.; Luppe, S.; Evans, D.R.; Harding, K.; Loveless, S.; Robertson, N.P.; Morgan, B.P. Plasma complement biomarkers distinguish multiple sclerosis and neuromyelitis optica spectrum disorder. Mult. Scler. 2017, 23, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Ha, S.J. Generation of tolerogenic dendritic cells and their therapeutic applications. Immune Netw. 2016, 16, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase i (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burman, J.; Tolf, A.; Hagglund, H.; Askmark, H. Autologous haematopoietic stem cell transplantation for neurological diseases. J. Neurol. Neurosurg. Psychiatry 2018, 89, 147–155. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, B. Effect of autologous hematopoietic stem cell transplantation on multiple sclerosis and neuromyelitis optica spectrum disorder: A prisma-compliant meta-analysis. Bone Marrow Transplant. 2020, 55, 1928–1934. [Google Scholar] [CrossRef]
- Ceglie, G.; Papetti, L.; Valeriani, M.; Merli, P. Hematopoietic stem cell transplantation in neuromyelitis optica-spectrum disorders (nmo-sd): State-of-the-art and future perspectives. Int. J. Mol. Sci. 2020, 21, 5304. [Google Scholar] [CrossRef]
- Corcione, A.; Benvenuto, F.; Ferretti, E.; Giunti, D.; Cappiello, V.; Cazzanti, F.; Risso, M.; Gualandi, F.; Mancardi, G.L.; Pistoia, V.; et al. Human mesenchymal stem cells modulate b-cell functions. Blood 2006, 107, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Ye, D.; Qian, L.; Zhu, L.; Wang, C.; Guan, D.; Zhang, X.; Xu, Y. Human umbilical cord mesenchymal stem cell therapy on neuromyelitis optica. Curr. Neurovasc. Res. 2012, 9, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Dulamea, A.O.; Sirbu-Boeti, M.P.; Bleotu, C.; Dragu, D.; Moldovan, L.; Lupescu, I.; Comi, G. Autologous mesenchymal stem cells applied on the pressure ulcers had produced a surprising outcome in a severe case of neuromyelitis optica. Neural. Regen. Res. 2015, 10, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Korsen, M.; Kunz, R.; Schminke, U.; Runge, U.; Kohlmann, T.; Dressel, A. Dalfampridine effects on cognition, fatigue, and dexterity. Brain Behav. 2017, 7, e00559. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, X.; Liu, X.; Mu, W.; Yang, W.; Liu, Y.; Ge, P.; Li, H. Patient with neuromyelitis optica spectrum disorder combined with sjogren’s syndrome relapse free following tacrolimus treatment. Intern. Med. 2014, 53, 2377–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Wu, Q.; Ke, G.; Bu, B. Efficacy and safety of tacrolimus treatment for neuromyelitis optica spectrum disorder. Sci. Rep. 2017, 7, 831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, M.; Oji, S.; Tanaka, S.; Izaki, S.; Hashimoto, B.; Fukaura, H.; Nomura, K. Tacrolimus is effective for neuromyelitis optica spectrum disorders with or without anti-aqp4 antibody. Mult. Scler. Relat. Disord. 