Drug Conjugated and Bispecific Antibodies for Multiple Myeloma: Improving Immunotherapies off the Shelf


Abstract
:1. Immunotherapy in Multiple Myeloma
- Agents that reverse tumor-related immune paralysis like immunomodulatory drugs (IMIds) and checkpoint inhibitors; these strategies act as immune booster and enhance the natural anticancer immune response;
- Agents directly targeting neoplastic cells including monoclonal antibodies and antibodies-drug conjugates (ADCs); this strategy represents a targeted therapy acting as passive immunotherapy since they can directly kill neoplastic cells and not always require an intact patient’s cellular immunity to exert their antitumoral properties;
- Agents directly activating the immune system against neoplastic cells like Chimeric Antigen Receptor expressing T-cell (CAR-T), Bispecific T cell redirecting antibodies (BsAbs), and peptide-based vaccines [10].
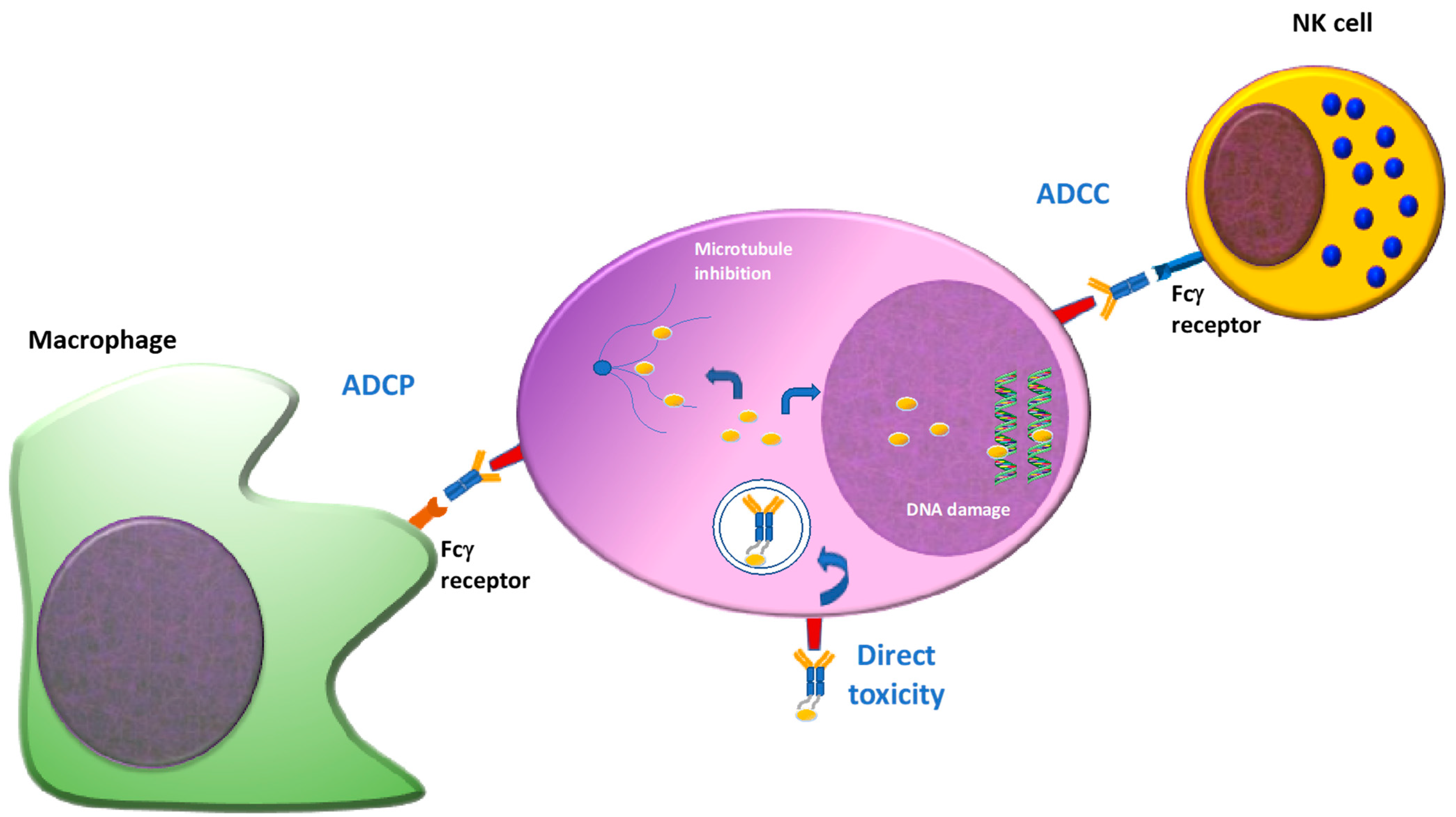
2. Antibodies Drug Conjugated
3. Antigen Target of ADC(s)
3.1. B Cell Maturation Antigen (BCMA)
3.2. CD74
3.3. CD46
3.4. SLAMF7/CS1
4. ADC(s) in Clinical Trials
4.1. Anti BCMA ADC(s)
4.1.1. Belantamab Mafodotin (Belamaf)
4.1.2. AMG224
4.1.3. MEDI2228 (M2)
4.1.4. CC-99712
4.1.5. HDP-101
4.2. ADC(s) Addressing Different Target from BCMA
4.2.1. STRO-001
4.2.2. FOR46
4.2.3. ABBV-838
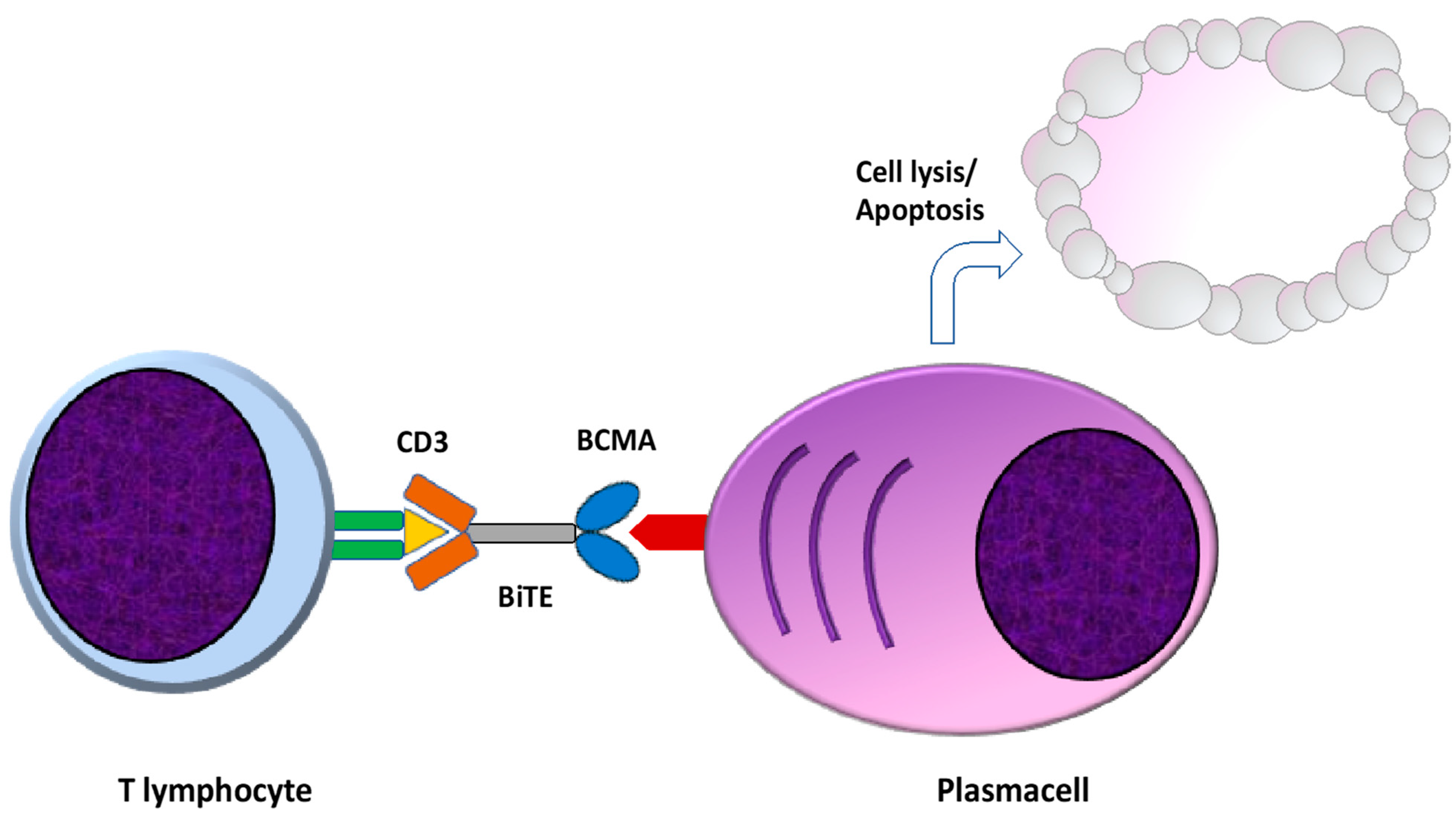
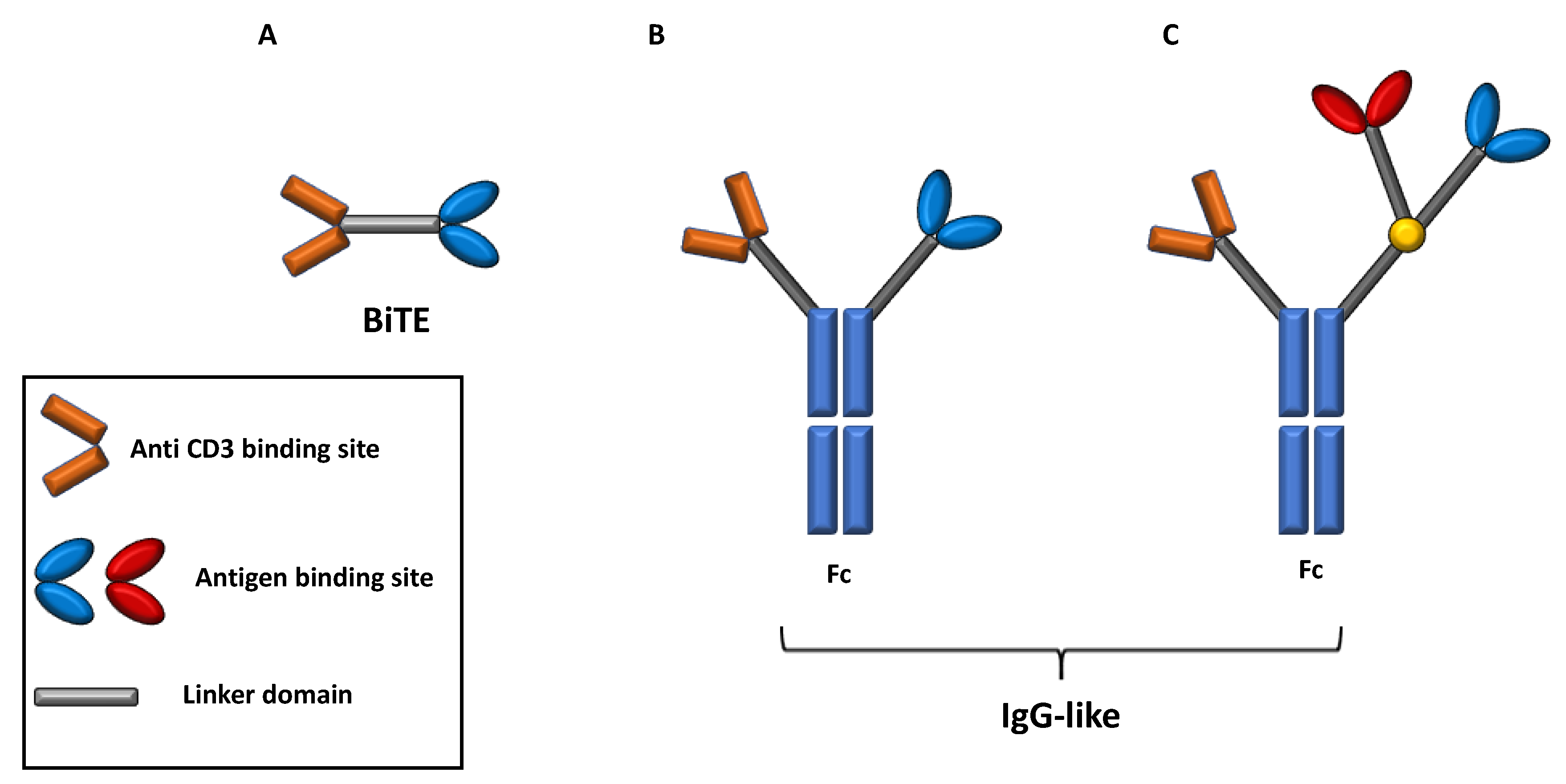
5. Bispecific Monoclonal Antibodies: Design and Mechanism of Action
5.1. BCMA Targeting Bispecific Monoclonal Antibodies
5.1.1. AMG420
5.1.2. AMG701
5.1.3. CC-93269
5.1.4. Teclistamab
5.1.5. REGN5458
5.1.6. TNB-383B
5.2. Bispecific Antibodies Addressing Different Targets from BCMA
5.2.1. BFCR4350A
5.2.2. Talquetamab
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of myeloma and their uses in immunotherapies. Blood Cancer J. 2015, 5, e306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillerey, C.; Nakamura, K.; Vuckovic, S.; Hill, G.R.; Smyth, M.J. Immune responses in multiple myeloma: Role of the natural immune surveillance and potential of immunotherapies. Cell Mol. Life Sci. 2016, 73, 1569–1589. [Google Scholar] [CrossRef]
- Minnie, S.A.; Hill, G.R. Immunotherapy of multiple myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef]
- Zeiser, R.; Finke, J. Allogeneic haematopoietic cell transplantation for multiple myeloma: Reducing transplant-related mortality while harnessing the graft-versus-myeloma effect. Eur. J. Cancer 2006, 42, 1601–1611. [Google Scholar] [CrossRef]
- Gay, F.; Engelhardt, M.; Terpos, E.; Wasch, R.; Giaccone, L.; Auner, H.W.; Caers, J.; Gramatzki, M.; van de Donk, N.; Oliva, S.; et al. From transplant to novel cellular therapies in multiple myeloma: European Myeloma Network guidelines and future perspectives. Haematologica 2018, 103, 197–211. [Google Scholar] [CrossRef]
- Gandolfi, S.; Prada, C.P.; Richardson, P.G. How I treat the young patient with multiple myeloma. Blood 2018, 132, 1114–1124. [Google Scholar] [CrossRef]
- Neri, P.; Bahlis, N.J.; Lonial, S. New strategies in multiple myeloma: Immunotherapy as a novel approach to treat patients with multiple myeloma. Clin. Cancer Res. 2016, 22, 5959–5965. [Google Scholar] [CrossRef] [Green Version]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Yu, B.; Liu, D. Antibody-drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J. Hematol. Oncol. 2019, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blattler, W.A.; Goldmacher, V.S. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef] [Green Version]
- Herrera, A.F.; Molina, A. Investigational antibody-drug conjugates for treatment of B-lineage malignancies. Clin. Lymphoma Myeloma Leuk. 2018, 18, 452–468 e454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolska-Washer, A.; Robak, T. Safety and tolerability of antibody-drug conjugates in cancer. Drug Saf. 2019, 42, 295–314. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Zhao, H.; Atkinson, J.; Gulesserian, S.; Zeng, Z.; Nater, J.; Ou, J.; Yang, P.; Morrison, K.; Coleman, J.; Malik, F.; et al. Modulation of macropinocytosis-mediated internalization decreases ocular toxicity of antibody-drug conjugates. Cancer Res. 2018, 78, 2115–2126. [Google Scholar] [CrossRef] [Green Version]
- Schonfeld, K.; Zuber, C.; Pinkas, J.; Hader, T.; Bernoster, K.; Uherek, C. Indatuximab ravtansine (BT062) combination treatment in multiple myeloma: Pre-clinical studies. J. Hematol. Oncol. 2017, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Ailawadhi, S.; Kelly, K.R.; Vescio, R.A.; Jagannath, S.; Wolf, J.; Gharibo, M.; Sher, T.; Bojanini, L.; Kirby, M.; Chanan-Khan, A. A phase I study to assess the safety and pharmacokinetics of single-agent lorvotuzumab mertansine (IMGN901) in patients with relapsed and/or refractory CD-56-positive multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 29–34. [Google Scholar] [CrossRef]