2019, 39, 101907. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, C.; Frati, F.; Ridolo, E.; Greco, A.; de Vincentiis, M.; Masieri, S.; Makri, E.; Incorvaia, C. The spectrum of therapeutic activity of mepolizumab. Expert Rev. Clin. Immunol. 2019, 15, 959–967. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Ratelade, J.; Zhang, H.; Verkman, A.S. Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti-aquaporin-4 immunoglobulin g into therapeutic antibody. Ann. Neurol. 2013, 73, 77–85. [Google Scholar] [CrossRef]
- Kappos, L.; Hartung, H.P.; Freedman, M.S.; Boyko, A.; Radu, E.W.; Mikol, D.D.; Lamarine, M.; Hyvert, Y.; Freudensprung, U.; Plitz, T.; et al. Atacicept in multiple sclerosis (atams): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014, 13, 353–363. [Google Scholar] [CrossRef]
- Hoffman, R.W.; Merrill, J.T.; Alarcon-Riquelme, M.M.; Petri, M.; Dow, E.R.; Nantz, E.; Nisenbaum, L.K.; Schroeder, K.M.; Komocsar, W.J.; Perumal, N.B.; et al. Gene expression and pharmacodynamic changes in 1,760 systemic lupus erythematosus patients from two phase iii trials of baff blockade with tabalumab. Arthritis Rheumatol. 2017, 69, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A.; Duggan, S.T. Belimumab: A review in systemic lupus erythematosus. Drugs 2018, 78, 355–366. [Google Scholar] [CrossRef]
- Dorner, T.; Posch, M.G.; Li, Y.; Petricoul, O.; Cabanski, M.; Milojevic, J.M.; Kamphausen, E.; Valentin, M.A.; Simonett, C.; Mooney, L.; et al. Treatment of primary sjogren’s syndrome with ianalumab (vay736) targeting b cells by baff receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann. Rheum. Dis. 2019, 78, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Varrin-Doyer, M.; Spencer, C.M.; Schulze-Topphoff, U.; Nelson, P.A.; Stroud, R.M.; Cree, B.A.; Zamvil, S.S. Aquaporin 4-specific t cells in neuromyelitis optica exhibit a th17 bias and recognize clostridium abc transporter. Ann. Neurol. 2012, 72, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541. [Google Scholar] [CrossRef] [PubMed]


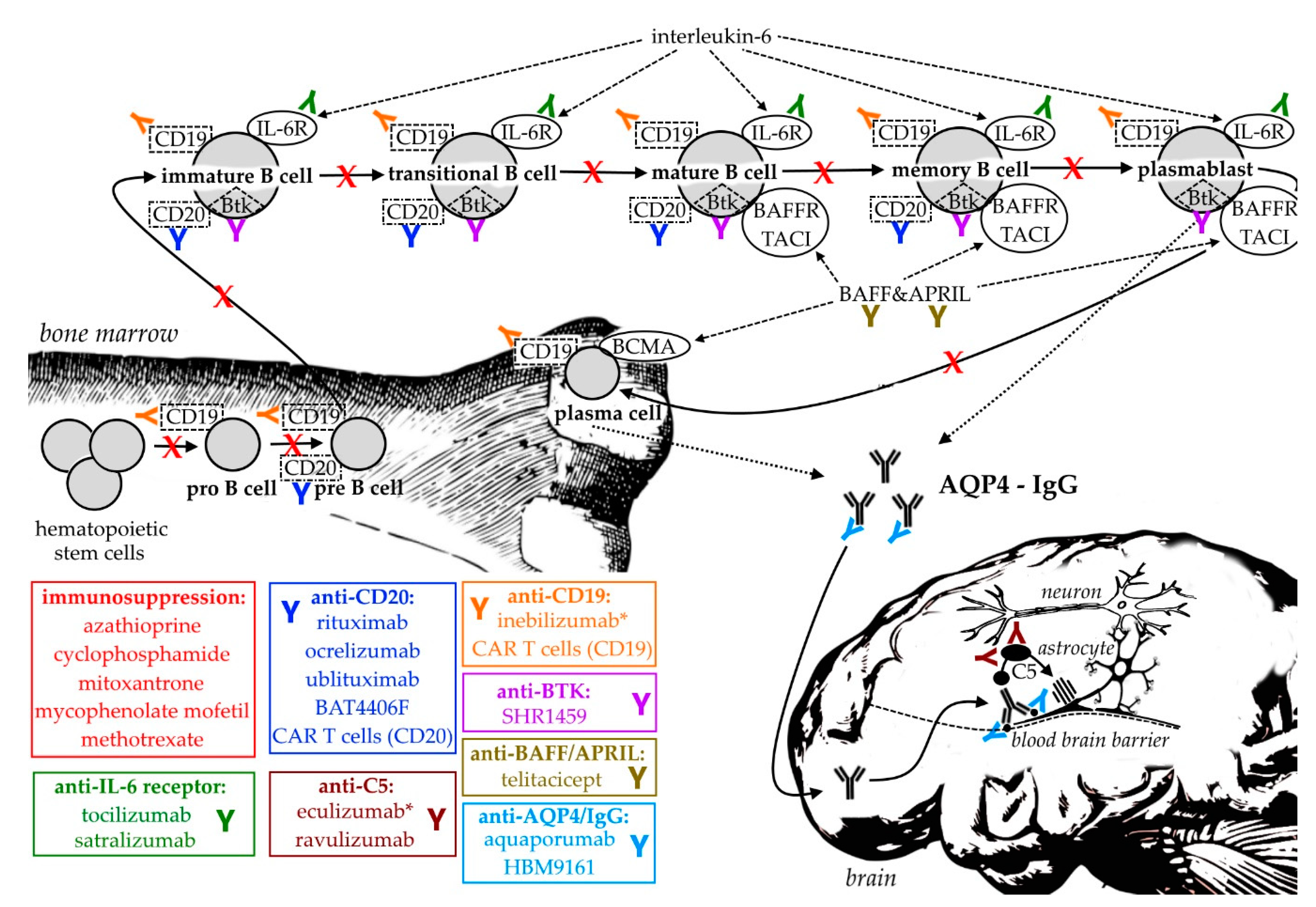{kind=link}
| Clinical Trials.gov Identifier | Phase | Year Study Started | Year Approved | Intervention | Target | Participants | Status (12/2020) | Results | Section |