- Willert, E.K.; Robinson, G.L.; Higgins, J.P.; Liu, J.; Lee, J.; Syed, S.; Zhang, Y.; Tavares, D.; Lublinsky, A.; Chattopadhyay, N.; et al. TAK-169, an exceptionally potent CD38 targeted engineered toxin body, as a novel direct cell kill approach for the treatment of multiple myeloma. Cancer Res. 2019, 79, 2384. [Google Scholar]
- Abrahams, C.L.; Li, X.; Embry, M.; Yu, A.; Krimm, S.; Krueger, S.; Greenland, N.Y.; Wen, K.W.; Jones, C.; DeAlmeida, V.; et al. Targeting CD74 in multiple myeloma with the novel, site-specific antibody-drug conjugate STRO-001. Oncotarget 2018, 9, 37700–37714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherbenou, D.W.; Aftab, B.T.; Su, Y.; Behrens, C.R.; Wiita, A.; Logan, A.C.; Acosta-Alvear, D.; Hann, B.C.; Walter, P.; Shuman, M.A.; et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J. Clin. Investig. 2016, 126, 4640–4653. [Google Scholar] [CrossRef]
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, E.; Tanenbaum, E.J.; Patil, S.; Li, M.; Soof, C.M.; Vidisheva, A.; Waterman, G.N.; Hekmati, T.; Tang, G.; Wang, C.S.; et al. The clinical significance of B-cell maturation antigen as a therapeutic target and biomarker. Expert Rev. Mol. Diagn. 2018, 18, 319–329. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Lemancewicz, D.; Bolkun, L.; Jablonska, E.; Kulczynska, A.; Bolkun-Skornicka, U.; Kloczko, J.; Dzieciol, J. Evaluation of TNF superfamily molecules in multiple myeloma patients: Correlation with biological and clinical features. Leuk. Res. 2013, 37, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, A.; Hassan, H.; Iftikhar, N.; Mushtaq, A.; Sohail, A.; Rosko, N.; Chakraborty, R.; Razzaq, F.; Sandeep, S.; Valent, J.N.; et al. Investigational monoclonal antibodies in the treatment of multiple myeloma: A systematic review of agents under clinical development. Antibodies 2019, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Bonello, F.; Mina, R.; Boccadoro, M.; Gay, F. Therapeutic monoclonal antibodies and antibody products: Current practices and development in multiple myeloma. Cancers 2019, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef] [Green Version]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.A.; Lonial, S.; White, D.; Moreau, P.; Palumbo, A.; San-Miguel, J.; Shpilberg, O.; Anderson, K.; Grosicki, S.; Spicka, I.; et al. Elotuzumab plus lenalidomide/dexamethasone for relapsed or refractory multiple myeloma: ELOQUENT-2 follow-up and post-hoc analyses on progression-free survival and tumour growth. Br. J. Haematol. 2017, 178, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus pomalidomide and dexamethasone for multiple myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Anderson, L.D., Jr.; Sutherland, H.J.; Yong, K.; et al. Targeting B-cell maturation antigen with GSK2857916 antibody-drug conjugate in relapsed or refractory multiple myeloma (BMA117159): A dose escalation and expansion phase 1 trial. Lancet Oncol. 2018, 19, 1641–1653. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Weisel, J.; Hopkins, T.G.; Fecteau, D.; Bao, W.; Quigley, C.; Jewell, R.C.; Nichols, M.; Opalinska, J. Dreamm-3: A phase 3, open-label, randomized study to evaluate the efficacy and safety of belantamab mafodotin (GSK2857916) monotherapy compared with pomalidomide plus low-dose dexamethasone (Pom/Dex) in participants with relapsed/refractory multiple myeloma (RRMM). Blood 2019, 134, 1900. [Google Scholar]
- Richardson, P.G.; Biswas, S.; Holkova, B.; Jackson, N.; Netherway, T.; Bao, W.; Ferron-Brady, G.; Yeakey, A.; Shelton, C.; Montes De Oca, R.; et al. Dreamm-5: Platform trial evaluating belantamab mafodotin (a BCMA-directed Immuno-conjugate) in combination with novel agents in relapsed or refractory multiple myeloma (RRMM). Blood 2019, 134, 1857. [Google Scholar] [CrossRef]
- Lee, H.C.; Raje, N.S.; Landgren, O.; Upreti, V.V.; Wang, J.; Avilion, A.A.; Hu, X.; Rasmussen, E.; Ngarmchamnanrith, G.; Fujii, H.; et al. Phase 1 study of the anti-BCMA antibody-drug conjugate AMG 224 in patients with relapsed/refractory multiple myeloma. Leukemia 2021, 35, 255–258. Available online: https://www.nature.com/articles/s41375-020-0834-9 (accessed on 9 December 2020). [CrossRef] [PubMed]
- Kinneer, K.; Flynn, M.; Thomas, S.B.; Meekin, J.; Varkey, R.; Xiao, X.; Zhong, H.; Breen, S.; Hynes, P.G.; Fleming, R.; et al. Preclinical assessment of an antibody-PBD conjugate that targets BCMA on multiple myeloma and myeloma progenitor cells. Leukemia 2019, 33, 766–771. [Google Scholar] [CrossRef]
- Xing, L.; Lin, L.; Yu, T.; Li, Y.; Cho, S.F.; Liu, J.; Wen, K.; Hsieh, P.A.; Kinneer, K.; Munshi, N.; et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia 2020, 34, 2150–2162. [Google Scholar] [CrossRef]
- Kumar, S.K.; Migkou, M.; Bhutani, M.; Spencer, A.; Ailawadhi, S.; Kalff, A.; Walcott, F.; Pore, N.; Gibson, D.; Wang, F.; et al. Phase 1, first-in-human study of MEDI2228, a BCMA-targeted ADC in patients with relapsed/refractory multiple myeloma. In Proceedings of the 62nd ASH annual Meeting and Exposition, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Singh, R.K.; Jones, R.J.; Hong, S.; Shirazi, F.; Wang, H.; Kuiatse, I.; Pahl, A.; Orlowski, R.Z. HDP101, a novel B-cell maturation antigen (BCMA)-targeted antibody conjugated to α-Amanitin, is active against myeloma with preferential efficacy against pre-clinical models of deletion 17p. Blood 2018, 132, 593. [Google Scholar] [CrossRef]