|---|---|---|---|---|---|---|---|---|---|
| Phase 1 | |||||||||
| NCT00501748 | 1 | 2004 | n/a | Rituximab | CD20 | 20 | completed | n/a | Section 3.2.1 |
| NCT01759602 | 1 | 2013 | n/a | C1 esterase inhibitor | C1 esterase | 10 | completed | [106] | Section 3.8.6 |
| NCT01777412 | 1 | 2013 | n/a | Bevacizumab | VEGF | 10 | completed | [107] | Section 3.8.1 |
| NCT02087813 | 1 | 2014 | n/a | Alpha1-antitrypsin | n/a | 0 | withdrawn | n/a | Section 3.8.11 |
| NCT02276963 | 1 | 2016 | n/a | Ublituximab | CD20 | 6 | completed | [108] | Section 3.2.4 |
| NCT02283671 | 1 | 2015 | n/a | Tolerogenic dendritic cells | n/a | 10 | completed | [109] | Section 3.8.7 |
| NCT03605238 | 1 | 2018 | n/a | Tandem CAR T cells | CD19/CD20 | 0 | withdrawn | n/a | Section 3.3.2 |
| NCT04146285 | 1 | 2019 | n/a | BAT4406F | CD20 | 48 | not yet recruiting | n/a | Section 3.2.5 |
| NCT04227470 | 1 | 2020 | n/a | HBM9161 | CD20 | 12 | recruiting | n/a | Section 3.7.2 |
| Phase 1/2 | |||||||||
| NCT00787722 | 1/2 | 2009 | n/a | Autologous stem cells | n/a | 13 | completed | [110] | Section 3.8.8 |
| NCT00904826 | 1/2 | 2009 | 2019 | Eculizumab | C5 | 14 | completed | [111] | Section 3.6.1 |
| NCT01339455 | 1/2 | 2011 | n/a | Autologous stem cells | n/a | 3 | terminated | n/a | Section 3.8.8 |
| NCT01364246 | 1/2 | 2010 | n/a | Mesenchymal stromal cells | n/a | 20 | unknown | n/a | Section 3.8.9 |
| NCT02865018 | 1/2 | 2014 | n/a | Cetirizine | H1 | 16 | completed | [112] | Section 3.8.3 |
| NCT03062579 | 1/2 | 2017 | n/a | Tocilizumab | IL-6 receptor | 10 | completed | n/a | Section 3.5.1 |
| Phase 2 | |||||||||
| NCT00716066 | 2 | 2008 | n/a | Autologous stem cells | n/a | 40 | recruiting | n/a | Section 3.8.8 |
| NCT01845584 | 2 | 2013 | n/a | Intravenous IgG | n/a | 7 | completed | n/a | Section 3.1.6 |
| NCT02166346 | 2 | 2014 | n/a | Dalfampridine | K+ channel | 24 | completed | [113] | Section 3.8.10 |
| NCT02249676 | 2 | 2013 | n/a | Mesenchymal stromal cells | n/a | 15 | completed | n/a | Section 3.8.9 |
| NCT02893111 | 2 | 2015 | n/a | Bortezomib | proteasome | 5 | completed | [114] | Section 3.8.2 |
| NCT04064944 | 2 | 2019 | n/a | Immunoabsorption | n/a | 144 | not yet recruiting | n/a | Section 3.1.1 |
| NCT04670770 | 2 | 2020 | n/a | SHR1459 | BTK | 10 | not yet recruiting | n/a | Section 3.4.1 |
| Phase 2/3 | |||||||||
| NCT02200770 | 2/3 | 2015 | 2020 | Inebilizumab | CD19 | 231 | active, not recruiting | [115] | Section 3.3.1 |
| NCT03002038 | 2/3 | 2015 | n/a | Rituximab | CD20 | 76 | completed | [116] | Section 3.2.1 |