- Shah, N.N.; Krishnan, A.Y.; Shah, N.D.; Burke, J.M.; Melear, J.M.; Spira, A.I.; Popplewell, L.L.; Andreadis, C.B.; Chhabra, S.; Sharman, J.P.; et al. Preliminary results of a phase 1 dose escalation study of the first-in-class anti-CD74 antibody drug conjugate (ADC), STRO-001, in patients with advanced B-cell malignancies. Blood 2019, 134, 5329. [Google Scholar] [CrossRef]
- Vij, R.; Nath, R.; Afar, D.E.H.; Mateos, M.V.; Berdeja, J.G.; Raab, M.S.; Guenther, A.; Martinez-Lopez, J.; Jakubowiak, A.J.; Leleu, X.; et al. First-in-human phase I study of ABBV-838, an antibody-drug conjugate targeting SLAMF7/CS1 in patients with relapsed and refractory multiple myeloma. Clin. Cancer Res. 2020, 26, 2308–2317. [Google Scholar] [CrossRef] [Green Version]
- Goebeler, M.E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- Caraccio, C.; Krishna, S.; Phillips, D.J.; Schurch, C.M. Bispecific antibodies for multiple myeloma: A review of targets, drugs, clinical trials, and future directions. Front. Immunol. 2020, 11, 501. [Google Scholar] [CrossRef]
- Dahlen, E.; Veitonmaki, N.; Norlen, P. Bispecific antibodies in cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2018, 6, 3–17. [Google Scholar] [CrossRef]
- Appleman, L.J.; Boussiotis, V.A. T cell anergy and costimulation. Immunol. Rev. 2003, 192, 161–180. [Google Scholar] [CrossRef]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef]
- Lejeune, M.; Kose, M.C.; Duray, E.; Einsele, H.; Beguin, Y.; Caers, J. Bispecific, T-cell-recruiting antibodies in B-cell Malignancies. Front. Immunol. 2020, 11, 762. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Klinger, M.; Brandl, C.; Zugmaier, G.; Hijazi, Y.; Bargou, R.C.; Topp, M.S.; Gokbuget, N.; Neumann, S.; Goebeler, M.; Viardot, A.; et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012, 119, 6226–6233. [Google Scholar] [CrossRef]
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific antibody based therapeutics: Strengths and challenges. Blood Rev. 2018, 32, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Einsele, H.; Rasche, L.; Topp, M.S.; Martin Kortum, K.; Duell, J. The use of bispecific antibodies to optimize the outcome of patients with acute leukemia, lymphoma and multiple myeloma after SCT. Bone Marrow Transplant. 2019, 54, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Porter, D.L. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Khadka, R.H.; Sakemura, R.; Kenderian, S.S.; Johnson, A.J. Management of cytokine release syndrome: An update on emerging antigen-specific T cell engaging immunotherapies. Immunotherapy 2019, 11, 851–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Kronke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.F.; Lin, L.; Xing, L.; Liu, J.; Yu, T.; Wen, K.; Hsieh, P.; Munshi, N.; Anderson, K.; Tai, Y.T. Anti-BCMA BiTE® AMG 701 potently induces specific T cell lysis of human multiple myeloma (MM) cells and immunomodulation in the bone marrow microenvironment. Blood 2018, 132, 592. [Google Scholar] [CrossRef]
- Harrison, J.S.; Minnema, C.M.; Lee, H.C.; Spencer, A.; Kapoor, P.; Madduri, D.; Larsen, J.; Ailawadhi, S.; Kaufman, J.L.; Raab, M.S.; et al. A phase 1 first in human (FIH) study of AMG 701, an anti-B-cell maturation antigen (BCMA) half-life extended (HLE) BiTE® (bispecific T-cell engager) molecule, in relapsed/refractory (RR) multiple myeloma (MM). In Proceedings of the 62nd ASH Annual Meeting and Exposition, Virtual Online, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; de la Rubia, J.; Mateos, M.V.; Ocio, E.O.; Rodríguez-Otero, P.; San-Miguel, J.; Li, S.; Sarmiento, R.; et al. First clinical study of the B-cell maturation antigen (BCMA) 2+1 T Cell engager (TCE) CC-93269 in patients (Pts) with relapsed/refractory multiple myeloma (RRMM): Interim results of a phase 1 multicenter trial. Blood 2019, 134, 143. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Powers, G.; Luistro, L.; Babich, A.; Baldwin, E.; Li, Y.; Zhang, X.; Mendonca, M.; Majewski, N.; Nanjunda, R.; et al. Teclistamab is an active T cell-redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020, 4, 4538–4549. [Google Scholar] [CrossRef]
- Garfall, A.; Usmani, S.Z.; Mateos, M.V.; Nahi, H.; Van De Donk, N.W.C.J.; San-Miguel, J.F.; Oriol Rocafiguera, A.; Rosinol, L.; Chari, A.; Bhutani, M.; et al. Updated phase 1 results of teclistamab, a B-cell maturation antigen (BCMA)×CD3 Bispecific antibody, in relapsed and/or refractory multiple myeloma (RRMM). In Proceedings of the 62nd ASH Annual Meeting and Exposition, Virtual Online, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Madduri, D.; Rosko, A.; Brayer, J.; Zonder, J.; Bensinger, W.I.; Li, J.; Xu, L.; Adriaens, L.; Chokshi, D.; Zhang, W.; et al. REGN5458, a BCMA × CD3 bispecific monoclonal