| NCT03350633 | 2/3 | 2017 | n/a | Tocilizumab | IL-6 receptor | 118 | completed | [117] | Section 3.5.1 |
| NCT03829566 | 2/3 | 2019 | n/a | Autologous stem cells | n/a | 0 | withdrawn | n/a | Section 3.8.8 |
| NCT04155424 | 2/3 | 2020 | 2019 | Eculizumab | C5 | 15 | recruiting | n/a | Section 3.6.1 |
| Phase 3 | |||||||||
| NCT00004645 | 3 | 1995 | n/a | Plasma exchange | n/a | 22 | unknown | n/a | Section 3.1.1 |
| NCT01892345 | 3 | 2014 | 2019 | Eculizumab | C5 | 143 | terminated | [111] | Section 3.6.1 |
| NCT02003144 | 3 | 2015 | 2019 | Eculizumab | C5 | 119 | active, not recruiting | n/a | Section 3.6.1 |
| NCT02028884 | 3 | 2014 | n/a | Satralizumab | IL-6 receptor | 83 | active, not recruiting | [118] | Section 3.5.2 |
| NCT02073279 | 3 | 2014 | n/a | Satralizumab | IL-6 receptor | 95 | active, not recruiting | [119] | Section 3.5.2 |
| NCT02398994 | 3 | 2015 | n/a | Intravenous IgG | n/a | 2 | terminated | [120] | Section 3.1.6 |
| NCT03330418 | 3 | 2017 | n/a | Telitacicept | APRIL/BAFF | 118 | recruiting | n/a | Section 3.8.5 |
| NCT04201262 | 3 | 2019 | n/a | Ravulizumab | C5 | 55 | recruiting | n/a | Section 3.6.2 |
| NCT04660539 | 3 | 2021 | n/a | Satralizumab | IL-6 receptor | 127 | not yet recruituing | n/a | Section 3.5.2 |
| Phase 4 | |||||||||
| NCT00304291 | 4 | 2001 | n/a | Mitoxantrone | n/a | 5 | completed | [67] | Section 3.1.4 |
| NCT02021825 | 4 | 2009 | n/a | Mitoxantrone | n/a | 50 | unknown | n/a | Section 3.1.4 |
| NCT02809079 | 4 | 2016 | n/a | Mycophenolate mofetil | n/a | 100 | unknown | [121] | Section 3.1.5 |
| NCT04256252 | 4 | 2014 | n/a | Rituximab | CD20 | 100 | terminated | n/a | Section 3.2.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traub, J.; Husseini, L.; Weber, M.S. B Cells and Antibodies as Targets of Therapeutic Intervention in Neuromyelitis Optica Spectrum Disorders. Pharmaceuticals 2021, 14, 37. https://doi.org/10.3390/ph14010037
Traub J, Husseini L, Weber MS. B Cells and Antibodies as Targets of Therapeutic Intervention in Neuromyelitis Optica Spectrum Disorders. Pharmaceuticals. 2021; 14(1):37. https://doi.org/10.3390/ph14010037
Chicago/Turabian StyleTraub, Jan, Leila Husseini, and Martin S. Weber. 2021. "B Cells and Antibodies as Targets of Therapeutic Intervention in Neuromyelitis Optica Spectrum Disorders" Pharmaceuticals 14, no. 1: 37. https://doi.org/10.3390/ph14010037
APA StyleTraub, J., Husseini, L., & Weber, M. S. (2021). B Cells and Antibodies as Targets of Therapeutic Intervention in Neuromyelitis Optica Spectrum Disorders. Pharmaceuticals, 14(1), 37. https://doi.org/10.3390/ph14010037