antibody, induces deep and durable responses in patients with relapsed/refractory multiple myeloma (RRMM). In Proceedings of the 62nd ASH Annual Meeting and Exposition, Virtual Online, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Rodriguez, C.; D’Souza, A.; Shah, N.; Voorhees, P.M.; Buelow, B.; Vij, R.; Kumar, S.K. Initial results of a phase I study of TNB-383B, a BCMA × CD3 bispecific T-cell redirecting antibody, in relapsed/refractory multiple myeloma. In Proceedings of the 62nd ASH Annual Meeting and Exposition, Virtual Online, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Cohen, A.D.; Harrison, S.J.; Krishnan, A.; Fonseca, R.; Forsberg, P.A.; Spencer, A.; Berdeja, J.G.; Laubach, J.P.; Li, M.; Choeurng, V.; et al. Initial clinical activity and safety of BFCR4350A, a FcRH5/CD3 T-cell-engaging bispecific antibody, in relapsed/refractory multiple myeloma. In Proceedings of the 62nd ASH Annual Meeting and Exposition, Virtual Online, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Pillarisetti, K.; Edavettal, S.; Mendonca, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-cell-redirecting bispecific G-protein-coupled receptor class 5 member D × CD3 antibody to treat multiple myeloma. Blood 2020, 135, 1232–1243. [Google Scholar] [CrossRef]
- Chari, A.; Berdeja, J.G.; Oriol, A.; Van De Donk, N.W.C.J.; Rodriguez, P.; Askari, A.; Mateos, M.V.; Minnema, M.C.; Verona, R.; Girgis, S.; et al. A Phase 1, first-in-human Study of talquetamab, a G protein-coupled receptor family C group 5 member D (GPRC5D) × CD3 bispecific antibody, in patients with relapsed and/or refractory multiple myeloma (RRMM). In Proceedings of the 62nd ASH Annual Meeting and Exposition, Virtual Online, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Wudhikarn, K.; Mailankody, S.; Smith, E.L. Future of CAR T cells in multiple myeloma. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 272–279. [Google Scholar] [CrossRef]
- Bruins, W.S.C.; Zweegman, S.; Mutis, T.; van de Donk, N. Targeted therapy with immunoconjugates for multiple myeloma. Front. Immunol. 2020, 11, 1155. [Google Scholar] [CrossRef]
- Lancman, G.; Richter, J.; Chari, A. Bispecifics, trispecifics, and other novel immune treatments in myeloma. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 264–271. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | Target | References |
|---|---|---|---|
| Belantamab mafodotin | monomethyl auristatin-F ADC | BCMA | [34,35,36] |
| AMG224 | Mertansine ADC | BCMA | [39] |
| MEDI2228 | Pyrrolobenzodiazepine ADC | BCMA | [41,42] |
| CC-99712 | Undisclosed ADC | BCMA | |
| HDP-101 | Amanitin ADC | BCMA | [43] |
| STRO-001 | Maytansinoid ADC | CD74 | [44] |
| FOR46 | monomethyl auristatin-F ADC | CD46 | |
| ABBV-838 | monomethyl auristatin-E ADC | SLAMF7/CS1 | [45] |
| AMG420 | BCMA × CD3 BiTe | BCMA | [58] |
| AMG701 | BCMA × CD3 BiTe | BCMA | [59,60] |
| CC-93269 | BCMA × CD3 trivalent BsAb | BCMA | [61,62] |
| Teclistamab | BCMA × CD3 BsAb | BCMA | [63,64] |
| REGN5458 | BCMA × CD3 BsAb | BCMA | [65] |
| TNB-383B | BCMA × CD3 BiTe | BCMA | [66] |
| BFCR4350A | FcRH5 × CD3 BiTe | FcRH5 | [67] |
| Talquetamab | GPRC5D × CD3 BsAb | GPRC5D | [68] |
| Antibody-Drug Conjugate(s) | Bispecific Antibodies | CAR-T | |
|---|---|---|---|
| Pros | “Off the shelf“ product | “Off the shelf” product | High response in the relapsed refractory setting |
| Independent from host immune function | High response in the relapsed refractory setting | Only one treatment required | |
| No delay in administration | No delay in administration | ||
| Can be given in the community setting | Can be given in the community setting? | ||
| Cons | High cost | High cost | High cost |
| Continuous therapy | Continuous therapy | Long production time (4–6 weeks) | |
| Higher doses may be required for antigen downmodulation | CRS and ICANs toxicity | CRS and ICANs toxicity | |
| Payload mediated toxicity | Requires conditioning therapy | ||
| Potential lower response rate | Require adequate lymphocyte count and function |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barilà, G.; Rizzi, R.; Zambello, R.; Musto, P. Drug Conjugated and Bispecific Antibodies for Multiple Myeloma: Improving Immunotherapies off the Shelf. Pharmaceuticals 2021, 14, 40. https://doi.org/10.3390/ph14010040
Barilà G, Rizzi R, Zambello R, Musto P. Drug Conjugated and Bispecific Antibodies for Multiple Myeloma: Improving Immunotherapies off the Shelf. Pharmaceuticals. 2021; 14(1):40. https://doi.org/10.3390/ph14010040
Chicago/Turabian StyleBarilà, Gregorio, Rita Rizzi, Renato Zambello, and Pellegrino Musto. 2021. "Drug Conjugated and Bispecific Antibodies for Multiple Myeloma: Improving Immunotherapies off the Shelf" Pharmaceuticals 14, no. 1: 40. https://doi.org/10.3390/ph14010040
APA StyleBarilà, G., Rizzi, R., Zambello, R., & Musto, P. (2021). Drug Conjugated and Bispecific Antibodies for Multiple Myeloma: Improving Immunotherapies off the Shelf. Pharmaceuticals, 14(1), 40. https://doi.org/10.3390/ph14010040